

Abstract
Human airway smooth muscle cells (HASMC) contribute to asthma pathophysiology through an increased smooth muscle mass and elevated cytokine/chemokine output. Little is known about how HASMC and the airway epithelium interact to regulate chronic airway inflammation and remodeling. Amphiregulin is a member of the family of epidermal growth factor receptor (EGFR) agonists with cell growth and proinflammatory roles and increased expression in the lungs of asthma patients. Here we show that bradykinin (BK) stimulation of HASMC increases amphiregulin secretion in a mechanism dependent on BK-induced COX-2 expression, increased PGE2 output, and the stimulation of HASMC EP2 and EP4 receptors. Conditioned medium from BK treated HASMC induced CXCL8, VEGF, and COX-2 mRNA and protein accumulation in airway epithelial cells, which were blocked by anti-amphiregulin antibodies and amphiregulin siRNA, suggesting a paracrine effect of HASMC-derived amphiregulin on airway epithelial cells. Consistent with this, recombinant amphiregulin induced CXCL8, VEGF, and COX-2 in airway epithelial cells. Finally, we found that conditioned media from amphiregulin-stimulated airway epithelial cells induced amphiregulin expression in HASMC and that this was dependent on airway epithelial cell COX-2 activity. Our study provides evidence of a dynamic axis of interaction between HASMC and epithelial cells that amplifies CXCL8, VEGF, COX-2, and amphiregulin production.
Keywords: amphiregulin, COX-2, CXCL8, epithelial, mesenchymal
inflammation and airway remodeling are central to the pathology of chronic asthma. In asthma key features are airway epithelial damage, abnormal repair, and a greater airway smooth muscle mass with amplified cytokine secretion, both enhancing chronic airway inflammation and remodeling (4, 18, 22). A great deal of information has emerged on how these cells function individually; however, there is much less information on how they interact and the role these interactions play in the inflammation and remodeling processes.
The epidermal growth factor receptor family (EGFR) plays a central role in maintaining the airway epithelium as an environmental barrier and as a trigger for inflammation (2, 3, 6, 28). In the asthmatic lung there is increased epithelial EGFR expression and activity (36, 37). The natural ligands of EGFR include epidermal growth factor (EGF), transforming growth factor-α (TGF-α), heparin binding epidermal growth factor (HB-EGF), betacellulin, epiregulin, amphiregulin, and the neuregulins 1 to 4. Two recent clinical studies demonstrated that the asthmatic lung contains more amphiregulin than the normal lung (15, 20). Amphiregulin has several properties relevant to asthma. It has been classified as a type-2 cytokine by virtue of its essential role in TH2 lymphocytes' inflammatory resistance to nematode infection (50). Amphiregulin expression is rapidly induced by allergen treatment of mast cells derived from asthma patients but not from normal subjects (32). Furthermore, rhinoviral and respiratory syncytial virus (RSV)-induced release of CXCL8 from the airway epithelium is dependent on protease triggered amphiregulin release (26, 28, 31).
Airway epithelial and smooth muscle cells both express high levels of prostaglandin endoperoxide synthase-2 (cyclooxygenase-2 or COX-2) (12, 40, 46). COX-2 products have a number of functional roles in asthma. High levels of inhaled PGE2 can reverse airway constriction during acute asthma attacks, and genetic ablation or pharmacological inhibition of COX-2 results in increased lung inflammation. However, PGE2 may also have a proinflammatory role. Genetic ablation of the PGE2 synthase (mPGES1) gene in mice results in a decrease in allergen-induced lung inflammation (11) and a reduction in systemic inflammation (49).
Bradykinin (BK) is a peptide produced by protease degradation of high-molecular-weight or low-molecular-weight kininogens. Kininogen levels are increased in the asthmatic lung (9, 10), and rhinoviral exacerbation of asthma is accompanied by a rise in kininogen activity (8).
Our studies have shown that COX-2 products can act as intermediates in the regulation of chemokine and growth factor expression in smooth muscle cells by BK. For example, BK induced secretion of CXCL8 and VEGF is COX-2 and PGE2 dependent (21, 47, 51) in human airway smooth muscle cells (HASMC), whereas endothelin-1-induced amphiregulin secretion is dependent on COX-2 activity and PGI2 production in pulmonary vascular smooth muscle cells. To date our studies have looked in single cell types without considering cross talk between the epithelium and the underlying mesenchyme.
Here we have explored the complexity of cross talk between airway epithelial and airway smooth muscle cells focusing on amphiregulin, COX-2, CXCL8, and VEGF. We hypothesized that BK-induced COX-2 activity would increase amphiregulin expression and secretion from HASMC and that, because of the presence of EGFR on the airway epithelium, HASMC-derived amphiregulin would provide the mechanism for interaction between the mesenchyme and the epithelium. Consistent with this we found that stimulation of HASMC with BK induced secretion of amphiregulin by HASMC. Furthermore, the resulting increase in amphiregulin in HASMC culture supernatants induced COX-2 expression, CXCL8, and VEGF secretion in airway epithelium in a paracrine manner.
These are the first studies to show that HASMC acts as a source of amphiregulin, that HASMC-derived amphiregulin can modify epithelial CXCL8 and VEGF synthesis and secretion, and that HASMC-derived amphiregulin can increase airway epithelial cell COX-2 expression. Because amphiregulin, EGFR activity, COX-2, CXCL8, and VEGF expression are all increased in the airways of asthmatic lungs, it is likely that these interactions play a role in driving airway remodeling and chronic airway inflammation in asthma.
MATERIALS AND METHODS
Reagents.
COX-2 antibodies were obtained from Cayman Chemicals. EGFR antibodies and EGFR phospho-tyrosine (Y845) antibodies were obtained from Cell Signaling Technology (New England BioLabs). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies were purchased from Santa Cruz Biotechnology. Anti-amphiregulin blocking antibody (Clone 31221) and mouse preimmune IgG were purchased from R&D Systems. Recombinant amphiregulin was purchased from R&D Systems. BK, PGE2, indomethacin, NS398, and AG1478 were purchased from Sigma Aldrich. ONO AEI329, ONO AEI 259, ONO AEI 248, and ONO AEI D1 004, were a kind gift from Ono Pharmaceutical Japan.
Human airway smooth muscle cell culture.
HASMC were explant cultured from human tracheas obtained postmortem, grown in DMEM supplemented with 4 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal bovine serum (GIBCO, Life Technologies) at 37°C with 5% CO2 and 100% humidity and used at passage 6 (34). HASMC were characterized by immunocytochemical analysis for positive α-smooth muscle actin expression and negative cytokeratin-18 and as described in (33). HASMC were 95% pure. Ethical approval for this study was obtained from the North Nottinghamshire Research Ethics Committee.
Human bronchial epithelial cell culture.
Immortalized human bronchial epithelial cells (HBEC) were a kind gift from Professor Jerry Shay, University of Texas, South Western (38). HBEC were seeded at a density of 5 × 104 ml−1 on 24-well or 6-well tissue culture plates and grown to confluence in complete KSFM medium (Invitrogen) at 37°C with 5% CO2 and 100% humidity. Confluent HBEC were washed twice in PBS-Dulbecco's (Sigma Aldrich) and cultured for 24 h in KSFM without bovine pituitary extract or EGF addition (serum free) prior to further experimentation.
Enzyme-linked immunosorbent assay.
Cell lines were plated to 24-well plates and grown to 100% confluence, and the medium was replaced with DMEM, with l-glutamine only, for 24 h. ELISA for amphiregulin, CXCL8, or VEGF165 (R&D Systems) was performed according to the manufacturer's protocol. All assay points were performed in triplicate on 24-well plates in a final medium volume of 500 μl. ELISAs were normalized to subsequent cell counts to give picograms per milliliter for a standard 1 × 104 cells.
RNA isolation and QPCR.
RNA extraction, first-strand cDNA synthesis, and quantitative PCR (QPCR) were performed as described previously (14). QPCR was performed with the following primers sets: amphiregulin forward: CGATGATTCAGTCAGAGTTGAACAGG, amphiregulin reverse: CCGTTCACCGAAATATTCTTGC; CXCL8 forward: CCAAGCTGGCCGTGGC, CXCL8 reverse: GCTCTCTTCCATCAGAAAG; COX-2 forward: GGAACACAACAGAGTATGCG, COX-2 reverse: AAGGGGATGCCAGTGATAGA; VEGF165 forward: GAGCAAGACAAGAAAATCCC, VEGF165 reverse: CCTCGGCTTGTCACATCTG; β2-microglobulin (β2-MG) forward: GCCTGGAGGCTATCCAG, β2-MG reverse: CCAGTCCTTGCTGAAAGACAAG.
All gene-specific quantification was calculated as ΔCt [target Ct minus housekeeping (β2-MG) Ct (cycle threshold)] relative to control or untreated cell experiment control to give a final ΔCt (test)/ΔCt (basal). All Ct calculations were performed by Stratagene, MxPro 3.2 software (Agilent).
siRNA knockdown of amphiregulin or COX-2.
siRNA (4 nM), either scrambled control (All-Stars, Qiagen) amphiregulin or COX-2 (GE Dharmacon, ON-TARGETplus siRNA) were incubated for 10 min with 3 μl of HiPerFect (Qiagen) in 100 μl of serum-free DMEM. siRNA complexes were incubated with 100 μl of HASMC at 6 × 105 ml−1 per well in a 24-well plate for 4 h. Complete medium was added to a total volume of 600 μl and the resulting plates were incubated at 37°C with 5% CO2 and 100% humidity for 48 h. siRNA-transfected cells were serum starved for the last 24 h then treated with the relevant agonist prior to RNA isolation or conditioned medium harvesting.
Protein isolation and Western blot analysis.
Protein extraction and Western blot analysis of COX-2, GAPDH, EGFR and EGFR phospho-tyrosine (Y845) were performed as described previously (13).
Statistical analysis of data.
Triplicate assay data are presented as standard error of the mean and comparative data were analyzed with a two-tailed paired t-test using GraphPad Prism (GraphPad, San Diego, CA). P values were scored as significant for 0.01–0.05 (*), 0.001–0.01 (**), and <0.001 (***).
RESULTS
Human airway smooth muscle cells secrete amphiregulin in response to BK via a COX-2/PGE2 dependent pathway.
Potential stimuli of amphiregulin secretion from HASMC were studied in a survey of asthma-related cytokines including IL-4, IL-13, IL-9, IL-1, TNF-α, TGF-β and BK. Only BK was capable of stimulating amphiregulin secretion from all human airway smooth muscle cell lines in a 24-h period (Fig. 1A) with a rapid 100-fold increase in amphiregulin mRNA accumulation (Fig. 1B). To determine whether endogenous cyclooxygenase-derived prostanoids were involved in amphiregulin production we looked at the effect of a nonselective COX inhibitor (indomethacin) and a selective COX-2 inhibitor [NS398 (39)] on BK-induced amphiregulin mRNA expression and protein secretion (Fig. 2, A and B). Both indomethacin and NS398 prevented amphiregulin mRNA accumulation and protein secretion from HASMC; furthermore, knockdown of COX-2 with siRNA (Fig. 2C) eliminated BK-induced amphiregulin mRNA accumulation (Fig. 2D). Consistent with a role for endogenous PGE2 in amphiregulin release, exogenously applied PGE2 also increased amphiregulin mRNA accumulation and amphiregulin secretion from HASMC (Fig. 2, E and F). Neither COX inhibitor prevented PGE2-induced amphiregulin secretion (Fig. 2G), implying that PGE2 induction of amphiregulin expression in HASMC is via a direct mechanism. Because PGE2 can act via EP2 or EP4 receptors in airway smooth muscle cells (5), we assessed EP2 or EP4 receptor dependence using selective EP receptor agonists. EP4 (ONOAEI 329) and EP2 (ONO AEI 259) receptor-selective agonists both-induced amphiregulin secretion from HASMC (Fig. 2H), suggesting that both EP receptor subtypes were involved. Collectively these findings show that BK-induced amphiregulin mRNA accumulation and protein section from HASMC via a COX-2/PGE2/EP2, EP4 autocrine loop. It is worth noting that HASMC can produce a baseline of secreted amphiregulin during the period of serum starvation (100 pg/ml per 1 × 104 cells). Previous studies from this group have established that there is a baseline of PGE2 production from HASMC and that this would be capable of stimulating amphiregulin expression without a proinflammatory stimulus.
Fig. 1.
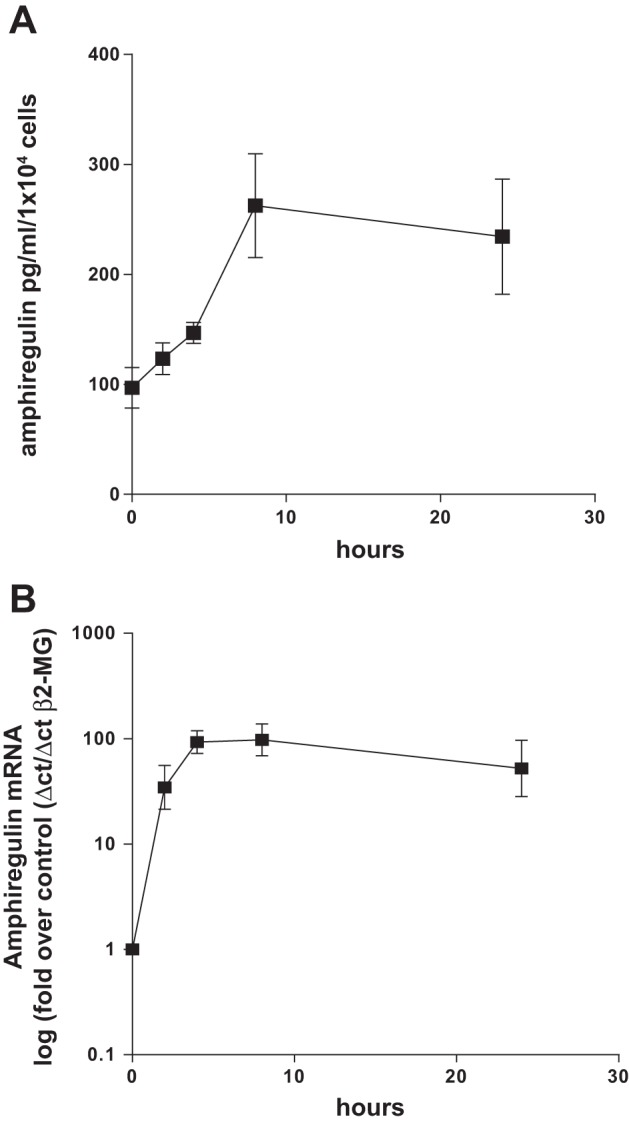
Bradykinin (BK) induced amphiregulin mRNA accumulation and protein secretion from airway smooth muscle cells. Three human airway smooth muscle (HASM) cell lines were treated with BK (1 × 10−5 M) in a time course of up to 24 h and the culture supernatants were analyzed by ELISA for amphiregulin (A). Three HASM cell lines were treated with BK (1 × 10−5 M) in a time course of up to 24 h and the accumulation of amphiregulin mRNA analyzed by quantitative PCR (QPCR) (B). β2-MG, β2-microglobulin. All measurements represent means ± SE of 3 independent experiments.
Fig. 2.

Bradykinin-induced amphiregulin mRNA accumulation and protein secretion from HASM cells (HASMC) is COX-2, PGE2, and EP2/EP4 receptor dependent. COX-2 inhibitors block BK-induced amphiregulin protein secretion and mRNA accumulation. Three HASM cell lines were treated with indomethacin (1 × 10−6 M) or NS398 (1 × 10−6 M) for 30 min prior to treatment with BK for 24 h. Culture supernatants were analyzed by ELISA for amphiregulin (A). Three HASM cell lines were treated with indomethacin (1 × 10−6 M) or NS398 (1 × 10−6 M) for 30 min prior to treatment with BK for 8 h with amphiregulin mRNA accumulation analyzed by QPCR (B). Three HASM cell lines were incubated with control siRNA (SC and solid bars) or COX-2 specific siRNA (I and open bars) for 48 h prior to treatment with BK (1 × 10−5 M) in a time course of up to 24 h. Protein samples were analyzed by Western blot to confirm COX-2 protein knockdown (C) and parallel RNA samples analyze by RT-QPCR for amphiregulin mRNA accumulation at 0 and 4 h post-BK addition. Each cell line was also incubated with amphiregulin siRNA (shaded bars) (D). PGE2 induces amphiregulin mRNA accumulation and amphiregulin secretion from HASM cells. Three HASM cell lines were treated with PGE2 (1 × 10−6 M) for up to 24 h and the accumulation of amphiregulin mRNA analyzed by QPCR (E); the experiment was repeated and amphiregulin secretion from HASM cells was monitored by amphiregulin ELISA (F). PGE2-induced amphiregulin secretion is not COX-2 dependent. Three HASM cell lines were treated with indomethacin (1 × 10−6 M) or NS398 (1 × 10−6 M) for 30 min prior to PGE2 addition (1 × 10−6 M) for 24 h followed by an amphiregulin ELISA of the culture supernatants (G). Selective EP2/EP4 receptor agonists-induced amphiregulin secretion from HASMC. Three HASM cell lines were treated with ONO AEI 329 (EP4), ONO AEI 259 (EP2), ONO AEI 248 (EP3), or ONO AEI D1 004 (EP1) for 24 h followed by an amphiregulin ELISA (H). All measurements represent means ± SE of 3 independent experiments. ***P < 0.001.
BK conditioned HASMC medium stimulates CXCL8 secretion from airway epithelial cells via an amphiregulin dependent mechanism.
Because increased airway smooth muscle mass is a key feature of remodeling in the asthmatic lung we originally considered the role of amphiregulin in HASMC growth. We found negligible growth (either BrdU incorporation or cell counting) of HASMC in response to recombinant amphiregulin or HASMC conditioned medium after 24 or 48 h of BK addition (data not shown). With no obvious role for amphiregulin in HASMC growth we considered the role of amphiregulin in airway inflammation. Release of amphiregulin from airway epithelial cell membranes is a known stimulus of CXCL8 expression and secretion (28, 31). To test whether HBEC cells could mount an inflammatory response to amphiregulin, HBEC were treated with amphiregulin for 24 h resulting in increased CXCL8 protein (Fig. 3A) and mRNA (Fig. 3B) levels. BK conditioned and control HASMC conditioned media were obtained with the addition of BK (1 × 10−5 M) or vehicle control to confluent HASMC for 24 h. Following serum starvation, confluent HBEC were treated with BK conditioned medium or control conditioned medium from HASMC, resulting in an increase in CXCL8 secretion (Fig. 3C) and de novo CXCL8 mRNA accumulation (Fig. 3D). HASMC conditioned medium may contain growth factors other than amphiregulin that are capable of inducing epithelial CXCL8 expression. To prove that amphiregulin in HASMC medium was responsible for the increase in epithelial CXCL8 expression, we pretreated BK conditioned HASMC medium with an amphiregulin blocking antibody or control preimmune mouse IgG1. Addition of anti-amphiregulin IgG to conditioned HASMC medium blocked CXCL8 mRNA accumulation in HBEC (Fig. 3E) and CXCL8 protein secretion from HBEC treated with control HASMC conditioned medium or BK HASMC conditioned medium (Fig. 3F). siRNA knockdown of amphiregulin in HASMC prevented BK conditioned medium from inducing 70% of the CXCL8 mRNA accumulation in HBEC compared with HASMC transfected with a scrambled siRNA control (Fig. 3G).
Fig. 3.

Recombinant amphiregulin and bradykinin conditioned cell culture medium from airway smooth muscle cells can induce CXCL8 mRNA accumulation and CXCL8 secretion from human airway epithelial cells. HASMC conditioned medium-induced CXCL8 expression in human bronchial epithelial cells (HBEC) is amphiregulin dependent. HBEC were treated with recombinant amphiregulin (10 ng/ml) for 24 h followed by a CXCL8 ELISA of the culture supernatant (A). HBEC were treated with recombinant amphiregulin for 24 h and CXCL8 mRNA accumulation analyzed by QPCR (B). Cell culture medium from HASMC treated for 24 h with BK (1 × 10−6 M) (BK con med) or a vehicle control (control con med) were added to HBEC for 24 h and the resulting HBEC supernatants and HASMC conditioned media were analyzed by CXCL8 ELISA, with HASMC conditioned medium CXCL8 levels subtracted from HBEC medium levels (C). HASMC BK conditioned medium and control conditioned media were used to treat HBEC for a time course of up to 24 h. CXCL8 mRNA accumulation was analyzed in HBEC by QPCR (D). HBEC were treated with BK conditioned HASMC medium (BK con med) for 24 h with either anti-amphiregulin blocking antibody (10 μg/ml) (anti-AR) or mouse IgG1 (10 μg/ml) (mouse IgG1) and CXCL8 mRNA levels analyzed by QPCR (E). HBEC were treated with HASMC control conditioned medium (con med) or HASMC BK conditioned medium (BK con med) for 24 h with either anti-amphiregulin blocking antibody (anti-AR) at 10 μg/ml or mouse IgG1 (anti-mouse) at 10 μg/ml, and secreted CXCL8 protein levels were analyzed by ELISA (F). HASM cells were transfected with control scrambled siRNA (solid bars) or amphiregulin siRNA (open bars) for 48 h, treated with BK for 24 h and the conditioned medium transferred to HBEC cells for 24 h, CXCL8 mRNA levels were analyzed by QPCR (G). All measurements represent means ± SE of 3 independent experiments. **P = 0.001–0.01; ***P < 0.001.
EGFR activity is required for HASMC-derived amphiregulin-induced CXCL8 expression in airway epithelial cells. EGFR plays a key role in both epithelial barrier repair and airway inflammatory responses. For this reason we sought to establish whether amphiregulin derived from HASMC was increasing CXCL8 expression from airway epithelial cells via the EGFR receptor. We found that pretreatment of HBEC with the EGFR inhibitor AG1478 (27) prevented recombinant amphiregulin induction of both CXCL8 mRNA accumulation and CXCL8 protein secretion from HBECs (Fig. 4, A and B). AG1478 also prevented BK-treated conditioned HASMC medium from inducing CXCL8 mRNA expression and CXCL8 protein secretion from HBECs (Fig. 4, C and D). EGFR activity is dependent on EGFR tyrosine kinase transphosphorylation of each EGFR subunit (42). We found that both recombinant amphiregulin (Fig. 4E) and BK conditioned HASMC medium induced EGFR tyrosine 845 transphosphorylation (Fig. 4F).
Fig. 4.

Amphiregulin and HASMC conditioned medium induction of CXCL8 mRNA expression and CXCL8 protein secretion in HBEC were blocked by EGFR inhibition with AG1478. Amphiregulin, BK conditioned HASMC medium, or control conditioned HASMC medium increased EGFR phosphorylation in HBEC was inhibited by AG1478. HBEC were assayed for CXCL8 mRNA (QPCR) after 2-h addition of amphiregulin (10 ng/ml) (solid bars) with or without pretreatment with AG1478 (1 × 10−6 M) (A). HBEC supernatants were assayed for CXCL8 protein by ELISA 24 h after addition of amphiregulin (10 ng/ml) (solid bars) with or without pretreatment with AG1478 (1 × 10−6 M) (B). HBEC were assayed for CXCL8 mRNA (QPCR) after incubation with BK conditioned HASMC medium (BK con med) or HASMC control conditioned medium (con med) (all solid bars) after 8 h, with or without pretreatment with AG1478 (1 × 10−6 M) (open bars are treated 0-h controls) (C). HBEC culture supernatants were assayed by ELISA for CXCL8 protein after pretreatment with AG1478 followed by treatment with HASMC control conditioned medium (con med) or BK conditioned medium (BK con med) with or without AG1478 (1 × 10−6 M) (D). Recombinant amphiregulin was added to HBEC in a time course, cell lysates were analyzed by Western blot for EGFR tyrosine-845 phosphorylation (EGFR-PY845), and blots were striped and reprobed with a “total” EGFR antibody (EGFR) (E). HBEC were treated with HASMC cell culture supernatants previously either untreated (con med) or treated with BK for 24 h (BK con med). Total cell lysates were analyzed by Western blot for EGFR tyrosine-845 phosphorylation (EGFR-PY845), then blots were striped and reprobed with anti-EGFR antibody (EGFR) (F). All measurements represent means ± SE of 3 independent experiments. Western blots are representative of 3 independent experiments. ***P < 0.001.
Amphiregulin in HASMC-derived conditioned medium increases COX-2 expression in airway epithelial cells. There is increased expression of COX-2 in the asthmatic airway epithelium (40), and, since a previous study suggests that EGFR activity is required to induce COX-2 expression in the airway epithelium (29), we tested the hypothesis that HASMC interact with the airway epithelium via amphiregulin to increase COX-2 expression. Recombinant amphiregulin rapidly increased both COX-2 mRNA and COX-2 protein expression in HBECs (both reaching peak expression at 2 h) (Fig. 5, A and B). Both control conditioned medium from HASMC and BK conditioned medium from HASMC increased COX-2 mRNA accumulation in HBECs (Fig. 5C). BK conditioned HASMC medium had a greater ability to induce COX-2 protein expression than control conditioned medium (Fig. 5D). To establish that amphiregulin in HASMC conditioned medium was inducing COX-2 expression in HBECs, HBECs were treated with HASMC medium containing amphiregulin blocking antisera or control mouse preimmune IgG1 for 4 h. Amphiregulin antibodies blocked both COX-2 mRNA and COX-2 protein expression in HBECs induced by HASMC conditioned medium (Fig. 6, A and B). To confirm that amphiregulin in HASMC conditioned medium was inducing COX-2 mRNA expression in HBEC, HBEC were treated with conditioned medium from scrambled control siRNA transfected HASMC or amphiregulin siRNA-transfected HASMC (Fig. 6C). Amphiregulin knockdown in HASMC was capable of reducing the accumulation of COX-2 mRNA in HBEC.
Fig. 5.

Recombinant amphiregulin and BK conditioned medium from airway smooth muscle cells increased COX-2 mRNA accumulation and COX-2 protein expression in HBEC. HBEC were treated with amphiregulin (10 ng/ml) in a time course of up to 24 h and assayed for COX-2 mRNA by QPCR (A). HBEC were treated with amphiregulin (10 ng/ml) in a time course of up to 24 h and the total protein extracts analyzed by Western blot for COX-2 protein (B). HBEC were treated with HASMC culture supernatants conditioned for 24 h with BK (▲, BK con med) or HASMC culture supernatants conditioned with a vehicle control (■, control con med) in a time course of up to 24 h. COX-2 mRNA levels were quantified by QPCR (C), COX-2 protein levels were quantified by Western blot (D). Blots were stripped and reprobed for GAPDH. Western blots are representative of 3 independent experiments.
Fig. 6.
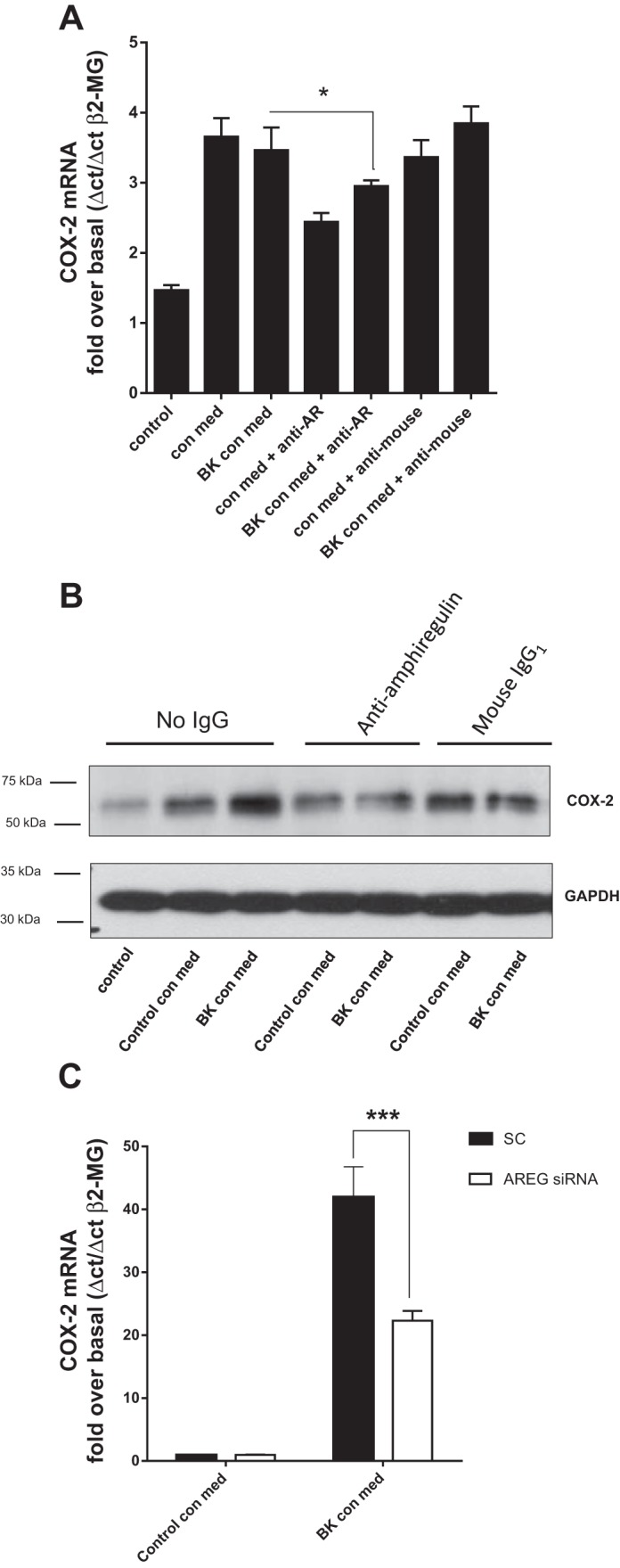
Amphiregulin blocking antibodies and amphiregulin siRNA decrease COX-2 mRNA accumulation and protein expression in HBEC exposed to BK treated airway smooth muscle cell conditioned medium. HBEC were cultured for 4 h with control HASMC conditioned medium (Control con med), or BK HASMC conditioned medium (BK con med) with either anti-amphiregulin antibody (anti-AR) or control mouse IgG1 antibody (anti-mouse). COX-2 mRNA expression was quantified by QPCR (A) and COX-2 protein by Western blot (B). Western blots were reprobed with antisera to GAPDH. Western blots are representative of 3 independent experiments. HASM cells were transfected with control scrambled siRNA (SC; solid bars) or amphiregulin siRNA (AREG siRNA; open bars) for 48 h and treated with BK for 24 h; the conditioned media were transferred to HBEC cells for 24 h, and COX-2 mRNA accumulation in HBEC was analyzed by QPCR (C). All other measurements represent means ± SE of 3 independent experiments. *P = 0.01–0.05; ***P < 0.001.
EGFR activity is required for amphiregulin and HASMC conditioned medium induction of HBEC COX-2 expression.
To demonstrate that the EGFR is required for amphiregulin induction of COX-2 expression in HBECs we pretreated HBEC with AG1478 and induced HBEC with recombinant amphiregulin for 4 h. AG1478 blocked amphiregulin-induced COX-2 mRNA accumulation (Fig. 7A) and COX-2 protein accumulation (Fig. 7B). These experiments were repeated with conditioned medium from HASMC. AG1478 blocked COX-2 mRNA induction (Fig. 7C) and COX-2 protein accumulation in HBECs in response to HASMC conditioned medium (Fig. 7D).
Fig. 7.

The EGFR inhibitor AG1478 blocks amphiregulin and BK conditioned HASMC medium induction of COX-2 mRNA and COX-2 protein accumulation in airway epithelial cells. HBEC were treated with amphiregulin (AR) for 4 h (solid bars) with or without AG1478 (1 × 10−6 M). COX-2 mRNA levels were quantified by QPCR (A). HBEC were treated with 10 ng/ml of AR in a time course of up to 24 h with or without AG1478 (1 × 10−6 M). Total protein extracts were analyzed by Western blot for COX-2 expression (B). HBEC were treated for 4 h with HASMC control conditioned medium (control con med) or BK conditioned (BK con med) smooth muscle medium with or without AG1478 (1 × 10−6 M) followed by QPCR for COX-2 mRNA (C). Total protein extracts from the same treatments at 4 and 8 h were Western blotted for COX-2 protein (D). All blots were reprobed with GAPDH antisera. Western blots are representative of 3 independent experiments. All other measurements represent means ± SE of 3 independent experiments. ***P < 0.001.
Amphiregulin in HASMC-derived conditioned medium increases VEGF expression in airway epithelial cells.
Asthma patients have increased levels of VEGF in their airways and airway cells (1, 17, 30) where VEGF plays a critical role in both airway remodeling (angiogenesis) and inflammation (23, 24). We tested the hypothesis that HASMC interact with the airway epithelium via amphiregulin to increase VEGF expression. Recombinant amphiregulin increased VEGF protein secretion and VEGF mRNA accumulation in HBECs (Fig. 8, A and B). Both control and BK conditioned medium from HASMC increased VEGF protein secretion from HBEC and VEGF mRNA accumulation in HBEC (Fig. 8, C and D). To establish that amphiregulin in HASMC conditioned medium was inducing VEGF expression in HBECs, HASMC medium containing amphiregulin blocking antisera or control mouse preimmune IgG1 was incubated with HBEC for 24 h. Amphiregulin antibodies blocked both VEGF mRNA accumulation and VEGF protein expression in HBECs induced by HASMC conditioned medium (Fig. 8, E and F). To confirm that amphiregulin in HASMC conditioned medium was inducing VEGF mRNA accumulation in HBEC, HBEC were treated with conditioned medium from scrambled control-transfected HASMC or amphiregulin siRNA-transfected HASMC (Fig. 8G). Amphiregulin knockdown in HASMC was capable of reducing HBEC VEGF expression. It is worth noting that amphiregulin chelation with a specific antibody in HASMC conditioned medium had a greater effect on VEGF expression and secretion from HBEC than siRNA ablation of amphiregulin expression in HASMC. Amphiregulin antisera may be a more efficient method of blocking the interaction between HASMC and HBEC than the depletion of amphiregulin mRNA in HASMC.
Fig. 8.

Recombinant amphiregulin and bradykinin conditioned cell culture medium from airway smooth muscle cells can induce VEGF165 mRNA accumulation and VEGF165 secretion from human airway epithelial cells. HASMC conditioned medium induced VEGF165 expression in HBEC is amphiregulin dependent. HBEC were treated with recombinant amphiregulin (10 ng/ml) for 24 h followed by a VEGF165 ELISA of the culture supernatant (A). HBEC were treated with recombinant amphiregulin for 24 h and VEGF165 mRNA accumulation was analyzed by QPCR (B). Cell culture medium from HASMC treated for 24 h with BK (1 × 10−6 M) (BK con med) or a vehicle control (control con med) were added to HBEC for 24 h and the resulting HBEC supernatants and HASMC conditioned medium were analyzed by VEGF165 ELISA, with HASMC conditioned medium VEGF165 levels subtracted from HBEC medium levels (C). HASMC BK conditioned medium and control conditioned media were used to treat HBEC for a time course of up to 24 h, VEGF165 mRNA accumulation was analyzed in HBEC by QPCR (D). HBEC were treated with BK conditioned HASMC medium (BK con med) for 24 h with either anti-amphiregulin blocking antibody (10 μg/ml) (anti-AR) or mouse IgG1 (10 μg/ml) and VEGF165 mRNA levels were analyzed by QPCR (E). HBEC were treated with HASMC control conditioned medium (con med) or HASMC BK con med for 24 h with either anti-AR at 10 μg/ml or mouse IgG1 (anti-mouse) at 10 μg/ml, and secreted VEGF165 protein levels were analyzed by ELISA (F). HASM cells were transfected with control scrambled siRNA (solid bars) or amphiregulin siRNA (open bars) for 48 h and treated with BK for 24 h, the conditioned media were transferred to HBEC cells for 24 h, and VEGF165 mRNA levels were analyzed by QPCR (G). All measurements represent means ± SE of 3 independent experiments. *P = 0.01–0.05; ***P < 0.001.
HBEC-derived supernatants induce amphiregulin expression in HASMC.
Since BK-induced amphiregulin expression in HASMC is dependent on a COX-2/PGE2 autocrine loop, we hypothesized that HBEC would amplify HASMC amphiregulin expression when COX-2 expression in HBEC increases. HBEC were stimulated with recombinant amphiregulin for 24 h with or without indomethacin. HBEC conditioned medium was then applied to HASMC for 4 h and amphiregulin mRNA accumulation analyzed by RT-QPCR. Conditioned medium from amphiregulin-treated HBEC-induced amphiregulin expression in HASMC, and addition of indomethacin during HBEC conditioning eliminated the ability of HBEC conditioned medium to induce HASMC amphiregulin expression (Fig. 9A). A summary of our findings is shown in a representative diagram highlighting the interaction between HASMC and HBEC, with COX-2 activity required to drive HASMC amphiregulin secretion, leading to amphiregulin costimulation of CXCL8 and VEGF secretion, with increased COX-2 expression in the epithelium, that is capable of “back-inducing” amphiregulin expression in airway in HASMC (Fig. 9B).
Fig. 9.

Amphiregulin-treated HBEC supernatants stimulate airway smooth muscle amphiregulin mRNA accumulation. Amphiregulin links reciprocal COX-2 in airway epithelial/smooth muscle inflammation. Culture supernatants from HBEC, untreated (HBEC con med) treated with amphiregulin (10 ng/ml) (AR con HBEC med) with indomethacin (1 × 10−6 M) (AR con med + indo), or serum free media only (CM−) for 8 h were then added to HASMC for 4 h, the HASMC RNA were assayed for amphiregulin expression by QPCR (A), and 10 ng/ml of recombinant amphiregulin was added as a control directly to the HASMC (AR direct). All other measurements represent means ± SE of 3 independent experiments. Reciprocal, COX-2-dependent, relationship between HASMC and HBEC: BK-induced HASMC amphiregulin acts as a proinflammatory stimulus resulting in COX-2, CXCL8, and VEGF production from HBEC and HBEC stimulation of the HASMC leading to amphiregulin growth factor production (B). BK1 and BK2, bradykinin receptors 1 and 2; AREG, amphiregulin; EP, EP class G-protein coupled receptors. ***P < 0.001.
DISCUSSION
There are a number of novel findings in this study. Firstly, we found that amphiregulin was expressed and secreted from HASMC in response to BK via a COX-2/PGE2 autocrine loop. Secondly, HASMC-derived amphiregulin stimulated the expression and secretion of CXCL8 and VEGF from airway epithelial cells. Thirdly, amphiregulin was a very rapid inducer of COX-2 expression in the airway epithelial cells, and, finally, prostanoids produced, by airway epithelial cells in response to amphiregulin, feedback to amplify amphiregulin expression in the airway smooth muscle cells. This bidirectional cross talk adds important information on how these two cell types interact to cascade inflammatory and remodeling signals in asthma.
There are previous examples of prostanoid-induced amphiregulin expression in cell systems other than the lung: amphiregulin secretion from colon and breast cancer cells is dependent on PGE2-induced gene expression (7, 43), as is gut subepithelial myofibroblast amphiregulin production (44). These studies have focused on cell growth or epithelial wound repair responses to amphiregulin, whereas our study is the first to link PGE2-dependent amphiregulin secretion to cross talk between the mesenchymal and epithelial cells, resulting in increased inflammatory gene expression, in this case CXCL-8 and VEGF.
Our experiments showed that the application of BK conditioned HASMC medium resulted in proinflammatory chemokine (CXCL8, VEGF) secretion and COX-2 expression in airway epithelial cells. Experiments with amphiregulin blocking antibodies and the EGFR inhibitor AG1478 suggested that the response was dependent on amphiregulin in the HASMC supernatants and the EGFR receptor on epithelial cells. It is worth noting, however, that although HASMC conditioned medium induced CXCL8, VEGF, and COX-2 expression in airway epithelial cells, amphiregulin blocking antibodies did not fully eliminate CXCL8 or COX-2 expression whereas the EGFR inhibitor AG1478 did. This may be explained by HASM cells expressing additional EGFR agonists. Our own studies have shown that HASMC express HB-EGF and epiregulin; however, the magnitude of effect of the amphiregulin blocking antibodies suggests that amphiregulin is the main EGFR agonist responsible for stimulating EGFR activity when airway epithelial cells are exposed to HASMC conditioned medium.
This study demonstrated that HASMC-derived amphiregulin induces rapid COX-2 expression in epithelial cells via EGFR. It has been proposed that continuous inflammation and remodeling resulting from epithelial damage and repair is a key part of asthma pathophysiology (16). Because both COX-2 and active EGFR are required for epithelial wound repair (41), with a broken epithelial barrier amphiregulin from underlying mesenchymal cells could cause both EGFR activation and consequent COX-2 expression as part of the airway epithelial repair mechanism.
We used the proinflammatory peptide BK to stimulate HASMC because the asthmatic lung possesses greater BK receptor coupling (35), higher levels of BK releasing kallikrein (9), more amphiregulin (15, 20), and increased EGFR expression (36). Our findings show that a BK/AR/EGFR mechanism coupling HASMC and the epithelium provides a link between these disease-related changes, possibly resulting in the higher levels of CXCL8, VEGF, and COX-2 seen in the asthmatic lung. These findings may also be relevant to viral exacerbations of asthma. The severity of asthma symptoms is greatly increased during viral infection. RSV and rhinoviral infection of epithelial cells rapidly induces amphiregulin release coupled with EGFR-dependent CXCL8 secretion (28, 31). Rhinoviral airway infections also increase BK releasing kallikrein activity in the asthmatic lung, possibly linking rhinovirus, BK, amphiregulin, and CXCL8 secretion to viral exacerbations of asthma (8). Both RSV and rhinoviral infection of airway epithelial cells result in elevated VEGF expression and secretion (25, 26). The role of amphiregulin and EGFR in this process is yet to be investigated.
We have considered how our findings fit in with existing knowledge of the role of the EGFR in asthma pathophysiology. Immunohistochemical studies of lung biopsies found greater EGFR expression in the airway epithelium in asthma patients compared with healthy controls (36, 37). Studies of EGFR inhibition have been undertaken in allergic airway animal models with somewhat contrasting results. In rats, ovalbumin (OVA)-induced remodeling was prevented by the EGFR inhibitor AG1478 (48) and in another study AG1478 prevented OVA-induced smooth muscle hypertrophy and reduced methacholine-induced airway hyperresponsiveness (45). Amphiregulin knockout did not, however, prevent OVA-induced inflammation or remodeling in mice (19). These differences may reflect interspecies differences and it is unclear which if either can be extrapolated to human asthma.
In conclusion, the data from this study provide evidence that mesenchymal cell amphiregulin, epithelial cell EGFR, and COX-2 in both cell types provide a dynamic bidirectional axis of mesenchymal and epithelial interactions with the resultant changes in gene expression in both cell types providing mechanisms explaining multiple observations made in airway tissue in asthma patients. To date there have been no studies of EGFR inhibitors or anti-amphiregulin blockade in human asthma; however, our findings suggest that these approaches warrant future investigation as anti-inflammatory/remodeling therapies for chronic asthma.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
K.D. conception and design of research; K.D. performed experiments; K.D. analyzed data; K.D. interpreted results of experiments; K.D. prepared figures; K.D. drafted manuscript; K.D. and A.J.K. edited and revised manuscript; K.D. and A.J.K. approved final version of manuscript.
REFERENCES
- 1.Asai K, Kanazawa H, Kamoi H, Shiraishi S, Hirata K, Yoshikawa J. Increased levels of vascular endothelial growth factor in induced sputum in asthmatic patients. Clin Exp Allergy 33: 595–599, 2003. [DOI] [PubMed] [Google Scholar]
- 2.Barrow RE, Wang CZ, Evans MJ, Herndon DN. Growth factors accelerate epithelial repair in sheep trachea. Lung 171: 335–344, 1993. [DOI] [PubMed] [Google Scholar]
- 3.Basbaum C, Li D, Gensch E, Gallup M, Lemjabbar H. Mechanisms by which gram-positive bacteria and tobacco smoke stimulate mucin induction through the epidermal growth factor receptor (EGFR). Novartis Found Symp 248: 171–176; discussion 176–180, 277–182, 2002. [PubMed] [Google Scholar]
- 4.Berair R, Saunders R, Brightling CE. Origins of increased airway smooth muscle mass in asthma. BMC Med 11: 145, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bradbury D, Clarke D, Seedhouse C, Corbett L, Stocks J, Knox A. Vascular endothelial growth factor induction by prostaglandin E2 in human airway smooth muscle cells is mediated by E prostanoid EP2/EP4 receptors and SP-1 transcription factor binding sites. J Biol Chem 280: 29993–30000, 2005. [DOI] [PubMed] [Google Scholar]
- 6.Burgel PR, Nadel JA. Epidermal growth factor receptor-mediated innate immune responses and their roles in airway diseases. Eur Respir J 32: 1068–1081, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Chang SH, Ai Y, Breyer RM, Lane TF, Hla T. The prostaglandin E2 receptor EP2 is required for cyclooxygenase 2-mediated mammary hyperplasia. Cancer Res 65: 4496–4499, 2005. [DOI] [PubMed] [Google Scholar]
- 8.Christiansen SC, Eddleston J, Bengtson SH, Jenkins GR, Sarnoff RB, Turner RB, Gwaltney JM Jr, Zuraw BL. Experimental rhinovirus infection increases human tissue kallikrein activation in allergic subjects. Int Arch Allergy Immunol 147: 299–304, 2008. [DOI] [PubMed] [Google Scholar]
- 9.Christiansen SC, Proud D, Cochrane CG. Detection of tissue kallikrein in the bronchoalveolar lavage fluid of asthmatic subjects. J Clin Invest 79: 188–197, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christiansen SC, Proud D, Sarnoff RB, Juergens U, Cochrane CG, Zuraw BL. Elevation of tissue kallikrein and kinin in the airways of asthmatic subjects after endobronchial allergen challenge. Am Rev Respir Dis 145: 900–905, 1992. [DOI] [PubMed] [Google Scholar]
- 11.Church RJ, Jania LA, Koller BH. Prostaglandin E2 produced by the lung augments the effector phase of allergic inflammation. J Immunol 188: 4093–4102, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Comer BS, Camoretti-Mercado B, Kogut PC, Halayko AJ, Solway J, Gerthoffer WT. Cyclooxygenase-2 and microRNA-155 expression are elevated in asthmatic airway smooth muscle cells. Am J Respir Cell Mol Biol 52: 438–447, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deacon K, Blank JL. Characterization of the mitogen-activated protein kinase kinase 4 (MKK4)/c-Jun NH2-terminal kinase 1 and MKK3/p38 pathways regulated by MEK kinases 2 and 3. MEK kinase 3 activates MKK3 but does not cause activation of p38 kinase in vivo. J Biol Chem 272: 14489–14496, 1997. [DOI] [PubMed] [Google Scholar]
- 14.Deacon K, Knox AJ. Endothelin-1 (ET-1) increases the expression of remodeling genes in vascular smooth muscle through linked calcium and cAMP pathways: role of a phospholipase A2(cPLA2)/cyclooxygenase-2 (COX-2)/prostacyclin receptor-dependent autocrine loop. J Biol Chem 285: 25913–25927, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Enomoto Y, Orihara K, Takamasu T, Matsuda A, Gon Y, Saito H, Ra C, Okayama Y. Tissue remodeling induced by hypersecreted epidermal growth factor and amphiregulin in the airway after an acute asthma attack. J Allergy Clin Immunol 124: 913–920.e1–e7, 2009. [DOI] [PubMed] [Google Scholar]
- 16.Holgate ST. Airway inflammation and remodeling in asthma: current concepts. Mol Biotechnol 22: 179–189, 2002. [DOI] [PubMed] [Google Scholar]
- 17.Hossny E, El-Awady H, Bakr S, Labib A. Vascular endothelial growth factor overexpression in induced sputum of children with bronchial asthma. Pediatr Allergy Immunol 20: 89–96, 2009. [DOI] [PubMed] [Google Scholar]
- 18.Johnson SR, Knox AJ. Synthetic functions of airway smooth muscle in asthma. Trends Pharmacol Sci 18: 288–292, 1997. [DOI] [PubMed] [Google Scholar]
- 19.Kajiwara N, Oboki K, Ohno T, Ishii A, Sunnarborg SW, Okumura K, Saito H, Nakae S. Amphiregulin is not essential for ovalbumin-induced acute airway inflammation in mice. Allergol Int 59: 207–211, 2010. [DOI] [PubMed] [Google Scholar]
- 20.Kim KW, Jee HM, Park YH, Choi BS, Sohn MH, Kim KE. Relationship between amphiregulin and airway inflammation in children with asthma and eosinophilic bronchitis. Chest 136: 805–810, 2009. [DOI] [PubMed] [Google Scholar]
- 21.Knox AJ, Corbett L, Stocks J, Holland E, Zhu YM, Pang L. Human airway smooth muscle cells secrete vascular endothelial growth factor: up-regulation by bradykinin via a protein kinase C and prostanoid-dependent mechanism. FASEB J 15: 2480–2488, 2001. [DOI] [PubMed] [Google Scholar]
- 22.Lazaar AL, Panettieri RA Jr. Airway smooth muscle: a modulator of airway remodeling in asthma. J Allergy Clin Immunol 116: 488–495; quiz 496, 2005. [DOI] [PubMed] [Google Scholar]
- 23.Lee CG, Link H, Baluk P, Homer RJ, Chapoval S, Bhandari V, Kang MJ, Cohn L, Kim YK, McDonald DM, Elias JA. Vascular endothelial growth factor (VEGF) induces remodeling and enhances TH2-mediated sensitization and inflammation in the lung. Nat Med 10: 1095–1103, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee CG, Ma B, Takyar S, Ahangari F, Delacruz C, He CH, Elias JA. Studies of vascular endothelial growth factor in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc 8: 512–515, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee CG, Yoon HJ, Zhu Z, Link H, Wang Z, Gwaltney JM, Landry M, Elias JA. Respiratory syncytial virus stimulation of vascular endothelial cell growth factor/vascular permeability factor. Am J Respir Cell Mol Biol 23: 662–669, 2000. [DOI] [PubMed] [Google Scholar]
- 26.Leigh R, Oyelusi W, Wiehler S, Koetzler R, Zaheer RS, Newton R, Proud D. Human rhinovirus infection enhances airway epithelial cell production of growth factors involved in airway remodeling. J Allergy Clin Immunol 121: 1238–1245.e4, 2008. [DOI] [PubMed] [Google Scholar]
- 27.Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science 267: 1782–1788, 1995. [DOI] [PubMed] [Google Scholar]
- 28.Liu K, Gualano RC, Hibbs ML, Anderson GP, Bozinovski S. Epidermal growth factor receptor signaling to Erk1/2 and STATs control the intensity of the epithelial inflammatory responses to rhinovirus infection. J Biol Chem 283: 9977–9985, 2008. [DOI] [PubMed] [Google Scholar]
- 29.Liu M, Yang SC, Sharma S, Luo J, Cui X, Peebles KA, Huang M, Sato M, Ramirez RD, Shay JW, Minna JD, Dubinett SM. EGFR signaling is required for TGF-beta 1 mediated COX-2 induction in human bronchial epithelial cells. Am J Respir Cell Mol Biol 37: 578–588, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez-Guisa JM, Powers C, File D, Cochrane E, Jimenez N, Debley JS. Airway epithelial cells from asthmatic children differentially express proremodeling factors. J Allergy Clin Immunol 129: 990–997.e6, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monick MM, Cameron K, Staber J, Powers LS, Yarovinsky TO, Koland JG, Hunninghake GW. Activation of the epidermal growth factor receptor by respiratory syncytial virus results in increased inflammation and delayed apoptosis. J Biol Chem 280: 2147–2158, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Okumura S, Sagara H, Fukuda T, Saito H, Okayama Y. FcepsilonRI-mediated amphiregulin production by human mast cells increases mucin gene expression in epithelial cells. J Allergy Clin Immunol 115: 272–279, 2005. [DOI] [PubMed] [Google Scholar]
- 33.Pang L, Knox AJ. Effect of interleukin-1 beta, tumour necrosis factor-alpha and interferon-gamma on the induction of cyclo-oxygenase-2 in cultured human airway smooth muscle cells. Br J Pharmacol 121: 579–587, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pang L, Knox AJ. PGE2 release by bradykinin in human airway smooth muscle cells: involvement of cyclooxygenase-2 induction. Am J Physiol Lung Cell Mol Physiol 273: L1132–L1140, 1997. [DOI] [PubMed] [Google Scholar]
- 35.Polosa R, Holgate ST. Comparative airway response to inhaled bradykinin, kallidin, and [des-Arg9]bradykinin in normal and asthmatic subjects. Am Rev Respir Dis 142: 1367–1371, 1990. [DOI] [PubMed] [Google Scholar]
- 36.Polosa R, Puddicombe SM, Krishna MT, Tuck AB, Howarth PH, Holgate ST, Davies DE. Expression of c-erbB receptors and ligands in the bronchial epithelium of asthmatic subjects. J Allergy Clin Immunol 109: 75–81, 2002. [DOI] [PubMed] [Google Scholar]
- 37.Puddicombe SM, Polosa R, Richter A, Krishna MT, Howarth PH, Holgate ST, Davies DE. Involvement of the epidermal growth factor receptor in epithelial repair in asthma. FASEB J 14: 1362–1374, 2000. [DOI] [PubMed] [Google Scholar]
- 38.Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, Peyton M, Zou Y, Kurie JM, Dimaio JM, Milchgrub S, Smith AL, Souza RF, Gilbey L, Zhang X, Gandia K, Vaughan MB, Wright WE, Gazdar AF, Shay JW, Minna JD. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res 64: 9027–9034, 2004. [DOI] [PubMed] [Google Scholar]
- 39.Range SP, Pang L, Holland E, Knox AJ. Selectivity of cyclo-oxygenase inhibitors in human pulmonary epithelial and smooth muscle cells. Eur Respir J 15: 751–756, 2000. [DOI] [PubMed] [Google Scholar]
- 40.Redington AE, Meng QH, Springall DR, Evans TJ, Creminon C, Maclouf J, Holgate ST, Howarth PH, Polak JM. Increased expression of inducible nitric oxide synthase and cyclo-oxygenase-2 in the airway epithelium of asthmatic subjects and regulation by corticosteroid treatment. Thorax 56: 351–357, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Savla U, Appel HJ, Sporn PH, Waters CM. Prostaglandin E2 regulates wound closure in airway epithelium. Am J Physiol Lung Cell Mol Physiol 280: L421–L431, 2001. [DOI] [PubMed] [Google Scholar]
- 42.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 103: 211–225, 2000. [DOI] [PubMed] [Google Scholar]
- 43.Shao J, Lee SB, Guo H, Evers BM, Sheng H. Prostaglandin E2 stimulates the growth of colon cancer cells via induction of amphiregulin. Cancer Res 63: 5218–5223, 2003. [PubMed] [Google Scholar]
- 44.Shao J, Sheng H. Amphiregulin promotes intestinal epithelial regeneration: roles of intestinal subepithelial myofibroblasts. Endocrinology 151: 3728–3737, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siddiqui S, Novali M, Tsuchiya K, Hirota N, Geller BJ, McGovern TK, Risse PA, Jo T, Zeroual MA, Martin JG. The modulation of large airway smooth muscle phenotype and effects of epidermal growth factor receptor inhibition in the repeatedly allergen-challenged rat. Am J Physiol Lung Cell Mol Physiol 304: L853–L862, 2013. [DOI] [PubMed] [Google Scholar]
- 46.Sousa A, Pfister R, Christie PE, Lane SJ, Nasser SM, Schmitz-Schumann M, Lee TH. Enhanced expression of cyclo-oxygenase isoenzyme 2 (COX-2) in asthmatic airways and its cellular distribution in aspirin-sensitive asthma. Thorax 52: 940–945, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stanford SJ, Pepper JR, Mitchell JA. Cyclooxygenase-2 regulates granulocyte-macrophage colony-stimulating factor, but not interleukin-8, production by human vascular cells: role of cAMP. Arterioscler Thromb Vasc Biol 20: 677–682, 2000. [DOI] [PubMed] [Google Scholar]
- 48.Tamaoka M, Hassan M, McGovern T, Ramos-Barbon D, Jo T, Yoshizawa Y, Tolloczko B, Hamid Q, Martin JG. The epidermal growth factor receptor mediates allergic airway remodelling in the rat. Eur Respir J 32: 1213–1223, 2008. [DOI] [PubMed] [Google Scholar]
- 49.Trebino CE, Stock JL, Gibbons CP, Naiman BM, Wachtmann TS, Umland JP, Pandher K, Lapointe JM, Saha S, Roach ML, Carter D, Thomas NA, Durtschi BA, McNeish JD, Hambor JE, Jakobsson PJ, Carty TJ, Perez JR, Audoly LP. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc Natl Acad Sci USA 100: 9044–9049, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zaiss DM, Yang L, Shah PR, Kobie JJ, Urban JF, Mosmann TR. Amphiregulin, a TH2 cytokine enhancing resistance to nematodes. Science 314: 1746, 2006. [DOI] [PubMed] [Google Scholar]
- 51.Zhu YM, Bradbury DA, Pang L, Knox AJ. Transcriptional regulation of interleukin (IL)-8 by bradykinin in human airway smooth muscle cells involves prostanoid-dependent activation of AP-1 and nuclear factor (NF)-IL-6 and prostanoid-independent activation of NF-kappaB. J Biol Chem 278: 29366–29375, 2003. [DOI] [PubMed] [Google Scholar]