

Abstract
Myelodysplastic syndromes (MDS) are a group of neoplasms characterized by ineffective myeloid hematopoiesis and various risks for leukemia. SRSF2, a member of the serine/arginine-rich (SR) family of splicing factors, is one of the mutation targets associated with poor survival in patients suffering from myelodysplastic syndromes. Here we report the biological function of SRSF2 in hematopoiesis by using conditional knockout mouse models. Ablation of SRSF2 in the hematopoietic lineage caused embryonic lethality, and Srsf2-deficient fetal liver cells showed significantly enhanced apoptosis and decreased levels of hematopoietic stem/progenitor cells. Induced ablation of SRSF2 in adult Mx1-Cre Srsf2flox/flox mice upon poly(I):poly(C) injection demonstrated a significant decrease in lineage− Sca+ c-Kit+ cells in bone marrow. To reveal the functional impact of myelodysplastic syndromes-associated mutations in SRSF2, we analyzed splicing responses on the MSD-L cell line and found that the missense mutation of proline 95 to histidine (P95H) and a P95-to-R102 in-frame 8-amino-acid deletion caused significant changes in alternative splicing. The affected genes were enriched in cancer development and apoptosis. These findings suggest that intact SRSF2 is essential for the functional integrity of the hematopoietic system and that its mutations likely contribute to development of myelodysplastic syndromes.
INTRODUCTION
Multiple classes of genetic aberrations have been suggested as the cause of myelodysplastic syndromes (MDS) (1, 2), including mutations in signal transduction, transcription factors, and epigenetic modifiers (3–5). Interestingly, recent genome-wide sequencing studies revealed that mutations in genes encoding splicing factors are commonly associated with MDS and other hematological malignancies (6–15). One of these newly identified genes codes for the SRSF2 splicing factor (also known as SC35), and its mutations have been linked to poor survival among MDS patients (16, 17). Most of the SRSF2 mutations occurred at proline 95, and the majority of these mutations changed this proline to histidine (P95H); less-frequent changes to leucine (P95L) and arginine (P95R) and in-frame deletion of 8 amino acids (aa) from P95 to R102 (Δ8aa) have also been reported previously (6, 16, 18–20). However, the causal effect of these mutations on MDS development remains to be established.
SRSF2 is one of the founding members of the serine/arginine-rich (SR) protein family of splicing factors (21). It is involved in both constitutive and regulated splicing. Homozygous germ line Srsf2 knockout (KO) mice are embryonically lethal (22), and conditional knockout (cKO) mice display various tissue-specific phenotypes (22–24). Importantly, Srsf2 downregulation in mouse embryonic fibroblasts results in G2/M cell cycle arrest and genomic instability (23). To date, systematic analysis of SRSF2 function in the blood system has not been reported except for its requirement in T cell development (24). Given the tight link of Srsf2 mutations to MDS, we aimed to directly test the hypothesis that SRSF2 plays an important role in normal hematopoiesis and that SRSF2 mutations induce specific changes in alternative splicing that favor disease progression.
Here we analyzed SRSF2 function in hematopoiesis on two mouse models by crossing cKO mice with blood cell-specific Vav-iCre mice and interferon-inducible Mx1-Cre mice. We also generated an inducible small hairpin RNA (shRNA)/cDNA expression system to replace endogenous SRSF2 with specific mutants in a MDS cell line to evaluate the splicing response to mutant SRSF2 by RNA-mediated oligonucleotide annealing, selection, and ligation coupled with next-generation sequencing (RASL-seq) (25, 26). We report that SRSF2 is essential for the survival of hematopoietic cells in developing embryos and adults and that its mutant forms switch the RNA splicing profile on a large panel of genes involved in cancer development and apoptosis. Together, these data suggest that SRSF2 mutations identified in MDS are not simply loss-of-function mutations but instead alter SRSF2 function in RNA splicing. Such changes may directly contribute to MDS development and later progression to more aggressive forms of leukemia.
MATERIALS AND METHODS
Mice.
C57BL/6 (CD45.2), congenic strain B6.SJL-Ptprca Pep3b/BoyJ (PEP3, CD45.1) mice, Vav-iCre mice, and Mx1-Cre mice were obtained from Jackson Laboratory. Conditional knockout Srsf2flox/flox mice of the C57BL/6 background were described previously (24). For embryo analyses, Srsf2flox/flox mice were mated with Vav-iCre+ Srsf2flox/+ mice. To collect peripheral blood (PB), embryos were bled from the umbilical cord into phosphate-buffered saline (PBS). Fetal livers (FLs) from embryonic day 14.5 (E14.5) mice were fixed in 4% formaldehyde–PBS, and the tissue section was stained with hematoxylin-eosin. Poly(I):poly(C) (Sigma) was injected intraperitoneally (i.p.) at either 250 μg/mouse every other day for a total of 3 injections or 600 μg/mouse as indicated. Age-matched adult mice (8 to 12 weeks old) were used for experiments. The day of the first injection was defined as day 0. The two protocols resulted in similar levels of knockdown efficiency. Genotyping PCR was performed using primers described previously (23). All the procedures were approved by the institutional animal care and use committee.
DNA constructs.
C-terminally hemagglutinin (HA)-tagged human SRSF2 was subcloned in the EcoRI site of murine stem cell virus (MSCV)-internal ribosome entry site (IRES)-green fluorescent protein (GFP) (MigR1), MSCV-IRES-puro (MIP), and pREV-tetracycline-inducible promoter (TRE) (23). P95H and Δ8aa mutants were made by PCR mutagenesis. Primers used for mutagenesis were as follows: P95H(F) (CAAATGGCGCGCTACGGCCGCCACCCGGACTCACACCACAGCCGC), P95H(R) (GCGGCTGTGGTGTGAGTCCGGGTGGCGGCCGTAGCGCGCCATTTG), Δ8aa(F) (CGGGTGCAAATGGCGCGCTACGGCCGCCGGGGACCGCCACCCCGCAGGTACGGGG), and Δ8aa(R) (CCCCGTACCTGCGGGGTGGCGGTCCCCGGCGGCCGTAGCGCGCCATTTGCACCCG). pTRIPZ-SRSF2 constructs were made using shRNA against the 3′ untranslated region (3′ UTR) of human wild-type (WT) SRSF2 (CTCTCCCGATTGCTCCTGTGTA) and human SRSF2 cDNA sequences with or without mutations.
Cell culture.
293T cells, mouse fetal liver (FL) cells (embryonic day 12.5 [E12.5] to E14.5), total bone marrow (BM) cells, and lineage-depleted BM cells (sorted by using a Lineage depletion kit from Miltenyi) were cultured as described before (27). To make a single-cell suspension of FL cells, FLs were sheared in PBS by pipetting, passed through 40-μm-pore-size cell strainers, and treated with ammonium-chloride-potassium (ACK) buffer (150 mM NH4Cl, 1 mM KHCO3, 0.1 mM EDTA) when necessary. MDS-L cells (28, 29) were kindly provided by Daniel Starczynowski (Cincinnati Children's Hospital Medical Center) and were cultured in RPMI medium supplemented with 10% fetal bovine serum, penicillin-streptomycin, and 10 ng/ml human interleukin-3 (hIL-3) (Peprotech). To induce the expression of shRNA and exogenous SRSF2 in MDS-L cell lines transduced with pTRIPZ vectors, 1 μg/ml doxycycline (Dox; Sigma) was added every day to the cells for 3 days. To reach 50% SRSF2 expression in shRNA-only cells, 2.5 μg/ml Dox was used in the assay. Cell growth was evaluated in duplicate using the trypan blue exclusion assay. As for CFU assays, specific numbers of cells (described in the figure legends) were seeded into M3434 medium (Stemcell Technologies). One week later, colonies were counted.
Flow cytometry.
Primary cells (FL, PB, and BM cells) from mice were treated with ACK buffer at room temperature for 5 min. For PB staining, a B cell lineage (allophycocyanin [APC]-conjugated B220), a T cell lineage (peridinin chlorophyll protein [PerCP]-Cy5.5-conjugated CD4 and CD8a), and myeloid lineages (phycoerythrin [PE]-conjugated Gr1 and CD11b) were used. For FL staining, the lineage cocktail consisted of CD3, CD4, CD8a, Gr1, B220, CD19, and Ter119. For the adult BM lineage cocktail, CD11b was added to that of FL. All of these antibodies are conjugated to PerCP-Cy5.5. Sca-1-APC, c-Kit–PECy7, CD48-PE, and CD150-biotin (all antibodies from eBioscience) together with streptavidin-APC-Alexa Fluor 750 (Invitrogen) were used for signaling lymphocytic activation molecule (SLAM) staining. Data were collected using FACSCanto or FACSCalibur (both from BD) and were analyzed using FACSDiva software (BD) or FlowJo software (Tree Star). For the apoptosis assay, an annexin V-APC apoptosis kit (BD) was used. The cell cycle was evaluated by pyronin Y and 7-amino actinomycin D (7AAD) staining (30).
RT and qPCR.
Total RNA was extracted by using TRIzol (Life Technologies) and was treated with DNase I (Qiagen). Reverse transcription (RT) reactions were carried out by using a qScript cDNA synthesis kit (Quanta Biosciences). Quantitative PCR (qPCR) was performed with a SYBR Fast qPCR kit (Kapa Biosystems). For validation of the results of RASL-seq, a OneStep RT-PCR kit (Qiagen) was used. The primers used for RT-PCR and qPCR were as follows: mSrsf2(F) (CGCGCTCCAGATCAACCTC), mSrsf2(R) (CTTGGACTCTCGCTTCGACAC), mGAPDH(F) (GAPDH, glyceraldehyde-3-phosphate dehydrogenase) (GGTGCTGAGTATGTCGTGGAGTCTA), mGAPDH(R) (AAAGTTGTCATGGATGACCTTGG), hSRSF2 3′-UTR(F) (GCACTAGGCGCAGTTGTGTA), hSRSF2 3′-UTR(R) (CAATCGGGAGAAAACAGGAA), hSRSF2 Exon2 CDS(F) (CTACAGCCGCTCGAAGTCTC), hSRSF2 Exon2 CDS(R) (TTGGATTCCCTCTTGGACAC), hGAPDH(F) (TCGCTCAGACACCATGGGGAAG), and hGAPDH(R) (GCCTTGACGGTGCCATGGAATTTG).
Western blotting.
Cell extracts from mouse bone marrow were prepared with Thermo Scientific Pierce immunoprecipitation (IP) lysis buffer (PI-87787), including protease inhibitor cocktail (PI-88665) and phosphatase inhibitor cocktail (PI-88667). Western blotting was performed following standard procedures. Protein samples were denatured in 1× loading buffer (10% glycerol, 2% SDS, 10 mM dithiothreitol [DTT], 50 mM Tris-HCl [pH 6.8]). The protein concentration was adjusted, and protein samples were loaded on SDS polyacrylamide gels after adding bromophenol blue (0.05%). Primary anti-SRSF2 antibody (ab28428; Abcam) and anti-β-actin antibody (A1978; Sigma-Aldrich) were used. Signals from fluorophore-conjugated secondary antibodies were detected with an Odyssey system (Li-COR).
Retrovirus infection.
The virus infection procedure was performed as described previously (27). For general retrovirus infection, 293T cells were transfected with 5 μg of retrovirus vectors and with 5 μg of Ecopac packaging vector using polyethylenimine (Polysciences Inc.). The 293T medium was changed from Dulbecco's modified Eagle's medium (DMEM) to Iscove's modified Dulbecco's medium (IMDM) 10 h posttransfection. Retroviral supernatants were harvested 48 h after transfection and filtered through a 0.45-μm-pore-size filter. The supernatant was added to primary bone marrow cells, along with 4% IL-3–carboxymethylcellulose (CM), 4% stem cell factor (SCF)-CM, 1% HEPES, and 0.1% Polybrene (final concentration, 4 μg/ml). The cells were spinoculated at 1,200 × g for 3 h at 32°C. Infections were performed twice on consecutive days. For overexpression of WT and mutant SRSF2, BM cells were transduced with MIP vector or MIP-SRSF2 expression retrovirus by two rounds of infection and were selected in 1 μg/ml puromycin for 3 days before assays.
For lentivirus production, 293T cells were transfected with pTRIPZ lentivirus vectors, pCMV-VSVG, and pCMV-dR8.2 using Lipofectamine 2000 (Life Technologies) for 6 h, and then the medium was changed. Forty-eight hours later, culture supernatant was harvested and filtered through a 0.45-μm-pore-size filter. For viral transduction, RPMI 1640 medium with 10 ng/ml hIL-3 was diluted with viral supernatant at a 1:1 ratio. The MDS-L cells were then cultured in the mixed medium with 8 μg/ml Polybrene overnight. The next day, medium was changed, and the stable cell lines with incorporated viral DNA segments were selected in 2 μg/ml puromycin for 7 days.
Stress hematopoiesis.
5-Fluorouracil (5-FU; GeneraMedix) was injected i.p. every week (150 mg/kg body weight). Survival was monitored every day. The combined results of independent two experiments are shown.
For sublethal irradiation, mice received 4 Gy total body irradiation. Cell counts were followed up.
BMT assay.
For bone marrow transplantation (BMT) assays of overexpressed-Srsf2 cells, donor C57BL/6 mice were injected i.p. with 150 mg 5-fluorouracil (5-FU)/kg of body weight 5 days prior to bone marrow harvest. BM cells were harvested, treated with ACK buffer, and washed with PBS. Cells were transduced with MigR1, MigR1-SRSF2WT, and MigR1-SRSF2P95H vectors. The GFP proportion was adjusted to 17% using uninfected cells. Infected BM cells were injected into lethally irradiated (9 Gy) recipient mice through the tail veins. Recipient mice were given acidic water (pH 4.0) for 3 weeks following BMT.
For competitive and noncompetitive BMT, total BM cells were harvested from indicated donor mice without treatment. In noncompetitive settings, 2 million test cells in 200 μl PBS were injected into lethally irradiated (9 Gy) recipient PEP3 mice. In competitive settings, test cells were mixed with competitor cells (PEP3 cells) at a 1:1 ratio, and 2 million cells were injected into lethally irradiated recipient PEP3 mice.
RASL-seq.
Dox was added at the indicated concentrations to MDS-L cell culture every day for 3 days. Total RNA was extracted from cells and used for RASL-seq. Analysis of splicing changes was performed as described previously (25, 26). Gene function and pathway analyses were performed using Qiagen's Ingenuity pathway analysis (IPA; Qiagen).
Statistics.
All the experiments were repeated at least twice. Results were represented as means ± standard deviations (SD) unless otherwise stated. Comparison of the results from the two groups was done by t test. Survival data were presented as Kaplan-Meier curves, and a log-rank test was performed. A P value of <0.05 was considered significant.
Accession number.
Original data from this work have been uploaded to the GEO database under accession no. GSE61052.
RESULTS
SRSF2 is essential for viability of blood cells in vitro.
To test the functional requirement of Srsf2 in blood cells, we first pursued an in vitro model by infecting bone marrow (BM) cells derived from adult Srsf2+/+ and Srsf2flox/flox mice with MSCV-Cre-IRES-enhanced yellow fluorescent protein (EYFP) (Cre) or empty vector (MSCV-IRES-EYFP) (EV) retrovirus. In the absence of Cre expression, Srsf2+/+ (+/+ EV) and Srsf2flox/flox (f/f EV) cells did not show any growth difference. However, in the presence of Cre expression, Srsf2flox/flox cells (f/f Cre cells) exhibited a clear growth disadvantage compared to Srsf2+/+ cells (+/+ Cre) (Fig. 1A). In addition, f/f Cre cells were significantly more apoptotic than the cells in other groups (Fig. 1B). These results demonstrate that SRSF2 is essential for blood cell growth and survival in vitro.
FIG 1.
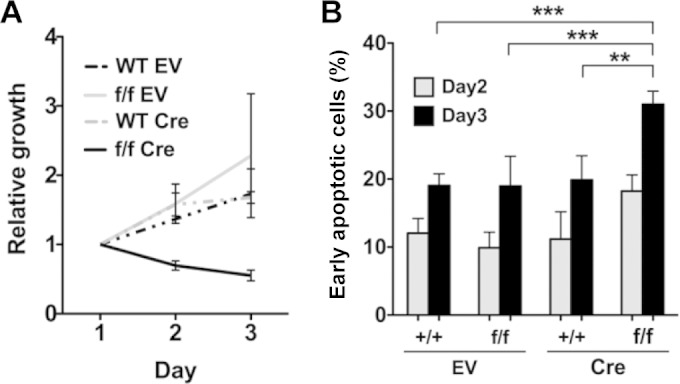
SRSF2 is essential for survival of adult BM cells in vitro. (A) MSCV-IRES-EYFP empty vector (EV) or MSCV-Cre-IRES-EYFP (Cre) retrovirus-infected Srsf2+/+ (WT) and Srsf2flox/flox (f/f) cells were seeded in duplicate, and cell numbers were counted with trypan blue staining. The number of retrovirus-infected cells was determined based on EYFP expression. Cre-expressing Srsf2 cKO cells (f/f Cre) showed growth suppression. (B) Apoptosis of EYFP+ cells on day 3. EYFP+ cells were gated, and early apoptotic cells were defined as annexin V+ 7AAD− cells. Cre+ Srsf2flox/flox (f/f) cells showed significantly enhanced apoptosis. Results of three independent experiments were used for statistical calculations. **, P < 0.005; ***, P < 0.001.
SRSF2 plays an indispensable role in the development of the hematopoietic system in mice.
To pursue the functional impact of Srsf2 deletion on hematopoietic cells under in vivo conditions, we first crossed Srsf2flox/flox mice with blood cell-specific Cre transgenic mice (Vav-iCre mice) (31). Fetal liver (FL) hematopoiesis was analyzed in multiple sets of littermates at embryonic day 12.5 (E12.5) and E14.5. Compared to those of the others, the Vav-iCre+ Srsfflox/flox (Srsf2Δ/Δ) embryos had paler and smaller FLs (Fig. 2A) which contained significantly fewer definitive blood cells (10% of the level seen with the wild-type controls at E14.5) (Fig. 2B). The remaining cells in Srsf2Δ/Δ FLs exhibited more apoptosis and quiescence (Fig. 2C and D), which is compatible with the in vitro data (Fig. 1B). A histological study performed on E14.5 FLs showed that erythroblasts and mature granulocytes were not detectable in Srsf2Δ/Δ FLs (Fig. 2E). Srsf2Δ/Δ embryos died during embryonic development between E16.5 and E18.5 with severe anemia and edema (Table 1 and data not shown). These results demonstrate the essential role of Srsf2 in hematopoiesis during embryonic development.
FIG 2.
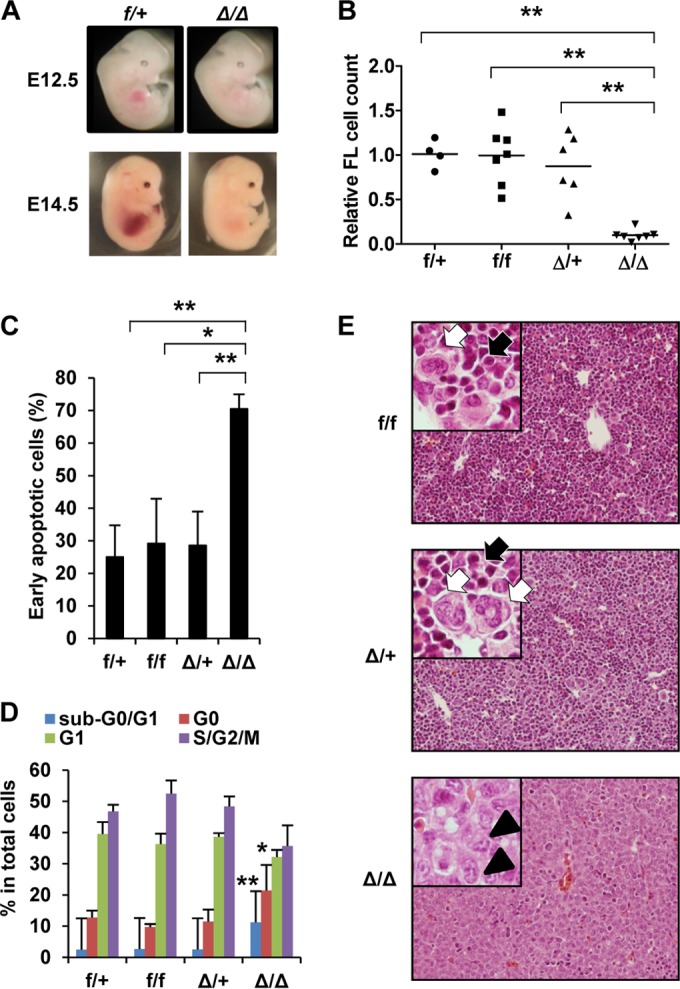
Fetal liver (FL) hematopoiesis is defective in Vav-iCre+ Srsf2flox/flox embryos. Vav-iCre+ Srsf2flox/+ mice were crossed with Srsf2flox/flox mice to generate Vav-iCre− Srsf2flox/+ (f/+), Vav-iCre+ Srsf2flox/+ (Δ/+), Vav-iCre− Srsf2flox/flox (f/f), and Vav-iCre+ Srsf2flox/flox (Δ/Δ) embryos. (A) Macroscopic appearance of E12.5 and E14.5 embryos. Δ/Δ embryos have smaller FLs than the controls. (B) Relative cell numbers of E14.5 fetal livers. Each cell number was normalized to the average of the numbers of f/+ FLs in the same litter. Horizontal bars show the average for each group. f/+, n = 4; f/f, n = 7; Δ/+, n = 6; Δ/Δ, n = 7. Δ/Δ FL cells were significantly fewer than those in other groups. (C) Apoptosis assay. Early apoptotic cells were defined as annexin V+ 7AAD− cells. f/+, n = 5; f/f, n = 2, Δ/+, n = 6; Δ/Δ, n = 6. Δ/Δ FLs had significantly more apoptotic cells than those in other groups. (D) Cell cycle analysis by pyronin Y and 7AAD staining. Δ/Δ FL cells show both a significant increase in the levels of apoptotic (sub-G0/G1) cells and a decrease in the levels of cycling (S/G2/M) cells compared to other groups. f/+, n = 5; f/f, n = 2; Δ/+, n = 6; Δ/Δ, n = 6. (E) Histology of E14.5 FL (hematoxylin-eosin stain). Δ/Δ FLs consist of hepatoblasts characterized by a large, pale-staining nucleus with distinct nucleoli (arrowheads). FLs of other genotypes are filled with hematopoietic cells, the majority of which are erythroblasts (black arrows), with scattered white blood cells (white arrows). Original magnification, ×200. Insets, ×400. Objective lenses, UPlanFL 20×, 0.50 numerical aperture (NA), and UPlanFL 40×, 0.75 NA (both from Olympus, Tokyo, Japan). Images were acquired at room temperature using an Olympus BX51 microscope equipped with a DP71 camera and DP controller/DP Manager software (Olympus, Tokyo, Japan). *, P < 0.05; **, P < 0.005.
TABLE 1.
Number of surviving and dead mice with different genotypes of Srsf2a
| Day | No. of live (dead) mice |
|||
|---|---|---|---|---|
| f/+ | f/f | Δ/+ | Δ/Δ | |
| E11.5 | 4 | 2 | 1 | 3 |
| E12.5 | 5 | 2 | 1 | 0 |
| E13.5 | 3 | 3 | 1 | 0 |
| E14.5 | 3 | 3 | 4 | 5 |
| E15.5 | 2 | 1 | 1 | 2 |
| E16.5 | 3 | 2 | 4 | 2 (4) |
| E17.5 | 5 | 5 | 6 | 0 (3) |
| E18.5 | 3 | 5 | 2 | 0 (6) |
| P21 | 7 | 4 | 6 | 0 |
f/+, Srsf2flox/+; f/f, Srsf2flox/flox; Δ/+, Srsf2Δ/+; Δ/Δ; Srsf2Δ/Δ; E, embryonic day; P, postnatal day.
To further characterize the defects in FL hematopoiesis, we next examined hematopoietic stem/progenitor cells (HSPCs) in FLs by flow cytometry. Importantly, we detected no HSPCs (lineage− c-Kit+) in E14.5 Srsf2Δ/Δ FL hematopoietic cells (Fig. 3A and B). In agreement with this result, a colony formation assay of E12.5 FL cells showed that Srsf2Δ/Δ cells had significantly lower clonogenicity (Fig. 3C). Differential counts of colonies did not show significant changes in any comparisons between groups (data not shown). In addition, peripheral blood (PB) of Vav-iCre+ Srsf2flox/flox (Srsf2Δ/Δ) embryos had significantly higher levels of primitive red blood cells as shown by the number of nucleated erythrocytes (Fig. 3D and E). Throughout these experiments, Δ/+ embryos did not show significant differences from +/+ embryos, suggesting that one allele is sufficient for normal hematopoietic stem cell (HSC) function. Thus, SRSF2 is essential for the survival of embryonic hematopoietic stem and progenitor cells (HSPCs).
FIG 3.
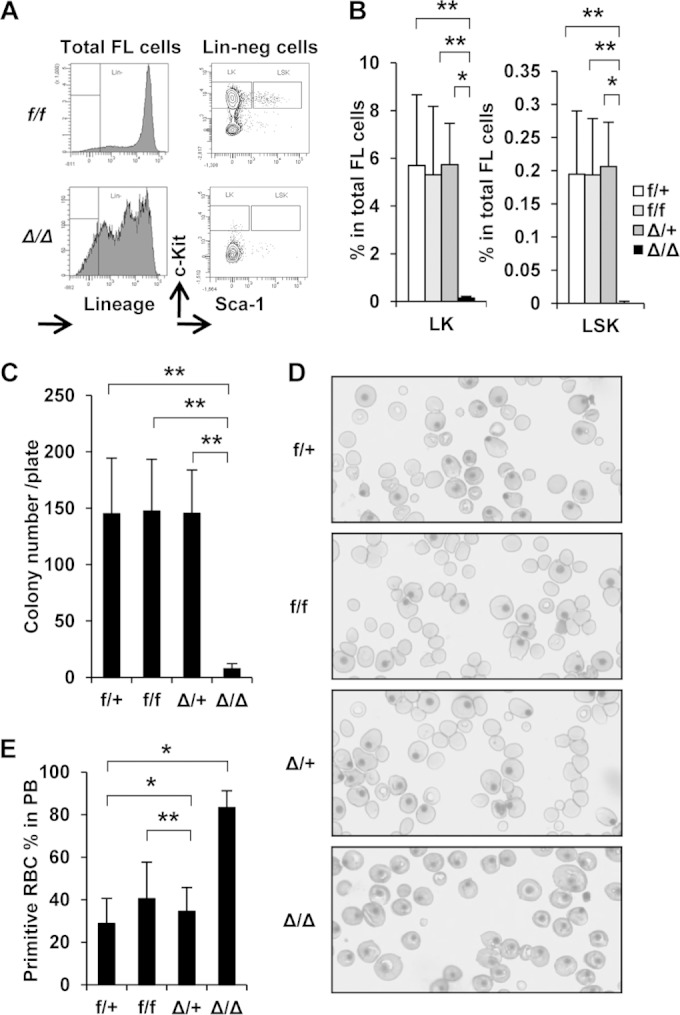
Srsf2 null fetal livers lack hematopoietic stem/progenitor cells. (A) Representative flow cytometry results from E14.5 FL cells. Lin-neg, lineage negative. Srsf2Δ/Δ FL lacked Lin-neg/c-Kit+ (LK) and Lin-neg/Sca1+/c-Kit+ (LSK) cells. (B) Quantification of the results presented in panel A. f/+, n = 10; f/f, n = 12; Δ/+, n = 5; Δ/Δ, n = 6. (C) Colony formation assay of E12.5 FL cells. Cells (2 × 104) were seeded in duplicate. f/+, n = 8; f/f, n = 24; Δ/+, n = 10; Δ/Δ, n = 6. Δ/Δ FL cells showed significantly lower colony-forming ability than others. (D) Representative results of Wright-Giemsa-stained cytospin processing of E14.5 PB. Δ/Δ PB lacked definitive red blood cells. (E) Quantification of the results presented in panel D. RBC, red blood cells. f/+, n = 4; f/f, n = 5; Δ/+, n = 8; Δ/Δ, n = 7. *, P < 0.05; **, P < 0.005.
SRSF2 is required for survival of adult BM cells.
Haploinsufficient expression of certain critical hematopoietic regulators, such as RUNX1 and PU.1, has been shown to affect both the number and distribution of different populations of blood cells (32, 33). Interestingly, most reported mutations of SRSF2 in MDS are monoallelic, suggesting potential haploinsufficiency of SRSF2 in disrupting hematopoiesis. To explore this possibility, we tested whether adult heterozygous (Vav-iCre+ Srsf2flox/+ [Δ/+]) mice had recordable phenotypes relative to control (Vav-iCre− Srsf2flox/+ [f/+]) mice. Srsf2 expression, measured by RT-qPCR and Western blot analysis, in Δ/+ BM cells was almost half the level seen with their flox/+ counterparts (data not shown). We detected little difference between WT and heterozygous mice on the basis of blood counts performed under either normal or stressed conditions (cell count recovery after sublethal irradiation [4 Gy] or weekly 5-FU injection [150 mg/kg], aging stress up to 10 months) (data not shown). These results suggest that the SRSF2 level in the heterozygous mice remained half of that in WT mice and that SRSF2 expression from one allele is sufficient for maintaining its normal cellular function.
Lack of SRSF2 in the developing heart resulted in dilated cardiomyopathy, while cKO of mature cardiomyocytes did not have any obvious phenotype (22, 23). Those reports suggest that loss of SRSF2 can result in different phenotypes at different developmental stages. Therefore, we also evaluated the effect of SRSF2 ablation in adult blood cells. We crossed Srsf2flox/flox mice with Mx1-Cre mice to generate poly(I):poly(C) [poly(IC)]-inducible Srsf2 knockout mice. Interestingly, Mx1-Cre+ Srsf2flox/flox mice stayed alive at least for 3 months after seven injections of 600 μg/dose poly(IC) (data not shown). Genotyping of blood and BM cells showed incomplete excision of the floxed Srsf2 allele in blood and BM cells from Mx1-Cre+ Srsf2flox/flox mice. In contrast, we detected nearly complete ablation of the floxed allele in blood and BM cells from heterozygous Mx1-Cre+ Srsf2flox/+ mice (Fig. 4A). Genotyping of colonies derived from single bone marrow cells confirmed incomplete knockout, showing both floxed and deleted bands for Mx1-Cre+ Srsf2flox/flox cells (data not shown). These results indicate a high level of selection pressure against the loss of Srsf2. On day 16, poly(IC)-treated Mx1-Cre+ Srsf2flox/flox mice had significantly decreased platelet counts in their PB (Fig. 4B). Other examined blood parameters, including neutrophil counts and percentages of myeloid (Gr1+ CD11b+), B (B220+), and T (CD4+ CD8+) cells, were normal (data not shown). Decreased counts of white blood cells and lymphocytes in Mx1-Cre+ Srsf2flox/+ and Mx1-Cre+ Srsf2flox/flox mice were observed (data not shown), which was possibly due to Cre expression. Importantly, BM showed a significant decrease in total BM cell counts, absolute numbers of lineage− Sca-1+ c-Kit+ (LSK) cells in total BM, and percentages of LSK cells in BM of Mx1-Cre+ Srsf2flox/flox mice compared to the Mx1-Cre− Srsf2flox/flox controls (Fig. 4C to E). SLAM staining using CD150 and CD48 antibodies did not record any significant change in the ratios of positive and negative cells (data not shown). These results suggest that the immature BM fraction (LSK) was more susceptible to the loss of SRSF2 than the further-differentiated cell populations.
FIG 4.

SRSF2 is indispensable for survival of adult BM cells. (A to D) Poly(IC) injection into adult Mx1-Cre+ or Mx1-Cre− Srsf2 cKO mice. Poly(IC) was injected i.p. every other day for three injections in total into cKO mice (600 μg/dose). The day of the first injection was defined as day 0. Mx1-Cre− Srsf2flox/+, n = 11. Mx1-Cre− Srsf2flox/flox, n = 23. Mx1-Cre+ Srsf2flox/+, n = 9. Mx1-Cre+ Srsf2flox/flox, n = 24. (A) Genotyping of PB and BM cells on day16. Mx1-Cre+ Srsf2flox/+ samples showed almost complete excision of the flox allele, while Mx1-Cre+ Srsf2flox/flox samples showed incomplete excision. (B) Platelet (Plt) count on day 16. (C) Total BM cells from femurs and tibias of both legs counted using trypan blue. (D) Absolute number of LSK cells in total BM measured by flow cytometry. (E) Percent LSK in total BM measured by flow cytometry. (F) Competitive BMT of Mx1-Cre cKO cells. One million test cells (CD45.2) were mixed with the same number of competitor cells (CD45.1) and transplanted. Chimerism in PB was evaluated by flow cytometry before (week 0) and after poly(IC) injection. Five mice were in each group. Mx1-Cre+ Srsf2flox/flox cells had lower engraftment efficiency than others and showed a continuous decrease in the level of donor chimerism after poly(IC) injection. *, P < 0.05; **, P < 0.005.
To further establish that the observed phenotypes are cell autonomous, we performed competitive bone marrow transplantation (BMT) of Mx1-Cre+ Srsf2flox/flox BM cells (Fig. 4F). One month after BMT, Mx1-Cre+ Srsf2flox/flox recipients showed significantly lower engraftment even before poly(IC) injection, possibly due to high interferon sensitivity of Mx1-Cre+ Srsf2flox/flox cells induced early in the BMT procedure (34). Poly(IC) injection caused a further decrease of donor chimerism in these mice. These results thus demonstrate the cell-autonomous effect of Srsf2 ablation on the survival of immature hematopoietic stem cells.
SRSF2 overexpression also causes a growth disadvantage.
Single-allele SRSF2 mutations have been commonly identified in patients suffering from MDS (6, 16, 17, 19). On the other hand, heterozygous deletion of Srsf2 in blood cells of Vav-iCre+ Srsf2flox/+ mice (Fig. 2 and 3) and poly(IC)-treated Mx1-Cre+ Srsf2flox/+ mice (Fig. 4) did not show obvious phenotypes. These results suggest that SRSF2 mutations in MDS are not simply loss-of-function mutations but rather reflect altered functions. We wished to further test the hypothesis of altered function under overexpression conditions by selecting the MDS-associated P95H missense mutation and in-frame 8-amino-acid deletion (Δ8aa [P95-to-R102 deletion]) for comparison with WT SRSF2. MIP vector and MIP-WT, -P95H, and -Δ8aa SRSF2 retroviruses were used to infect murine bone marrow cells. The level of exogenously expressed WT, P95H, and Δ8aa SRSF2 protein is 4-fold to 7-fold above the endogenous SRSF2 level (Fig. 5A). Liquid culture of infected cells showed growth suppression by both WT and mutant SRSF2 under such overexpression conditions (Fig. 5B). Importantly, the cells expressing the mutant SRSF2 showed a significant increase in apoptosis relative to their WT counterparts (Fig. 5C), though the cell cycle was not inhibited (data not shown). Consistent with the liquid culture data, the colony formation assay showed significantly lower colony and cell numbers in WT-infected cells, and the numbers were even lower in the P95H and Δ8aa cells than in the MIP-infected cells (Fig. 5D). The colony types (granulocyte, erythroid, macrophage, megakaryocyte [GEMM], burst-forming unit–erythroid [BFU-E], CFU-granulocyte [CFU-G], and CFU-granulocyte/macrophage [CFU-GM], surface markers (Gr-1 and CD11b), and cell morphologies were not different among the groups (data not shown). These results suggest that increased expression of SRSF2 mainly affects cell survival but does not disrupt myeloid cell differentiation. More importantly, overexpression of the P95H and Δ8aa mutants always showed stronger negative effects on cell survival than overexpression of the WT SRSF2.
FIG 5.

Overexpression of WT and overexpression of mutant SRSF2 result in similar phenotypes. (A) Protein expression of the overexpressed SRSF2 in mouse BM cells. β-Actin served as a loading control. Signal intensities of SRSF2 and the loading control were used to calculate the relative SRSF2 expression levels. (B) Growth curves of total BM cells in liquid culture. Means and SD of the results from three independent experiments are shown. All of the WT-, P95H-, and Δ8aa-expressing retrovirus-transduced cells showed a growth disadvantage compared to MIP control cells. (C) Apoptosis assay of cells on day 3. Combined data from independent three experiments are shown. P95H and Δ8aa cells showed enhanced apoptosis compared to MIP and WT cells. (D) Colony formation assay. Combined data from three independent experiments are shown. (E) BMT of SRSF2-overexpressing cells. Bone marrow cells were transduced with MigR1 vector, MigR1-SRSF2 WT, and MigR1-SRSF2 P95H retrovirus. Two days after the initial cell infection, cells were transplanted into lethally irradiated recipient mice. Five recipients were used in each group. Representative results of three independent experiments are shown. MigR1 recipients had a higher percentage of GFP in PB compared to their SRSF2 WT and P95H counterparts. *, P < 0.05; **, P < 0.005.
We next performed a transplantation of SRSF2-overexpressing bone marrow cells to evaluate the repopulation ability in vivo. The percentage of GFP+ cells in PB of WT and P95H cell recipients was lower than in MigR1 cell recipient mice. However, the difference between the WT and P95H groups was not significant (Fig. 5E). Lineage staining of GFP+ PB cells did not show lineage skewing (myeloid versus lymphoid) in response to overexpression of WT or mutant SRSF2 (data not shown). Furthermore, these cells did not show a difference in dysplastic morphology in PB and BM during the observation of 10 months (data not shown). Data in these experiments suggest that overexpressed WT and P95H both caused a significant growth disadvantage, which was also accompanied by induced apoptosis. However, neither appears to have had any recordable effect on cell differentiation either in vitro and in vivo.
SRSF2 mutants altered a large RNA splicing program.
Although our functional analysis of overexpressed SRSF2 is consistent with earlier reports of overexpressed SR proteins interfering with developmental processes (35–37), the data indicate a clear advantage in studying the mutant function of SRSF2 in cellular systems that may enable us to recapitulate SRSF2 single-allele mutations in MDS patients. We generated a relatively low-level expression system by lowering levels of endogenous SRSF2 with shRNA and simultaneously expressing the mutant version of the protein in a human MDS cell line (MDS-L) (28, 29). This cell line was derived from an MDS patient and has a chromosome 5q deletion but no mutation in splicing factor SRSF2 or splicing factor U2AF1 (confirmed by resequencing; data not shown). We constructed Tet-inducible lentivirus vectors to coexpress an shRNA targeting the 3′ UTR of endogenous SRSF2 and an shRNA-resistant form of SRSF2 cDNA (WT or mutant) (Fig. 6A). MDS-L cells were transduced with SRSF2 shRNA/cDNA lentivirus to establish pools of inducible cells expressing WT, P95H, and Δ8aa shRNA. Doxycycline (Dox) treatment reduced endogenous SRSF2 (shown by 3′-UTR expression) while inducing relatively low overexpression of total SRSF2 RNA (3-fold to 4-fold of the endogenous level) (shown by the exon 2 coding sequence [CDS]) (Fig. 6B). Under these conditions, mutant cells (P95H and Δ8aa) showed significant growth arrest and enhanced apoptosis compared to WT cells (Fig. 6C and D). However, cell cycle analysis did not show any obvious change except for a detectable increase in the levels of sub-G0/G1 cells (data not shown). These data indicate that the system closely mimics certain MDS conditions in a disease-relevant cell line.
FIG 6.
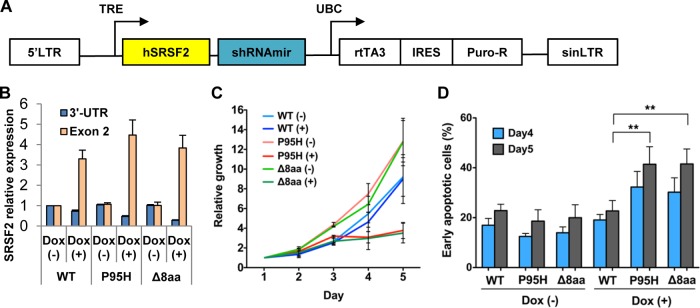
Inducible low-level expression of mutant SRSF2 causes apoptosis and growth arrest. (A) Structure of pTRIPZ-SRSF2 constructs. Expression of microRNA-adapted shRNA for endogenous SRSF2 (shRNAmir) and cDNA of shRNA-resistant WT or mutant human SRSF2 (hSRSF2) is driven by a tetracycline-inducible promoter (TRE). LTR, long terminal repeat; UBC, human ubiquitin C promoter; rtTA3, reverse tetracycline transactivator 3; IRES, internal ribosome entry site; Puro-R, puromycin-resistant gene; sinLTR, self-inactivating long terminal repeat. (B) Quantitative RT-PCR of SRSF2. The samples were measured in duplicate and normalized to GAPDH. Expression in untreated WT cells was set to 1. 3′-UTR, endogenous SRSF2 expression; Exon 2, exon 2 coding sequence, representing total SRSF2 expression. (C) Growth curves. Cells were seeded in triplicate in four independent experiments and counted using trypan blue. Dox-treated (+) P95H and Δ8aa cells showed significant growth suppression compared to other groups. (D) Apoptosis assay. Cells were seeded in duplicate in four independent experiments. Early apoptotic cells are defined as annexin V+ 7AAD− cells. Dox-treated P95H and Δ8aa cells showed significantly enhanced apoptosis compared to other groups. **, P < 0.005.
Although it is widely anticipated that SRSF2 mutations will perturb the splicing program in diseased cells, the global impact of SRSF2 mutations on regulated splicing has not been investigated. To reveal potential functional differences between WT and mutant SRSF2 that may be relevant to MDS development, we examined the splicing response by using the RASL-seq platform, which was designed to detect 5,530 annotated alternatively spliced mRNA isoforms in the human genome (26). MDS-L cells coexpressing Tet-inducible SRSF2 shRNA and WT, P95H, or Δ8aa SRSF2 were cultured with or without Dox for 3 days to modulate SRSF2 expression. Total RNA was isolated from these cells for splicing profiling.
Based on stringent cutoff values (a fold ratio change of >1.5 and P value of <0.05), we detected both overlapped and separated changes in splicing upon expression of WT and mutant SRSF2 (Fig. 7A and B; see also Table S1 to S5 in the supplemental material), indicating that MDS-associated mutations have a function shared with and an effect independent from WT SRSF2 with respect to splicing. Relative to WT cells, P95H and Δ8aa mutants induced a common set of 487 events, 470 of which were altered in the same direction, including 164 enhanced cassette exon inclusions and 306 enhanced cassette exon exclusions (Fig. 7C; see also Table S6). Importantly, the P95H and Δ8aa mutants induced 210 and 483 extra events, respectively. Ten commonly changed splicing events in genes related to hematopoiesis were selected and validated by RT-PCR (Fig. 7D). Similarly to the results seen with shRNA alone, MEIS1, UPF38, and PRKAA1 had increased ratios of short to long isoforms in SRSF2 mutant cells, suggesting a reduction of the SRSF2 function of SRSF2 mutants in these splicing events. RBM23, PDK1, PDE4DIP, MLL, and RNF34 had increased short-to-long-isoform ratios while CBFB and SMG7 had decreased ratios in SRSF2 mutant cells independently of shRNA alone, suggesting the gain-of-function effect of SRSF2 P95H and Δ8aa mutants in these splicing events. Furthermore, the change of the scale of exon inclusion or exclusion induced by Δ8aa mutant was over 1.5 times greater in 450 of the 470 common events relative to the P95H mutant. Together, these data suggest that, even though the two mutants showed certain overlapping effects on splicing, they were not functionally equivalent; the deletion mutation may have a more profound impact on regulated splicing than the P95H mutation.
FIG 7.

Target genes of WT and mutant SRSF2 by RASL-seq analysis. (A) Unsupervised hierarchical clustering showing that splicing events of WT, P95H, and Δ8aa (Δ8) cells in the absence of doxycycline (Dox −) are very similar and are separated from those of cells with activated shRNA/SRSF2 expression (Dox +) (left panel). Δ8aa and P95H data are grouped side by side, with Δ8aa showing a stronger signal. Results of unsupervised hierarchical clustering of splicing events consisting of shRNA-only cells cultured in the presence or absence of Dox are also shown (right panel). Green, less short isoform, more long isoform; red, more short isoform, less long isoform. (B) Venn diagram showing overlaps and differences of splicing events upon Dox-activated expression of WT, P95H, and Δ8aa SRSF2 compared to shRNA-only expression. (C) Venn diagram showing significant overlapped events (n = 487) from the P95H and Δ8aa groups relative to the WT SRSF2 group. Among these shared events, over 96% were oriented in the same direction. For the significant events that changed in the same direction, Δ8aa-induced changes in 450 of 470 events were over 1.5-fold greater than those corresponding to P95H-induced events. (D) Validation of 10 selected RASL-seq events. Upper panel, heat map demonstration of the ratios of 10 RASL-seq events (short isoform/long isoform). Green, less short isoform, more long isoform; red, more short isoform, less long isoform. Bottom panel, RT-PCR validation of 10 RASL-seq events.
To gain functional insights into such a dramatically altered splicing program, we reasoned that the altered splice events commonly affected by P95H and Δ8aa might be related more to MDS, while extra events induced by each mutant might be responsible for enhancing the disease phenotype. We thus focused on the commonly affected set of 470 genes by applying Ingenuity pathway analysis. Consistent with the functional consequences triggered by the mutations in our cellular and animal models, we observed cancer development and apoptosis pathways among the 10 top-ranked canonical (P95H induced) pathways (Fig. 8A). These findings support the possibility that the MDS-associated mutations in SRSF2 promote the development of the disease phenotype with potential to induce a cascade of events that lead to both disease progression and more-aggressive types of blood disorders.
FIG 8.
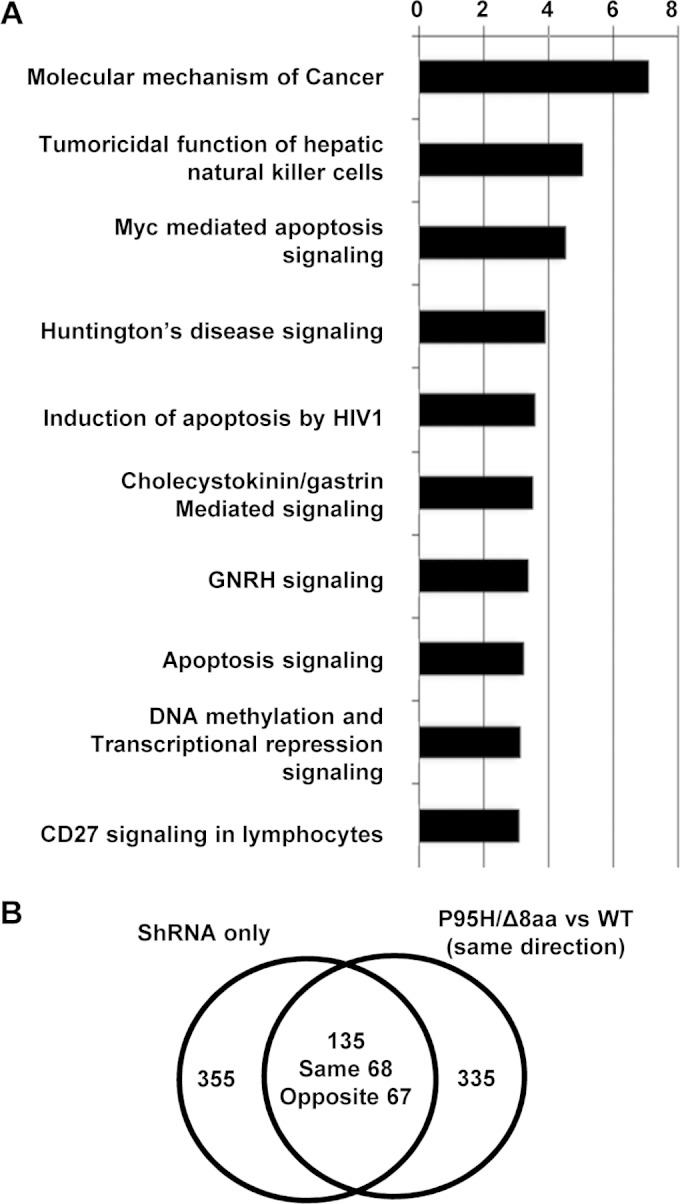
Pathway and correlation studies of target genes of WT and mutant SRSF2. (A) Top biological functions of the 470 common and same-direction targets of Δ8aa and P95H based on IPA. The −log(P value) (x axis) corresponds to the significance of the functions with respect to the data set. GNRH, gonadotropin-releasing hormone. (B) Venn diagram showing significant overlapped events among 470 common targets of P95H and Δ8aa cells versus WT SRSF2 cells relative to cells expressing only shRNA. Events that changed in the same direction or in opposite directions are also listed separately.
To analyze the possible loss or gain of function of the P95H and Δ8aa mutations, we also examined their 470 commonly affected splicing events in MDS-L cells that expressed only shRNA of SRSF2 but not any shRNA-resistant SRSF2. Upon Dox treatment, SRSF2 expression in these cells was knocked down nearly 50% (data not shown). A total of 135 events (29% of 470) were affected by the SRSF2 reduction (Fig. 8B; see also Table S7 in the supplemental material). Interestingly, we detected similar numbers of splicing events changed in the same direction and in opposite directions between the SRSF2 mutants and knockdown cells. The remaining 335 of 470 events were not affected by SRSF2 knockdown. These results suggest that P95H and Δ8aa mutations lead to loss of function in some splicing events (shared and in the same direction), enhanced function in some other WT SRSF2-involved splicing events (shared but in opposite directions), and also potentially gain of function in regulating new splicing events (not overlapped). It is possible that some of these combinatory loss and gain effects promote MDS development.
DISCUSSION
In this report, we examined the role of SRSF2 in hematopoiesis using the Srsf2 cKO mouse model. We also analyzed the cellular effect of expressing MDS-related mutant forms of SRSF2. Our analyses demonstrate that SRSF2 is essential for the survival of blood cells, including HSCs. In the absence of SRSF2, blood cells had increased senescence and apoptosis. However, unlike previous reports in several other cellular systems, we did not detect any significant delay of the cell cycle in SRSF2-deficient blood cells or observe an effect on differentiation of myeloid cells. Interestingly, ablation of SRSF2 in pituitary cells and thymocytes results in decreased organ size but no increase of apoptosis (23, 24). Developing thymocytes showed a block of differentiation from the CD4 CD8 double-negative stage to the double-positive stage (24). Ablation of SRSF2 in the embryonic heart showed normal development, but the mice later suffered from dilated cardiomyopathy (22). However, mice with the Srsf2 knockout in postmitotic cardiomyocytes stayed normal (23). These different phenotypes of SRSF2 in different cell types may result from alteration of cell type-specific targets of SRSF2. For example, in thymocytes lacking SRSF2, the defective splicing of lymphoid cell-specific CD45 RNA has been related to their abnormal differentiation (23).
SR proteins, including SRSF2, are known to autoregulate their own expression via an intron retention event in the 3′ UTR (38–40). In response to SRSF2 overexpression, splicing of this retained intron likely produces an exon junction complex (EJC), triggering nonsense-mediated mRNA decay. As the intron is mostly retained in normal cells, reduction of SRSF2 may have little effect on mRNA stability. Consistently, we detected a nearly 50% decrease of SRSF2 levels in Srsf2Δ/+ heterozygous blood cells. In light of a recent report that heterozygous Sf3b1-deficient mice showed haploinsufficiency in HSCs (41) and because SRSF2 mutations in MDS patients are generally heterozygous, we examined hematopoiesis in mice with Srsf2 heterozygous blood cells during development and under stress conditions. No obvious phenotypes were observed, suggesting that the MDS-related heterozygous SRSF2 mutations are not simply loss-of-function mutations and that MDS may not result from SRSF2 haploinsufficiency. Instead, our data suggest likely gain-of-function mutations in SRSF2, including potential dominant-negative effects that reduce total SRSF2 activity.
It is striking that SRSF2 mutations in MDS mainly occur at proline 95 (P95), suggesting a critical role of P95 in SRSF2. SRSF2 has an N-terminal RNA recognition motif (RRM) domain and a C-terminal arginine-and-serine-rich (RS) domain. The RRM domain is primarily involved in specific RNA recognition. The RS domain can be highly phosphorylated and is involved in both RNA binding and protein-protein interactions during spliceosome assembly (21). P95 is generally believed to be located in the hinge region between the RRM domain and the RS domain. A recent structural analysis of the N-terminal 101 amino acids of SRSF2 bound to 6-oligonucleotide RNA targets showed that P95 directly contacts the C3 and G3 nucleotides in RNA containing the UCCAGU and UGGAGU motifs, which represent two consensus SRSF2 binding sequences (42). It is possible that SRSF2 with mutations at P95 decreases the RNA binding specificity. This possibility is supported by a recent report that, unlike the WT SRSF2, the P95H mutant preferentially binds to a CCAG motif (43). Furthermore, the unique features of proline among the 20 protein-forming amino acids, including its cis-trans isomerization (44), CH/Pi hydrogen bond formation (45), and higher conformational rigidity in the secondary structure of proteins (44), may result in a different local structure to create a new surface for interaction with additional proteins and RNA sequences. These speculations are subject to future biochemical studies.
We compared WT SRSF2 and mutant (P95H and Δ8aa) SRSF2 by overexpressing them via MSCV retrovirus transduction. With this approach, exogenously expressed SRSF2 protein is 4-fold to 7-fold above its endogenous level. Similar inhibitory effects of the WT and both mutants on cell growth, colony formation, and in vivo blood cell repopulation were detected in these overexpression assays (Fig. 5). These results support the hypothesis presented above that WT SRSF2 and P95 mutant SRSF2 have certain similar functions and that, above normal levels, such functions disrupt hematopoiesis. Interestingly, the negative effects of overexpressing the WT SRSF2 and P95H SRSF2 on in vivo repopulation are nearly identical (Fig. 5D), suggesting that HSPCs are likely more sensitive to the level of SRSF2.
We also used a Tet-On system to induce coexpression of an SRSF2 shRNA and an shRNA-resistant WT or mutant SRSF2 to mimic expression levels in the MDS-L cell line in patients. In this experimental setting, we detected unique cell growth-inhibitory effects of mutant SRSF2 which were more likely due to the unique gain-or-loss functions of mutant SRSF2.
Using the RASL-seq platform, we observed that apoptosis- and cancer-related pathways were among the 10 top-ranked canonical pathways affected by SRSF2 mutations, which is consistent with the pathology of MDS cells of patients (1, 2). MDS cells were noted to be highly apoptotic at early stages, likely reflecting a key mechanism to eliminate cells, and eventually become malignant (1, 2, 46). Thus, mutant forms of SRSF2 in MDS seem to trigger genomic instability and to promote accumulation of unwanted oncogenic mutations (47). RASL-seq (25, 26) is a convenient way to screen the alternative splicing changes and has multiple advantages, including (i) the ability to use limited amounts of total RNA (typically from 0.1 to 1 μg of total RNA) for the analysis, (ii) a considerable tolerance of a range of RNA quality to obtain robust data, (iii) the sensitivity to obtain quantitative information on alternative splicing events in less-abundant transcripts, and (iv) the ability to perform highly parallel analyses of biological samples in a cost-effective manner. However, compared to transcriptome sequencing (RNA-seq), RASL-seq is limited to analysis of annotated alternative splicing events and cannot detect other aberrant RNA processing events. Despite these limitations, we successfully applied this technique to identify a large cohort of alternative splicing events uniquely caused by mutant SRSF2, suggesting that the mutant splicing factor has the capacity to dramatically alter the splicing program in diseased cells. It is unclear whether mutant SRSF2 also affects certain constitutive splicing events.
In conclusion, we demonstrated that SRSF2 is essential for the survival of hematopoietic cells and that its loss results in apoptosis and growth suppression. No obvious phenotypes were detected in Srsf2 heterozygous blood cells, but growth arrest and increased apoptosis were clearly detected in cells expressing either P95H or Δ8aa mutants. Together, these results support the idea that MDS-related SRSF2 mutations are not simply loss-of-function mutations. Furthermore, RASL-seq analysis demonstrated that MDS mutant forms of SRSF2 dysregulate RNA splicing. Future work will be focused on understanding the molecular mechanism of SRSF2 mutations in MDS development, including their effects on RNA splicing and other aspects of regulated gene transcription (48, 49). Future studies will also examine how mutations in SRSF2 may act in synergy with mutations in other key leukemia genes, such as those encoding TET2, IDH2, and RUNX1, to propel MDS progression to more-aggressive blood cell disorders.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIHR01DK098808. Y.K. was supported partially by Sumitomo Life Social Welfare Services Foundation and Mochida Memorial Foundation for Medical and Pharmaceutical Research. Y.-J.H. is supported partially by a scholarship from Ministry of Education from the Republic of China.
We thank Amanda Scholl for editing the manuscript, Omar Abdel-Wahab for critical review, Daniel Starczynowski for providing MDS-L cells, and Gregory Hannon for providing pTRIPZ vector.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00202-15.
REFERENCES
- 1.Ades L, Itzykson R, Fenaux P. 20 March 2014, posting date Myelodysplastic syndromes. Lancet doi: 10.1016/S0140-6736(13)61901-7. [DOI] [PubMed] [Google Scholar]
- 2.Malcovati L, Hellstrom-Lindberg E, Bowen D, Ades L, Cermak J, Del Canizo C, Della Porta MG, Fenaux P, Gattermann N, Germing U, Jansen JH, Mittelman M, Mufti G, Platzbecker U, Sanz GF, Selleslag D, Skov-Holm M, Stauder R, Symeonidis A, van de Loosdrecht AA, de Witte T, Cazzola M. 2013. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood 122:2943–2964. doi: 10.1182/blood-2013-03-492884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kulasekararaj AG, Mohamedali AM, Mufti GJ. 2013. Recent advances in understanding the molecular pathogenesis of myelodysplastic syndromes. Br J Haematol 162:587–605. doi: 10.1111/bjh.12435. [DOI] [PubMed] [Google Scholar]
- 4.Walter MJ, Shen D, Shao J, Ding L, White BS, Kandoth C, Miller CA, Niu B, McLellan MD, Dees ND, Fulton R, Elliot K, Heath S, Grillot M, Westervelt P, Link DC, DiPersio JF, Mardis E, Ley TJ, Wilson RK, Graubert TA. 2013. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia 27:1275–1282. doi: 10.1038/leu.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D, Ebert BL. 2011. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 364:2496–2506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, Chalkidis G, Suzuki Y, Shiosaka M, Kawahata R, Yamaguchi T, Otsu M, Obara N, Sakata-Yanagimoto M, Ishiyama K, Mori H, Nolte F, Hofmann WK, Miyawaki S, Sugano S, Haferlach C, Koeffler HP, Shih LY, Haferlach T, Chiba S, Nakauchi H, Miyano S, Ogawa S. 2011. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 7.Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, Krysiak K, Harris CC, Koboldt DC, Larson DE, McLellan MD, Dooling DJ, Abbott RM, Fulton RS, Schmidt H, Kalicki-Veizer J, O'Laughlin M, Grillot M, Baty J, Heath S, Frater JL, Nasim T, Link DC, Tomasson MH, Westervelt P, DiPersio JF, Mardis ER, Ley TJ, Wilson RK, Walter MJ. 2012. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet 44:53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshida K, Ogawa S. 2014. Splicing factor mutations and cancer. Wiley Interdiscip Rev RNA 5:445–459. doi: 10.1002/wrna.1222. [DOI] [PubMed] [Google Scholar]
- 9.Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, Godfrey AL, Rapado I, Cvejic A, Rance R, McGee C, Ellis P, Mudie LJ, Stephens PJ, McLaren S, Massie CE, Tarpey PS, Varela I, Nik-Zainal S, Davies HR, Shlien A, Jones D, Raine K, Hinton J, Butler AP, Teague JW, Baxter EJ, Score J, Galli A, Della Porta MG, Travaglino E, Groves M, Tauro S, Munshi NC, Anderson KC, El-Naggar A, Fischer A, Mustonen V, Warren AJ, Cross NC, Green AR, Futreal PA, Stratton MR, Campbell PJ. 2011. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 365:1384–1395. doi: 10.1056/NEJMoa1103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Visconte V, Makishima H, Jankowska A, Szpurka H, Traina F, Jerez A, O'Keefe C, Rogers HJ, Sekeres MA, Maciejewski JP, Tiu RV. 2012. SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia 26:542–545. doi: 10.1038/leu.2011.232. [DOI] [PubMed] [Google Scholar]
- 11.Zhang SJ, Rampal R, Manshouri T, Patel J, Mensah N, Kayserian A, Hricik T, Heguy A, Hedvat C, Gonen M, Kantarjian H, Levine RL, Abdel-Wahab O, Verstovsek S. 2012. Genetic analysis of patients with leukemic transformation of myeloproliferative neoplasms shows recurrent SRSF2 mutations that are associated with adverse outcome. Blood 119:4480–4485. doi: 10.1182/blood-2011-11-390252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damm F, Nguyen-Khac F, Fontenay M, Bernard OA. 2012. Spliceosome and other novel mutations in chronic lymphocytic leukemia and myeloid malignancies. Leukemia 26:2027–2031. doi: 10.1038/leu.2012.86. [DOI] [PubMed] [Google Scholar]
- 13.Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, Ramsay AJ, Bea S, Pinyol M, Martinez-Trillos A, Lopez-Guerra M, Colomer D, Navarro A, Baumann T, Aymerich M, Rozman M, Delgado J, Gine E, Hernandez JM, Gonzalez-Diaz M, Puente DA, Velasco G, Freije JM, Tubio JM, Royo R, Gelpi JL, Orozco M, Pisano DG, Zamora J, Vazquez M, Valencia A, Himmelbauer H, Bayes M, Heath S, Gut M, Gut I, Estivill X, Lopez-Guillermo A, Puente XS, Campo E, Lopez-Otin C. 2012. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 44:47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 14.Rossi D, Bruscaggin A, Spina V, Rasi S, Khiabanian H, Messina M, Fangazio M, Vaisitti T, Monti S, Chiaretti S, Guarini A, Del Giudice I, Cerri M, Cresta S, Deambrogi C, Gargiulo E, Gattei V, Forconi F, Bertoni F, Deaglio S, Rabadan R, Pasqualucci L, Foa R, Dalla-Favera R, Gaidano G. 2011. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood 118:6904–6908. doi: 10.1182/blood-2011-08-373159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dolnik A, Engelmann JC, Scharfenberger-Schmeer M, Mauch J, Kelkenberg-Schade S, Haldemann B, Fries T, Kronke J, Kuhn MW, Paschka P, Kayser S, Wolf S, Gaidzik VI, Schlenk RF, Rucker FG, Dohner H, Lottaz C, Dohner K, Bullinger L. 2012. Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood 120:e83–e92. doi: 10.1182/blood-2011-12-401471. [DOI] [PubMed] [Google Scholar]
- 16.Thol F, Kade S, Schlarmann C, Loffeld P, Morgan M, Krauter J, Wlodarski MW, Kolking B, Wichmann M, Gorlich K, Gohring G, Bug G, Ottmann O, Niemeyer CM, Hofmann WK, Schlegelberger B, Ganser A, Heuser M. 2012. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood 119:3578–3584. doi: 10.1182/blood-2011-12-399337. [DOI] [PubMed] [Google Scholar]
- 17.Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC, Pellagatti A, Shlien A, Groves MJ, Forbes SA, Raine K, Hinton J, Mudie LJ, McLaren S, Hardy C, Latimer C, Della Porta MG, O'Meara S, Ambaglio I, Galli A, Butler AP, Walldin G, Teague JW, Quek L, Sternberg A, Gambacorti-Passerini C, Cross NC, Green AR, Boultwood J, Vyas P, Hellstrom-Lindberg E, Bowen D, Cazzola M, Stratton MR, Campbell PJ. 2013. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 122:3616–3627, quiz 3699. doi: 10.1182/blood-2013-08-518886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirabayashi S, Flotho C, Moetter J, Heuser M, Hasle H, Gruhn B, Klingebiel T, Thol F, Schlegelberger B, Baumann I, Strahm B, Stary J, Locatelli F, Zecca M, Bergstraesser E, Dworzak M, van den Heuvel-Eibrink MM, De Moerloose B, Ogawa S, Niemeyer CM, Wlodarski MW. 2012. Spliceosomal gene aberrations are rare, coexist with oncogenic mutations, and are unlikely to exert a driver effect in childhood MDS and JMML. Blood 119:e96–e99. doi: 10.1182/blood-2011-12-395087. [DOI] [PubMed] [Google Scholar]
- 19.Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, Przychodzen B, Bupathi M, Guinta K, Afable MG, Sekeres MA, Padgett RA, Tiu RV, Maciejewski JP. 2012. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 119:3203–3210. doi: 10.1182/blood-2011-12-399774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu SJ, Kuo YY, Hou HA, Li LY, Tseng MH, Huang CF, Lee FY, Liu MC, Liu CW, Lin CT, Chen CY, Chou WC, Yao M, Huang SY, Ko BS, Tang JL, Tsay W, Tien HF. 2012. The clinical implication of SRSF2 mutation in patients with myelodysplastic syndrome and its stability during disease evolution. Blood 120:3106–3111. doi: 10.1182/blood-2012-02-412296. [DOI] [PubMed] [Google Scholar]
- 21.Zhou Z, Fu XD. 2013. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 122:191–207. doi: 10.1007/s00412-013-0407-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding JH, Xu X, Yang D, Chu PH, Dalton ND, Ye Z, Yeakley JM, Cheng H, Xiao RP, Ross J, Chen J, Fu XD. 2004. Dilated cardiomyopathy caused by tissue-specific ablation of SC35 in the heart. EMBO J 23:885–896. doi: 10.1038/sj.emboj.7600054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao R, Sun Y, Ding JH, Lin S, Rose DW, Rosenfeld MG, Fu XD, Li X. 2007. Splicing regulator SC35 is essential for genomic stability and cell proliferation during mammalian organogenesis. Mol Cell Biol 27:5393–5402. doi: 10.1128/MCB.00288-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang HY, Xu X, Ding JH, Bermingham JR Jr, Fu XD. 2001. SC35 plays a role in T cell development and alternative splicing of CD45. Mol Cell 7:331–342. doi: 10.1016/S1097-2765(01)00181-2. [DOI] [PubMed] [Google Scholar]
- 25.Li H, Qiu J, Fu XD. 2012. RASL-seq for massively parallel and quantitative analysis of gene expression. Curr Protoc Mol Biol Chapter 4:4.13.1–4.13.9. doi: 10.1002/0471142727.mb0413s98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou Z, Qiu J, Liu W, Zhou Y, Plocinik RM, Li H, Hu Q, Ghosh G, Adams JA, Rosenfeld MG, Fu XD. 2012. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol Cell 47:422–433. doi: 10.1016/j.molcel.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komeno Y, Yan M, Matsuura S, Lam K, Lo MC, Huang YJ, Tenen DG, Downing JR, Zhang DE. 25 April 2014, posting date Runx1 exon 6-related alternative splicing isoforms differentially regulate hematopoiesis in mice. Blood doi: 10.1182/blood-2013-08-521252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsuoka A, Tochigi A, Kishimoto M, Nakahara T, Kondo T, Tsujioka T, Tasaka T, Tohyama Y, Tohyama K. 2010. Lenalidomide induces cell death in an MDS-derived cell line with deletion of chromosome 5q by inhibition of cytokinesis. Leukemia 24:748–755. doi: 10.1038/leu.2009.296. [DOI] [PubMed] [Google Scholar]
- 29.Drexler HG, Dirks WG, Macleod RA. 2009. Many are called MDS cell lines: one is chosen. Leuk Res 33:1011–1016. doi: 10.1016/j.leukres.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 30.Toba K, Winton EF, Koike T, Shibata A. 1995. Simultaneous three-color analysis of the surface phenotype and DNA-RNA quantitation using 7-amino-actinomycin D and pyronin Y. J Immunol Methods 182:193–207. doi: 10.1016/0022-1759(95)00050-K. [DOI] [PubMed] [Google Scholar]
- 31.de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, Norton T, Williams K, Roderick K, Potocnik AJ, Kioussis D. 2003. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol 33:314–325. doi: 10.1002/immu.200310005. [DOI] [PubMed] [Google Scholar]
- 32.Iwasaki H, Somoza C, Shigematsu H, Duprez EA, Iwasaki-Arai J, Mizuno S, Arinobu Y, Geary K, Zhang P, Dayaram T, Fenyus ML, Elf S, Chan S, Kastner P, Huettner CS, Murray R, Tenen DG, Akashi K. 2005. Distinctive and indispensable roles of PU.1 in maintenance of hematopoietic stem cells and their differentiation. Blood 106:1590–1600. doi: 10.1182/blood-2005-03-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun W, Downing JR. 2004. Haploinsufficiency of AML1 results in a decrease in the number of LTR-HSCs while simultaneously inducing an increase in more mature progenitors. Blood 104:3565–3572. doi: 10.1182/blood-2003-12-4349. [DOI] [PubMed] [Google Scholar]
- 34.Cong X, Yan M, Yin X, Zhang DE. 2010. Hematopoietic cells from Ube1L-deficient mice exhibit an impaired proliferation defect under the stress of bone marrow transplantation. Blood Cells Mol Dis 45:103–111. doi: 10.1016/j.bcmd.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lopato S, Kalyna M, Dorner S, Kobayashi R, Krainer AR, Barta A. 1999. atSRp30, one of two SF2/ASF-like proteins from Arabidopsis thaliana, regulates splicing of specific plant genes. Genes Dev 13:987–1001. doi: 10.1101/gad.13.8.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kraus ME, Lis JT. 1994. The concentration of B52, an essential splicing factor and regulator of splice site choice in vitro, is critical for Drosophila development. Mol Cell Biol 14:5360–5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Labourier E, Bourbon HM, Gallouzi IE, Fostier M, Allemand E, Tazi J. 1999. Antagonism between RSF1 and SR proteins for both splice-site recognition in vitro and Drosophila development. Genes Dev 13:740–753. doi: 10.1101/gad.13.6.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sureau A, Gattoni R, Dooghe Y, Stevenin J, Soret J. 2001. SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J 20:1785–1796. doi: 10.1093/emboj/20.7.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE. 2007. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 446:926–929. doi: 10.1038/nature05676. [DOI] [PubMed] [Google Scholar]
- 40.Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O'Brien G, Shiue L, Clark TA, Blume JE, Ares M Jr. 2007. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev 21:708–718. doi: 10.1101/gad.1525507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsunawa M, Yamamoto R, Sanada M, Sato-Otsubo A, Shiozawa Y, Yoshida K, Otsu M, Shiraishi Y, Miyano S, Isono K, Koseki H, Nakauchi H, Ogawa S. 18 February 2014, posting date Haploinsufficiency of Sf3b1 leads to compromised stem cell function but not to myelodysplasia. Leukemia doi: 10.1038/leu.2014.73. [DOI] [PubMed] [Google Scholar]
- 42.Daubner GM, Clery A, Jayne S, Stevenin J, Allain FH. 2012. A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J 31:162–174. doi: 10.1038/emboj.2011.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, Cho H, Kim MK, Zebari AS, Aumann S, Park CY, Buonamici S, Smith PG, Deeg HJ, Lobry C, Aifantis I, Modis Y, Allain FH, Halene S, Bradley RK, Abdel-Wahab O. 2015. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell 27:617–630. doi: 10.1016/j.ccell.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.MacArthur MW, Thornton JM. 1991. Influence of proline residues on protein conformation. J Mol Biol 218:397–412. doi: 10.1016/0022-2836(91)90721-H. [DOI] [PubMed] [Google Scholar]
- 45.Ozawa T, Okazaki K, Kitaura K. 2011. Importance of CH/pi hydrogen bonds in recognition of the core motif in proline-recognition domains: an ab initio fragment molecular orbital study. J Comput Chem 32:2774–2782. doi: 10.1002/jcc.21857. [DOI] [PubMed] [Google Scholar]
- 46.Zhou T, Hasty P, Walter CA, Bishop AJ, Scott LM, Rebel VI. 2013. Myelodysplastic syndrome: an inability to appropriately respond to damaged DNA? Exp Hematol 41:665–674. doi: 10.1016/j.exphem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Montecucco A, Biamonti G. 2013. Pre-mRNA processing factors meet the DNA damage response. Front Genet 4:102. doi: 10.3389/fgene.2013.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ji X, Zhou Y, Pandit S, Huang J, Li H, Lin CY, Xiao R, Burge CB, Fu XD. 2013. SR proteins collaborate with 7SK and promoter-associated nascent RNA to release paused polymerase. Cell 153:855–868. doi: 10.1016/j.cell.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mo S, Ji X, Fu XD. 2013. Unique role of SRSF2 in transcription activation and diverse functions of the SR and hnRNP proteins in gene expression regulation. Transcription 4:251–259. doi: 10.4161/trns.26932. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.