

Abstract
Respiratory infections are a threat to health and economies worldwide, yet the basis for striking variation in the severity of infection is not completely understood. Environmental exposures during development are associated with increased severity and incidence of respiratory infection later in life. Many of these exposures include ligands of the aryl hydrocarbon receptor (AHR), a transcription factor expressed by immune and nonimmune cells. In adult animals, AHR activation alters CD4+ T cells and changes immunopathology. Developmental AHR activation impacts CD4+ T-cell responses in lymphoid tissues, but whether skewed responses are also present in the infected lung is unknown. To determine whether pulmonary CD4+ T-cell responses are modified by developmental AHR activation, mice were exposed to the prototypical AHR ligand 2,3,7,8-tetrachlorodibenzo-p-dioxin during development and infected with influenza virus as adults. Lungs of exposed offspring had greater bronchopulmonary inflammation compared with controls, and activated, virus-specific CD4+ T cells contributed to the infiltrating leukocytes. These effects were CD4+ T cell subset specific, with increases in T helper type 1 and regulatory T cells, but no change in the frequency of T helper type 17 cells in the infected lung. This is in direct contrast to prior reports of suppressed conventional CD4+ T-cell responses in the lymph node. Using adoptive transfers and manipulating the pathogen properties, we determined that developmental exposure influenced factors intrinsic and extrinsic to CD4+ T cells and may involve developmentally induced changes in signals from infected lung epithelial cells. Thus developmental exposures lead to context-dependent changes in pulmonary CD4+ T-cell subsets, which may contribute to differential responses to respiratory infection.
Keywords: aryl hydrocarbon receptor, developmental exposure, influenza virus, CD4 T cell, respiratory infection
respiratory infections are a continuous burden to public health and the global economy (46). One aspect surrounding respiratory infections that is poorly understood is why there is such variation in the severity of illness among infected individuals. Environmental exposures, especially early in life, are likely overlooked factors, as exposures during development correlate with increased severity of respiratory infections later in life (12, 13, 15, 20, 22, 38). However, the cellular and molecular targets by which early life exposures have lasting effects on respiratory defenses are largely unexplored.
Some of the developmental insults that have been associated with altered responses to infection later in life contain ligands for the aryl hydrocarbon receptor (AHR). The AHR is an environment-sensing transcription factor in the Per-Arnt-Sim protein superfamily that is broadly expressed in immune and nonimmune cells, including cells of the lung (16). Although endogenous AHR ligands are not fully elucidated, numerous exogenously derived chemicals bind to the AHR. Among its ligands are many persistent environmental contaminants, such as dioxins and some polychlorinated biphenyls. In addition, some pharmaceuticals, natural products derived from dietary sources, and compounds produced by some microorganisms contain AHR ligands (24, 29, 31). In many different disease models, AHR activation in adult animals has profound effects on the magnitude and nature of immune responses (16, 26, 35). Although less extensively probed, several studies indicate that AHR signaling also influences the development of the immune system (9, 24). Moreover, inappropriate AHR activation during development alters immune responses in the adult offspring, although the majority of these effects have been characterized outside of the lung (e.g., lymphoid organs and blood) (44). A subset of these reports showed persistent changes in host responses to infection in developmentally exposed offspring (6, 21, 39, 41), yet effects on responses in the lung during infection have not been fully investigated.
One common cell lineage that is functionally modified after direct AHR activation in adult animals is CD4+ T cells (35). CD4+ T cells play vital roles in respiratory host defenses; however, many of their functions, if poorly controlled, can also cause serious immunopathology (14). During respiratory infection, signals derived from the pathogen and host tissue cause naive CD4+ T cells to become activated and differentiate into effector cells, which contribute to pathogen elimination and influence the onset and resolution of inflammation and tissue damage. Among the effector CD4+ cells present in the infected lung are conventional helper cells, including T helper type 1 (Th1), Th17, and Th2 cells. Th1 cells are the primary CD4+ subset involved in responses to pathogens that replicate within host cells, such as influenza viruses, whereas Th2 cells are important in combatting infection by some extracellular agents. Less is known about the role of Th17 cells during respiratory infections. They are important effector cells at mucosal sites, including the lung, and are detected in severe influenza virus infections, suggesting a role in tissue pathology (11, 14, 19, 27). Another subset of effector CD4+ T cells found in the infected lung is regulatory CD4+ T cells (Tregs). Their precise roles in the lung during infection are not yet fully understood, but they control the magnitude of immune responses to infectious and noninfectious agents in other tissues, suggesting they execute similar functions in the infected lung (2, 5, 11). Activation of the AHR in adult animals skews the differentiation pattern of these CD4+ T-cell subsets by altering the ratio of conventional effector CD4+ T cells to Tregs, thereby influencing immune mediated pathologies in nonpulmonary tissues (35).
It was recently reported that activation of the AHR during development alters the differentiation of CD4+ T cells later in life, such that there are fewer conventional CD4+ T cells and more Tregs in lymphoid organs (6). However, it is unknown whether developmental exposures result in similar changes in the infected lung. Prior reports support that the pulmonary immune response to infection may be deregulated by activation of the AHR during development, as there are more neutrophils, but fewer CD8+ T cells, in airways of adult mice infected with influenza A virus (21, 41). The present study further characterizes how developmental activation of the AHR modulates pulmonary immune responses by examining the CD4+ T-cell response in the lung during influenza virus infection. To better define the origins of the changes observed, we probed the consequences of developmental exposure on CD4+ T-cell responses in the lung by altering properties of the pathogen and by using reciprocal adoptive transfers. The findings presented here extend our understanding of how early life activation of the AHR contributes to increased severity of respiratory infection later in life.
METHODS
Animal treatments.
C57BL/6 (B6, CD90.2+CD45.2+) mice and B6-LY5.2/Cr (CD90.2+CD45.1+) mice (age 5 wk) were purchased from the NCI Mouse Repository (Frederick, MD). B6.PL-Thy1a/CyJ (CD90.1+CD45.2+) mice were obtained from Jackson Laboratory (Bar Harbor, ME). Nulliparous females were housed with males and checked daily for the presence of a vaginal plug, which was designated as day 0 of gestation. Impregnated mice were treated with 1 μg 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)/kg body wt or the peanut oil vehicle control (vehicle) by gavage on days 0, 7, and 14 of gestation, and 2 days postparturition. TCDD (≥99% purity; Cambridge Isotope Laboratories, Woburn, MA) was dissolved in anisole and diluted in peanut oil. The vehicle control consisted of peanut oil containing an equivalent concentration of anisole (0.01%). The effects of TCDD exposure are solely mediated by the AHR, and developmental exposure to TCDD results in AHR activation in developmentally exposed offspring (31, 45). Offspring from treated dams were weaned at 20–21 days of age. All mice were housed in microisolator cages in a specific pathogen-free facility at the University of Rochester Medical Center and were provided food and water ad libitum.
Adult offspring of TCDD- or vehicle-treated dams (6–8 wk of age) were anesthetized by intraperitoneal injection of avertin (2,2,2-tribromoethanol; Sigma Aldrich, Milwaukee, WI) for pulmonary instillation of pathogens. Influenza virus strain A/HKx31 (HKx31; H3N2) was prepared, titered, and stored as previously described (43). Mice were given a sublethal intranasal infection with 120 hemagglutinating units of live HKx31 diluted in PBS. For experiments using inactivated influenza virus, the virus was inactivated by exposure to heat (65°C, 1 h) and then UV light (4), and mice were inoculated with 200 hemagglutinating units (∼60 μg) inactivated virus intranasally. Mycobacterium bovis strain bacillus Calmette-Guérin-Pasteur (BCG; passage 3) was grown in 7H9-ADS-TY (Middlebrook 7H9 medium supplemented with 0.5% bovine serum albumin fraction V, 0.2% dextrose, 0.85% NaCl, and 0.05% Tyloxypol) and then washed and resuspended in 7H9-ADS-TY with 25% glycerol and frozen in aliquots at −80°C. The frozen stocks were titered by viable plate counts, and, when needed for infection, a stock tube was thawed and diluted in PBS/0.05% Tween 80 for an intranasal dose of 1 × 106 colony-forming unit BCG in 25 μl. At specified points in time, lungs were perfused, and lungs, spleens, and lymph nodes were removed. Single-cell suspensions were prepared as previously described (23, 30, 42). In separate experiments, cells from the airways were collected by bronchoalveolar lavage (BAL), as previously described (43). Red blood cells were removed by hypotonic lysis. Blood was collected by cardiac puncture, and serum was separated and stored at −80°C. All animal treatments and work with infectious agents were conducted with prior approval of Institutional Animal Care and Use Committee and Institutional Biosafety Committee of the University of Rochester.
Lung histology and differential cell counts.
Lungs were inflation fixed with 10% neutral-buffered formalin, embedded in paraffin, and sectioned as previously described (32). Tissue sections were stained with hematoxylin and eosin to evaluate changes in bronchopulmonary inflammation. Stained sections were visualized with an Olympus BX51 light microscope (Olympus, Melville, NY). Images were captured with SPOT RT 4.6 (Diagnostic Instruments, New Hyde Park, NY). For fluorescent immunohistochemistry, lungs were inflated with Tissue-Tek O.C.T. compound (Sakura, Torrance, CA) and frozen. Lungs were cut in 8-μm slices using a Shandon Cryotome FSE Cryostat (Thermo Scientific, Waltham, MA) and fixed in acetone. Slices were stained with fluorescently conjugated rat anti-mouse CD4 (eBioscience, San Diego, CA) and unlabeled goat anti-collagen IV IgG (Southern Biotechnology, Birmingham, AL), followed by fluorescently conjugated donkey anti-goat IgG (Jackson Immunoresearch, West Grove, PA), and mounted using ProLong Gold antifade reagent with 4,6-diamidino-2-phenylindole (Life Technologies, Grand Island, NY). Fluorescence was visualized using an Olympus BX43 conventional fluorescence microscope (Olympus America, Center Valley, PA), Retiga 1300 camera (QImaging, Surrey, BC), and ImagePro Plus Software (Media Cybernetics, Rockville, MD). Images for each sample were taken using the same magnification and exposure settings.
In other experiments, lung airways were washed, and BAL cells were obtained, as previously described (43). BAL cells were counted using a Coulter counter (Beckman Coulter, Miami, FL), and 1 × 105 cells were transferred to coded microscope slides using a cytological centrifuge. Cells were fixed and stained with hematoxylin and eosin for differential cell counting.
Flow cytometry.
Isolated cells were incubated with previously determined optimal concentrations of fluorochrome-conjugated antibodies against the following cell surface antigens: CD3ε, CD4, CD25, CD44, CD62L, CD45.1, CD45.2, and CD90.2. Where designated, before cell surface staining, cells were incubated with fluorescently conjugated major histocompatibility class II tetramers containing a peptide of HKx31 (nucleoprotein, I-Ab/NP311–325, NIAID Tetramer Core Facility). To enumerate Th1, Th17, and Tregs, cells were incubated with fluorochrome-conjugated antibodies against CD4 and CD25, fixed and permeabilized using the forkhead box protein P3 (Foxp3) staining kit (eBioscience, San Diego, CA), and subsequently incubated with fluorochrome-conjugated antibodies against T-box transcription factor TBX21 (TBet), retinoid-related orphan receptor-γt (RORγt), and Foxp3 (6). Nonspecific staining was blocked by first incubating cells with an anti-mouse CD16/32 monoclonal antibody. All antibodies were purchased from BD Biosciences (San Diego, CA) or eBiosciences (San Diego, CA). Fluorescence-minus-one controls were used to determine nonspecific fluorescence. Data were collected using an LSRII flow cytometer (BD Bioscience, San Diego, CA) and analyzed using the FlowJo software program (TreeStar, Ashland, OR).
Adoptive transfers.
Naive CD4+ T cells from peripheral lymph nodes of adult offspring of vehicle and TCDD-treated dams were harvested and processed under aseptic conditions, as previously described (6). Briefly, CD4+ T cells were enriched and sorted (FACSAria, BD Bioscience, San Diego, CA) to obtain CD4+ CD44lo (naive) cells (≥95% purity). CD44loCD4+ T cells (5 × 105) were transferred intravenously into recipient mice. For dual adoptive transfers, CD44loCD4+ T cells from each treatment group were combined in a 1:1 ratio and cotransferred into the same recipient (i.e., recipients received 5 × 105 naive CD4+ T cells; 2.5 × 105 from each donor). Recipients were infected with influenza virus 36 h posttransfer, and cells were harvested from the lung 9 days later.
Statistical analysis.
The dam is defined as the statistical unit for all experiments, as the dam, not her pups, is directly treated with TCDD or vehicle control. Treatment groups were composed of adult offspring, each from a different treated dam. Data were analyzed using JMP software (SAS Software, Cary, NC). Differences between two groups were evaluated using a Student's t-test and considered significant when P values were ≤0.05. Error bars on all graphs represent the SE of the mean. All experiments were independently repeated at least once with similar results.
RESULTS
Developmental activation of the AHR enhances inflammation in the infected lung.
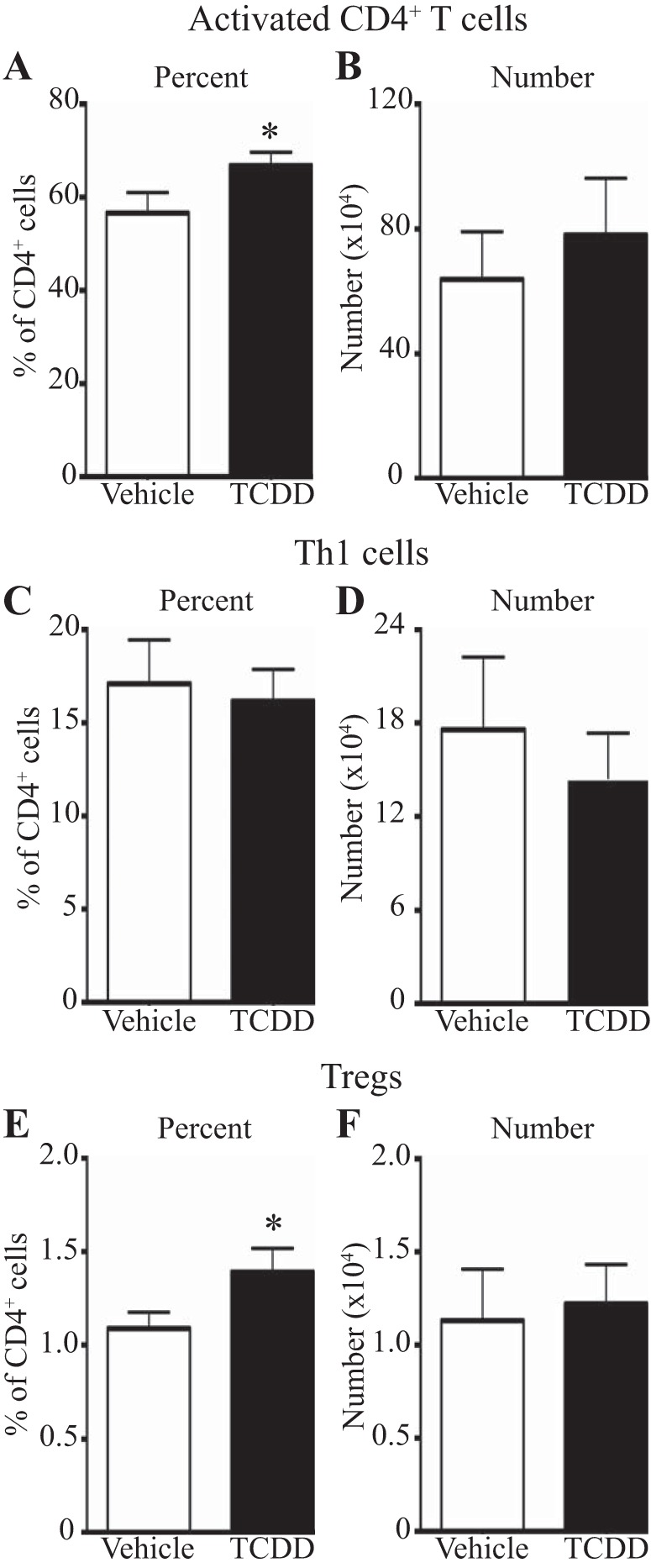
Adult mice that were developmentally exposed to TCDD or the vehicle control were infected with a sublethal dose of influenza A virus (HKx31, H3N2), and lung inflammation was examined. Infection with influenza virus results in an influx of leukocytes to the lung airways and alveolar spaces. Compared with offspring of control dams, infected offspring of TCDD-treated dams had an increase in the amount of infiltrating leukocytes in their lungs, both near the large airways and in alveolar regions (Fig. 1A). Differential cell counts were performed to examine the types of cells infiltrating the infected lungs of developmentally exposed mice. The number of neutrophils was significantly higher in lungs of offspring of TCDD-treated dams compared with control offspring, yet there was no change in the number of macrophages or lymphocytes (Fig. 1B). Two of the main lymphocyte populations recruited to the lung during influenza infection are CD4+ and CD8+ T cells (25). Previous work showed a reduction in the frequency of CD8+ T cells in the airways of adult mice that were developmentally exposed to TCDD (41). Therefore, we determined whether the lack of change in the number of total lymphocytes in the lungs of developmentally exposed mice was due to changes in the CD4+ T-cell response. Specifically, immunofluorescent histochemistry and flow cytometry were used to evaluate CD4+ T cells in the influenza virus-infected lung. CD4+ T cells were detected throughout the infected lung, and there were no differences in their distribution in situ (Fig. 1C). However, compared with infected offspring of control dams, infected mice that were developmentally exposed had approximately a 30% increase in the percentage (Fig. 1D) and number (Fig. 1E) of activated (CD44hiCD62Llo) CD4+ T cells in their lungs. The increase in activated CD4+ T cells likely reflects a greater number in both the airways and interstitial spaces, because, when each compartment was examined separately, the effect of developmental exposure was attenuated (data not shown). We also measured the abundance of virus-specific CD4+ T cells in the lungs of developmentally exposed mice using fluorescently conjugated major histocompatibility class II tetramers loaded with a viral peptide (I-AbNP311–325). While there was no difference in the percentage of virus-specific CD4+ T cells, there was a twofold increase in the number of these cells in lungs of infected offspring of TCDD-treated dams compared with vehicle controls (Fig. 1, F and G). Thus developmental activation of the AHR leads to increased bronchopulmonary inflammation after influenza virus infection, and activated, virus-specific CD4+ T cells contribute to the increase in infiltrating immune cells.
Fig. 1.

Developmental activation of the aryl hydrocarbon receptor (AHR) leads to increased bronchopulmonary inflammation after influenza virus infection in adult offspring. Mice were developmentally exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) or vehicle and infected (intranasally) with influenza A virus as adults, as described in methods. A: representative images of bronchopulmonary inflammation around the airways (top row) and in alveolar regions (bottom row) of mice developmentally exposed to vehicle (left) or TCDD (right) on day 9 postinfection are shown. B: the graph shows the number of monocyte/macrophages (Mac), lymphocytes (Lymph), and neutrophils (Neut) in bronchoalveolar lavage (BAL) fluid obtained from developmentally exposed mice on day 8 postinfection. V, vehicle; T, TCDD. C: representative images from lungs from mice developmentally exposed to vehicle (left) or TCDD (right) on day 9 postinfection. D–G: 9 days after infection, lung-derived immune cells were stained for flow cytometry. Activated CD4+ T cells and virus-specific CD4+ T cells were defined as CD44hiCD62LloCD3+CD4+ and I-AbNP311–325+CD3+CD4+, respectively. Representative dot plots show the percentage of activated (D) and virus-specific (F) CD4+ T cells. The bar graphs depict the number of activated (E) and virus-specific (G) CD4+ T cells. Values are means ± SE; for each experiment, n = 3–8 same-sex offspring per group from separately treated dams. *P value ≤ 0.05. DAPI, 4,6-diamidino-2-phenylindole.
Developmental exposure increases the frequency of pulmonary effector CD4+ T cells after influenza virus infection.
During influenza virus infection, CD4+ T cells can differentiate into conventional helper cells (e.g., Th1 and Th17 cells) or Tregs, which traffic from the lymph node to the infected lung (36). Developmental activation of the AHR alters the proportion of CD4+ T-cell subsets in lymphoid tissues after infection, leading to a decrease in activated and conventional CD4+ T-cell subsets, but an increase in Tregs (6). It is unknown whether this skewing translates to the response in the influenza virus-infected lung. Consequently, we determined the proportion of CD4+ T-cell subsets in the virally infected lung of developmentally exposed mice. Th1 cells are the most abundant CD4+ T-cell subset generated during primary influenza virus infection, are defined by the transcription factor responsible for driving their lineage (TBet), and reach their peak number in the lung on the 9th day after infection (10, 47). Activation of the AHR during development leads to a twofold increase in the percentage (Fig. 2A) and number (Fig. 2B) of Th1 cells in the infected lung. Th17 cells are defined by their expression of the transcription factor RORγt and are numerically the second most abundant subset found in the influenza virus-infected lung (19, 27). In contrast to enhanced Th1 cells, developmental exposure to TCDD does not significantly alter the percentage or number of Th17 cells in the lung 9 days after influenza virus infection (Fig. 2, C and D), nor any other point in time examined (data not shown). Tregs, which are defined as Foxp3+CD25+CD4+ T cells, are critical mediators in the lung during infection (1, 5, 17). While the percentage of Tregs in the infected lung is not significantly affected by developmental exposure to TCDD (Fig. 2E), the number of Tregs is doubled compared with that in control offspring (Fig. 2F). Together, these data show that developmental activation of the AHR increases the number of Th1 cells and Tregs, but not Th17 cells, in the influenza virus-infected lung.
Fig. 2.
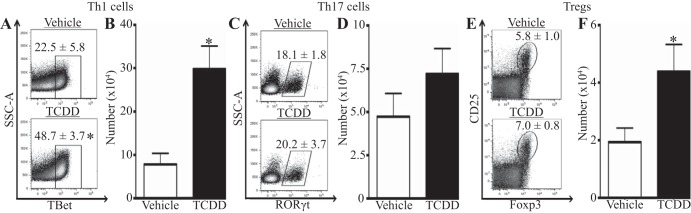
Activation of the AHR during development increases the frequency of T helper type 1 (Th1) cells and regulatory CD4+ T cells (Tregs) in the infected lung. Mice were developmentally exposed and infected with influenza virus at maturity. Cell suspensions of lung-derived immune cells were prepared and stained for flow cytometry. CD4+ T-cell subsets are defined as follows: Th1 cells: TBet+CD4+; Th17 cells: RORγt+CD4+; and Tregs: Foxp3+CD25+CD4+. Representative dot plots depict the percentage of Th1 cells (A), Th17 cells (C), and Tregs (E). The bar graph shows the number of Th1 cells (B), Th17 cells (D), and Tregs (F). Values are means ± SE; n = 5–6 offspring per treatment from separately treated dams. *P value ≤ 0.05. SSC-A, side scatter area; TBet, T-box transcription factor TBX21; RORγt, retinoid-related orphan receptor-γt; Foxp3, forkhead box protein P3.
Intrinsic and extrinsic effects of AHR activation on the CD4+ T-cell lineage influence their response to pulmonary infection.
Activation of the AHR during development does not alter the proportions of CD4+ T-cell subsets before infection (data not shown). This suggests that changes in CD4+ T cells imparted by developmental exposure to TCDD are revealed after the immune system is activated by influenza virus infection. However, the signals altered by developmental activation of the AHR that lead to changes in the pulmonary CD4+ T-cell response are unknown. To begin to further define infection-associated signals, we determined whether developmental activation of the AHR leads to an increase in CD4+ T cells in the lung via intrinsic or extrinsic changes in the CD4+ T-cell lineage. To examine whether factors extrinsic to CD4+ T cells contribute to their increase in the lung, we transferred naive, unexposed CD4+ T cells into recipients that were offspring of vehicle or TCDD-treated dams. Donor cells were distinguished using congenic markers, as donor cells were CD45.1+ and recipient cells were CD45.2+ (Fig. 3A). Developmental exposure of the recipients did not affect the success of the transfers, as determined by the number of transferred CD4+ T cells recovered from uninfected recipients (data not shown). However, 9 days after infection, there was a significant increase in the percentage, but not number, of activated CD4+ T cells (Fig. 3, B and E) and Th1 cells (Fig. 3, C and F) from unexposed donors in recipient mice developmentally exposed to TCDD compared with vehicle-exposed recipients. The proportion and number of transferred CD4+ T cells that were Tregs did not differ significantly between recipient exposure groups (Fig. 3, D and G).
Fig. 3.

The enhanced CD4+ T-cell response to infection is due to factors extrinsic and intrinsic to the CD4 lineage. CD90.2+CD45.1+ and CD90.2+CD45.2+ mice were exposed to vehicle or TCDD during development as described in methods. A: naive CD44loCD4+ T cells were isolated from unexposed and uninfected CD90.2+CD45.1+ mice and transferred into adult CD90.2+CD45.2+ offspring of vehicle or TCDD-treated dams. Donor and recipient cells were distinguished by congenic markers. Thirty-six hours posttransfer, recipients were infected with influenza virus. Lung-derived immune cells were stained for flow cytometry 9 days after infection. Cell populations were defined as in Figs. 1 and 2. B–D: the percentage of transferred CD4+ T cells that had an activated (B), Th1 (C), or Tregs (D) phenotype is graphed. E–G: the number of activated CD4+ T cells (E), Th1 cells (F), or Tregs (G) is shown. H: CD44loCD4+ cells were isolated from adult developmentally exposed offspring and transferred at a 1:1 ratio into unexposed CD45.2+CD90.1+ recipients. Cells from vehicle-exposed offspring were CD45.1+CD90.2+, and cells from offspring of TCDD-treated dams were CD45.2+CD90.2+. I–N: recipients were infected 36 h after transfer, and on day 9 postinfection lung cells were stained for flow cytometry as in Figs. 1 and 2. I–K: the graphs depict the percentage of activated CD4+ T cells (I), Th1 cells (J), and Tregs (K). L–N: the number of activated CD4+ T cells (L), Th1 cells (M), and Tregs (N) are shown. Values are means ± SE; n = 5–10 recipients used in each experiment. *P value ≤ 0.05.
Extrinsic influences of developmental activation of the AHR on CD4+ T cells may not be the only forces that contribute to alterations in the influenza virus-infected lung. In fact, developmental activation of the AHR has been shown to lead to intrinsic changes in CD4+ T cells in lymphoid tissue (6). Therefore, we investigated the contribution of intrinsic differences in CD4+ T cells after developmental exposure to TCDD on the pulmonary CD4+ T-cell response to influenza virus infection. Naive CD4+ T cells from offspring of vehicle and TCDD-treated dams were isolated, combined in a one-to-one ratio, and transferred into unexposed recipients. Donor cells were identified by differential expression of congenic markers. Naive CD4+ T cells from adult offspring developmentally exposed to vehicle were CD45.1+CD90.2+, those from offspring of TCDD-treated dams were CD45.2+CD90.2+, and recipients were CD45.2+CD90.1+ (Fig. 3H). Thirty-six hours after transfer, recipients were infected with influenza virus. There was no difference in the percentage (Fig. 3I) or number (Fig. 3L) of activated CD4+ T cells from donors that had been developmentally exposed to vehicle or TCDD in the lungs of recipient mice on day 9 postinfection. However, the percentage of Th1 cells was significantly increased when CD4+ T cells came from donors that were developmentally exposed to TCDD (Fig. 3J), although the number of Th1 cells derived from the two donor groups was not statistically different (Fig. 3M). Similar to the reciprocal adoptive transfers, neither the percentage (Fig. 3K) nor number (Fig. 3N) of Tregs was different between the two treatment groups of donor cells. Together, these data suggest that the increase in Th1 cells in the lungs of influenza virus-infected, developmentally exposed mice is due to changes that are both intrinsic and extrinsic to the CD4 lineage. The increase in activated CD4+ T cells arises from extrinsic effects of AHR activation during development, whereas the increase in Tregs requires a combination of effects of developmental AHR activation that is both intrinsic and extrinsic to CD4+ T cells.
A live, infectious pathogen is required for the enhanced pulmonary CD4+ T-cell response in developmentally exposed mice.
It is unknown whether the enhancement in CD4+ T cells in the lung of developmentally exposed mice is a generalized response to any pulmonary insult, or if it is specific to the response to influenza virus infection. To test whether the changes are influenza virus specific, we infected developmentally exposed mice with a different model respiratory pathogen that also elicits a strong Th1 response: Mycobacterium bovis BCG (3). Developmentally exposed mice were infected with BCG intranasally, and their activated CD4+ T-cell response was examined on day 28 postinfection, which is the peak of the CD4+ T-cell response (data not shown and Ref. 18). Similar to influenza virus infection, the percentage of activated CD4+ T cells was significantly higher in lungs of infected offspring of TCDD-exposed dams (Fig. 4A). However, in contrast to influenza virus, there was not a significant difference in the number of activated CD4+ T cells (Fig. 4B), nor in the percentage or number of Th1 cells (Fig. 4, C and D), in lungs of BCG-infected mice that were developmentally exposed to TCDD, or the vehicle control. Following BCG infection, the percentage of Tregs in the lung was significantly increased in offspring developmentally exposed to TCDD compared with vehicle control mice (Fig. 4E); however, the number of Tregs in the lung was the same in both groups (Fig. 4F). These observations suggest that the presence of a pulmonary infection that drives a strong Th1 response may be sufficient to reveal some aspects of the enhanced CD4+ T-cell response in the lungs of mice developmentally exposed to TCDD, but cannot be generalized to all aspects of the altered CD4+ T-cell response.
Fig. 4.
Developmental activation of the AHR increases the proportion of activated CD4+ T cells and Tregs, but not Th1 cells, after bacillus Calmette-Guérin-Pasteur (BCG) infection. Mice were developmentally exposed and infected (intranasally) with BCG, as described in methods. On day 28 postinfection, lung immune cell suspensions were stained for flow cytometry, as described in Figs. 1 and 2. The percentage (A, C, and E) and number (B, D, and F) of activated CD4+ T cells (A and B), Th1 cells (C and D), and Tregs (E and F) are shown. Values are means ± SE; n = 6–8 adult offspring from vehicle or TCDD-treated dams. *P value ≤ 0.05.
To determine whether an infectious, replicating pathogen is required for the increase in CD4+ T-cell responses in the lung after respiratory infection, developmentally exposed mice were inoculated with inactivated influenza virus intranasally, and the CD4+ T cells in the lung were measured. By day 9 postinoculation, CD4+ T cells have expanded at least 1.5- to 4-fold compared with the number of cells recovered from the lung of naive mice (data not shown). On day 9 postinoculation, the percentage (Fig. 5A) and number (Fig. 5B) of activated CD4+ T cells were reduced in mice developmentally exposed to TCDD compared with controls. There were no differences in the percentage (Fig. 5C) or number (Fig. 5D) of Th1 cells, nor were there changes in the percentage (Fig. 5E) or number (Fig. 5F) of Tregs. Therefore, the increased frequency of activated and effector CD4+ T cells in the lungs of mice that were developmentally exposed likely requires a live, replicating pathogen to be revealed.
Fig. 5.

The CD4+ T-cell response to inactivated influenza virus is not enhanced after developmental exposure to TCDD. Adult offspring of vehicle and TCDD treated dams were inoculated (intranasally) with inactivated influenza virus. Nine days later, lung-derived immune cells were stained for flow cytometry. The percentage (A, C, and E) and number (B, D, and F) of CD4+ T cells with an activated phenotype (A and B), Th1 cells (C and D), and Tregs (E and F) are shown. Values are means ± SE; n = offspring of 5–6 separately treated dams. *P value ≤ 0.05.
DISCUSSION
Despite vaccination and improvements to health care infrastructure, respiratory infections, such as influenza viruses, remain a public health and economic burden. Several epidemiological studies show that developmental exposures to pollutants that bind to the AHR correlate with increased incidence or severity of respiratory infections later in life, but why this occurs is unknown (12, 13, 15, 20, 22, 38). There is a growing appreciation for the role of CD4+ T cells in the pulmonary response to primary influenza virus infection, particularly when the CD8+ T-cell response is blunted (7), as is the case after developmental exposure to TCDD (21, 41). In this study, we used a mouse model to examine if the pulmonary immune response to influenza virus is modified after developmental exposure to the signature AHR ligand TCDD. We present the novel finding that the CD4+ T-cell response to infection is enhanced in lungs of mice developmentally exposed to TCDD. These findings could likely be extended to developmental exposure to other AHR ligands that result in sustained activation of the receptor. Modulation of the CD4+ T-cell response as a result of AHR activation during development could have a substantial impact on the progression and resolution of infection (14). Previous work showed that, in lymphoid tissues of developmentally exposed mice, the response of conventional CD4+ T cells during infection was suppressed rather than enhanced (6). Therefore, the consequences of developmental exposure on CD4+ T cells are context dependent. Moreover, these findings suggest that tissue-derived influences altered by developmental exposures have the ability to impact the immune response to a pathogen. The implications of these findings extend beyond immune response to infections, particularly because CD4+ T cells are implicated in the pathogenesis of both allergic and autoimmune responses (48).
Developmental activation of the AHR increases CD4+ T cells in the lung with an activated phenotype, Th1 cells, and Tregs after pulmonary infection. Yet the factors driving the enhancement of these three cell types varied. The increased proportion of activated CD4+ T cells is due to influences of developmental AHR activation extrinsic to these cells. This is similar to what has been reported for the enhanced pulmonary neutrophilia in developmentally exposed mice infected with influenza virus (21). Extrinsic factors likely to be involved in mediating changes in the CD4+ T-cell response to infection include changes in other immune cells, including dendritic cells, and altered signals from cells of the lung. All of these cells express the AHR and can be modulated by its activation in adult animals (16). The increase in Th1 cells can occur as a result of intrinsic changes in the CD4 lineage, or due to changes in cells extrinsic to the CD4 lineage after developmental AHR activation. However, a combination of intrinsic and extrinsic influences of developmental exposure is required for the increase in Tregs to occur. Studies examining the effect of direct AHR activation on CD4+ T cells in adult animals have reported a similar duality in the factors that influence skewed differentiation (16, 35). Furthermore, both intrinsic and extrinsic effects of AHR activation during development influence CD4+ T cells in lymphoid tissue (6). It is not surprising that each subset of CD4+ T cells responds distinctly to alterations in tissue-specific and infection-driven cues due to AHR activation, as the signals required for the commitment and maintenance of each lineage are very different (36, 47). The reduction of conventional CD4+ T-cell subsets in the lymph node of developmentally exposed mice, yet increase in these same cell subsets in the lung, suggests changes in cell death, recruitment, or proliferation. All of these factors could occur due to exposure-mediated influences intrinsic and extrinsic to the CD4 lineage, and each subset may be affected by a unique combination of one or more of these factors. Activation of the AHR in CD4+ T cells of adult animals has been shown to alter genes involved in lineage commitment, cell cycle regulation, and apoptosis (8, 28, 33, 34, 37), emphasizing the potential that one or more of these pathways may also be altered after developmental exposure in a lineage-specific fashion.
Active pulmonary infection is required for the increase in effector CD4+ T cells in the lung after developmental exposure to TCDD. However, all aspects of the enhanced frequency of CD4+ T cells after respiratory infection are not observed in both influenza virus and BCG infection. This may partly be due to the fact that the overall magnitude of the CD4+ T-cell response to BCG infection is smaller than the response to influenza virus infection, as differences in pathogenicity and antigen density have an impact on how CD4+ T cells function in pulmonary tissue (40). Additionally, the cells that interact with the pathogen play an important role in the initiation of an immune response (25), and they may be modified by developmental activation of the AHR. In fact, developmentally exposed mice given influenza virus intraperitoneally, where it cannot replicate and does not interact with cells of the lung, exhibited no changes in CD4+ T-cell number, activation status, or subset distribution in the spleen at the peak of their response (data not shown). Thus the signals derived from the antigen and the immunogenicity of each immunological challenge reveal important differences in how the response of CD4+ T cells is altered as a result of developmental AHR activation.
A deregulated immune response has profound consequences for the host, including immune-mediated pathology and further dissemination of the pathogen, both of which can lead to worsened disease and even death. In this study, we determined that developmental activation of the AHR causes an increase in CD4+ T cells in the lung after respiratory viral infection, which is due to a combination of effects that are intrinsic and extrinsic to the CD4 lineage. These findings suggest developmental exposures likely affect many cell types, which contribute to altered immune responses later in life. This emphasizes the importance of work examining the contribution of cell-type and tissue-specific consequences of developmental exposures on immune responses later in life.
GRANTS
This work was supported by research and training grants from the National Institutes of Health (R01-ES017250, R01-HL097141, T32-ES07026, T32-AI007285, T32-HL066988, R01-AI073772, and P30-ES01247) and funds from the University of Rochester.
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: L.A.B., B.W., K.L., B.A.V., and M.S.P. performed experiments; L.A.B., B.W., B.A.V., and B.P.L. analyzed data; L.A.B. and B.P.L. interpreted results of experiments; L.A.B. prepared figures; L.A.B. and B.P.L. drafted manuscript; L.A.B., D.J.T., M.S.P., and B.P.L. edited and revised manuscript; L.A.B., B.W., K.L., B.A.V., D.J.T., M.S.P., and B.P.L. approved final version of manuscript; B.P.L. conception and design of research.
ACKNOWLEDGMENTS
The authors thank Dr. Deborah Fowell for advice on CD4+ T-cell subset analyses, and Catherine Burke for thoughtful discussion and helpful critique of this manuscript. We are also grateful to Dr. Timothy Bushnell and the outstanding team at the URMC Flow Cytometry Core, and the staff at the NIAID Tetramer Core Facility.
REFERENCES
- 1.Antunes I, Kassiotis G. Suppression of innate immune pathology by regulatory T cells during Influenza A virus infection of immunodeficient mice. J Virol 84: 12564–12575, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bedoya F, Cheng GS, Leibow A, Zakhary N, Weissler K, Garcia V, Aitken M, Kropf E, Garlick DS, Wherry EJ, Erikson J, Caton AJ. Viral antigen induces differentiation of Foxp3+ natural regulatory T cells in influenza virus-infected mice. J Immunol 190: 6115–6125, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Behar SM, Carpenter SM, Booty MG, Barber DL, Jayaraman P. Orchestration of pulmonary T cell immunity during Mycobacterium tuberculosis infection: immunity interruptus. Semin Immunol 26: 559–577, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bender A, Bui LK, Feldman MA, Larsson M, Bhardwaj N. Inactivated influenza virus, when presented on dendritic cells, elicits human CD8+ cytolytic T cell responses. J Exp Med 182: 1663–1671, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Betts RJ, Prabhu N, Ho AW, Lew FC, Hutchinson PE, Rotzschke O, Macary PA, Kemeny DM. Influenza A virus infection results in a robust, antigen-responsive, and widely disseminated Foxp3+ regulatory T cell response. J Virol 86: 2817–2825, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boule LA, Winans B, Lawrence BP. Effects of developmental activation of the AhR on CD4+ T-cell responses to influenza virus infection in adult mice. Environ Health Perspect 122: 1201–1208, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown DM, Roman E, Swain SL. CD4 T cell responses to influenza infection. Semin Immunol 16: 171–177, 2004. [DOI] [PubMed] [Google Scholar]
- 8.Camacho IA, Singh N, Hegde VL, Nagarkatti M, Nagarkatti PS. Treatment of mice with 2,3,7,8-tetrachlorodibenzo-p-dioxin leads to aryl hydrocarbon receptor-dependent nuclear translocation of NF-kappaB and expression of Fas ligand in thymic stromal cells and consequent apoptosis in T cells. J Immunol 175: 90–103, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Casado FL, Singh KP, Gasiewicz TA. The aryl hydrocarbon receptor: regulation of hematopoiesis and involvement in the progression of blood diseases. Blood Cells Mol Dis 44: 199–206, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chapman TJ, Castrucci MR, Padrick RC, Bradley LM, Topham DJ. Antigen-specific and non-specific CD4+ T cell recruitment and proliferation during influenza infection. Virology 340: 296–306, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Chen K, Kolls JK. T cell-mediated host immune defenses in the lung. Annu Rev Immunol 31: 605–633, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dallaire F, Dewailly E, Muckle G, Vezina C, Jacobson SW, Jacobson JL, Ayotte P. Acute infections and environmental exposure to organochlorines in Inuit infants from Nunavik. Environ Health Perspect 112: 1359–1365, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dallaire F, Dewailly E, Vezina C, Muckle G, Weber JP, Bruneau S, Ayotte P. Effect of prenatal exposure to polychlorinated biphenyls on incidence of acute respiratory infections in preschool Inuit children. Environ Health Perspect 114: 1301–1305, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Damjanovic D, Small CL, Jeyanathan M, McCormick S, Xing Z. Immunopathology in influenza virus infection: uncoupling the friend from foe. Clin Immunol 144: 57–69, 2012. [DOI] [PubMed] [Google Scholar]
- 15.Dewailly E, Ayotte P, Bruneau S, Gingras S, Belles-Isles M, Roy R. Susceptibility to infections and immune status in Inuit infants exposed to organochlorines. Environ Health Perspect 108: 205–211, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esser C, Rannug A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol Rev 67: 259–279, 2015. [DOI] [PubMed] [Google Scholar]
- 17.Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nat Immunol 6: 331–337, 2005. [DOI] [PubMed] [Google Scholar]
- 18.Fulton SA, Martin TD, Redline RW, Henry Boom W. Pulmonary immune responses during primary mycobacterium bovis-Calmette-Guerin bacillus infection in C57Bl/6 mice. Am J Respir Cell Mol Biol 22: 333–343, 2000. [DOI] [PubMed] [Google Scholar]
- 19.Gallorini S, Taccone M, Bonci A, Nardelli F, Casini D, Bonificio A, Kommareddy S, Bertholet S, O'Hagan DT, Baudner BC. Sublingual immunization with a subunit influenza vaccine elicits comparable systemic immune response as intramuscular immunization, but also induces local IgA and TH17 responses. Vaccine 32: 2382–2388, 2014. [DOI] [PubMed] [Google Scholar]
- 20.Glynn A, Thuvander A, Aune M, Johannisson A, Darnerud PO, Ronquist G, Cnattingius S. Immune cell counts and risks of respiratory infections among infants exposed pre- and postnatally to organochlorine compounds: a prospective study. Environ Health 7: 62, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hogaboam JP, Moore AJ, Lawrence BP. The aryl hydrocarbon receptor affects distinct tissue compartments during ontogeny of the immune system. Toxicol Sci 102: 160–170, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jedrychowski W, Galas A, Pac A, Flak E, Camman D, Rauh V, Perera F. Prenatal ambient air exposure to polycyclic aromatic hydrocarbons and the occurrence of respiratory symptoms over the first year of life. Eur J Epidemiol 20: 775–782, 2005. [DOI] [PubMed] [Google Scholar]
- 23.Jin GB, Winans B, Martin KC, Lawrence BP. New insights into the role of the aryl hydrocarbon receptor in the function of CD11c cells during respiratory viral infection. Eur J Immunol 44: 1685–1698, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kiss EA, Vonarbourg C. Aryl hydrocarbon receptor: a molecular link between postnatal lymphoid follicle formation and diet. Gut Microbes 3: 577–582, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kohlmeier JE, Woodland DL. Immunity to respiratory viruses. Annu Rev Immunol 27: 61–82, 2009. [DOI] [PubMed] [Google Scholar]
- 26.Lawrence BP, Vorderstrasse BA. New insights into the aryl hydrocarbon receptor as a modulator of host responses to infection. Semin Immunopathol 35: 615–626, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maroof A, Yorgensen YM, Li Y, Evans JT. Intranasal vaccination promotes detrimental Th17-mediated immunity against influenza infection. PLoS Pathog 10: e1003875, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol 185: 3190–3198, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moura-Alves P, Fae K, Houthuys E, Dorhoi A, Kreuchwig A, Furkert J, Barison N, Diehl A, Munder A, Constant P, Skrahina T, Guhlich-Bornhof U, Klemm M, Koehler AB, Bandermann S, Goosmann C, Mollenkopf HJ, Hurwitz R, Brinkmann V, Fillatreau S, Daffe M, Tummler B, Kolbe M, Oschkinat H, Krause G, Kaufmann SH. AhR sensing of bacterial pigments regulates antibacterial defence. Nature 512: 387–392, 2014. [DOI] [PubMed] [Google Scholar]
- 30.Neff-LaFord H, Teske S, Bushnell TP, Lawrence BP. Aryl hydrocarbon receptor activation during influenza virus infection unveils a novel pathway of IFN-gamma production by phagocytic cells. J Immunol 179: 247–255, 2007. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol 21: 102–116, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O'Reilly MA, Marr SH, Yee M, McGrath-Morrow SA, Lawrence BP. Neonatal hyperoxia enhances the inflammatory response in adult mice infected with influenza A virus. Am J Respir Crit Care Med 177: 1103–1110, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oberg M, Bergander L, Hakansson H, Rannug U, Rannug A. Identification of the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole, in cell culture medium, as a factor that controls the background aryl hydrocarbon receptor activity. Toxicol Sci 85: 935–943, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Poland A, Glover E. Chlorinated biphenyl induction of aryl hydrocarbon hydroxylase activity: a study of the structure-activity relationship. Mol Pharmacol 13: 924–938, 1977. [PubMed] [Google Scholar]
- 35.Quintana FJ, Sherr DH. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol Rev 65: 1148–1161, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Semerad CL, Liu F, Gregory AD, Stumpf K, Link DC. G-CSF is an essential regulator of neutrophil trafficking from the bone marrow to the blood. Immunity 17: 413–423, 2002. [DOI] [PubMed] [Google Scholar]
- 37.Singh NP, Singh UP, Singh B, Price RL, Nagarkatti M, Nagarkatti PS. Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of FoxP3 and IL-17 expression and amelioration of experimental colitis. PLoS One 6: e23522, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stolevik SB, Nygaard UC, Namork E, Haugen M, Meltzer HM, Alexander J, Knutsen HK, Aaberge I, Vainio K, van Loveren H, Lovik M, Granum B. Prenatal exposure to polychlorinated biphenyls and dioxins from the maternal diet may be associated with immunosuppressive effects that persist into early childhood. Food Chem Toxicol 51: 165–172, 2013. [DOI] [PubMed] [Google Scholar]
- 39.Sugita-Konishi Y, Kobayashi K, Naito H, Miura K, Suzuki Y. Effect of lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin on the susceptibility to Listeria infection. Biosci Biotechnol Biochem 67: 89–93, 2003. [DOI] [PubMed] [Google Scholar]
- 40.Torabi-Parizi P, Vrisekoop N, Kastenmuller W, Gerner MY, Egen JG, Germain RN. Pathogen-related differences in the abundance of presented antigen are reflected in CD4+ T cell dynamic behavior and effector function in the lung. J Immunol 192: 1651–1660, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vorderstrasse BA, Cundiff JA, Lawrence BP. Developmental exposure to the potent aryl hydrocarbon receptor agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin Impairs the cell-mediated immune response to infection with influenza a virus, but enhances elements of innate immunity. J Immunotoxicol 1: 103–112, 2004. [DOI] [PubMed] [Google Scholar]
- 42.Vorderstrasse BA, Cundiff JA, Lawrence BP. A dose-response study of the effects of prenatal and lactational exposure to TCDD on the immune response to influenza a virus. J Toxicol Environ Health A 69: 445–463, 2006. [DOI] [PubMed] [Google Scholar]
- 43.Warren TK, Mitchell KA, Lawrence BP. Exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) suppresses the humoral and cell-mediated immune responses to influenza A virus without affecting cytolytic activity in the lung. Toxicol Sci 56: 114–123, 2000. [DOI] [PubMed] [Google Scholar]
- 44.Winans B, Humble MC, Lawrence BP. Environmental toxicants and the developing immune system: a missing link in the global battle against infectious disease? Reprod Toxicol 31: 327–336, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winans B, Nagari A, Chae M, Post CM, Ko CI, Puga A, Kraus WL, Lawrence BP. Linking the aryl hydrocarbon receptor with altered dna methylation patterns and developmentally induced aberrant antiviral CD8+ T cell responses. J Immunol 194: 4446–4457, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.World Health Organization. The Top 10 Causes of Death (Online). http://www.who.int/mediacentre/factsheets/fs310/en/index1.html [February 2015].
- 47.Yamane H, Paul WE. Early signaling events that underlie fate decisions of naive CD4(+) T cells toward distinct T-helper cell subsets. Immunol Rev 252: 12–23, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood 112: 1557–1569, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]