

Abstract
Accurate replication of DNA is imperative for the maintenance of genomic integrity. We identified Enhancer of Rudimentary Homolog (ERH) using a whole-genome RNA interference (RNAi) screen to discover novel proteins that function in the replication stress response. Here we report that ERH is important for DNA replication and recovery from replication stress. ATR pathway activity is diminished in ERH-deficient cells. The reduction in ATR signaling corresponds to a decrease in the expression of multiple ATR pathway genes, including ATR itself. ERH interacts with multiple RNA processing complexes, including splicing regulators. Furthermore, splicing of ATR transcripts is deficient in ERH-depleted cells. Transcriptome-wide analysis indicates that ERH depletion affects the levels of ∼1,500 transcripts, with DNA replication and repair genes being highly enriched among those with reduced expression. Splicing defects were evident in ∼750 protein-coding genes, which again were enriched for DNA metabolism genes. Thus, ERH regulation of RNA processing is needed to ensure faithful DNA replication and repair.
INTRODUCTION
Proper repair of DNA damage is crucial for the maintenance of genomic integrity. DNA is constantly bombarded by genotoxic factors, leading to the acquisition of multiple types of DNA damage, including oxidation and alkylation of bases, DNA cross-linking, formation of pyrimidine dimers, adduct formation, and DNA breaks.
Damage encountered during DNA replication is especially problematic, and cells possess tightly regulated mechanisms to ensure that DNA is replicated completely and accurately to maintain genomic integrity. The ataxia telangiectasia-mutated and Rad3-related (ATR) signaling pathway is a major mechanism by which cells respond to and repair replication-associated damage (1). DNA damage encountered by the replication machinery leads to replication fork stalling and/or collapse (2, 3). This is often accompanied by uncoupling of the DNA polymerase from the replicative helicase that unwinds the DNA at the fork, leading to the formation of single-stranded DNA (ssDNA) (1, 4). The ssDNA is then coated by replication protein A (RPA), followed by the binding of ATRIP to RPA, which recruits its binding partner ATR to the stalled replication fork (5–7). The RFC-RAD17 complex, the RAD9-RAD1-HUS1 (9-1-1) complex, and TOPBP1 are also recruited, contributing to the activation of ATR (1, 8, 9). Additional mechanisms operate to fine-tune ATR regulation (10). Once ATR is activated, it phosphorylates CHK1 and hundreds of other proteins, affecting a phosphorylation cascade that results in the activation of cell cycle checkpoints, stabilization of the replication fork, and repair of the DNA damage (1, 11).
Here we describe the function of Enhancer of Rudimentary Homolog (ERH), which we identified by a whole-genome small interfering RNA (siRNA) screen designed to find replication stress response genes. ERH is the homolog of the Drosophila melanogaster E(r) gene, which was found as an enhancer of the phenotype caused by mutations in rudimentary (r) (12). rudimentary encodes a protein required for pyrimidine biosynthesis, suggesting that ERH could have some function in nucleic acid metabolism (13).
Previous reports have proposed various functions for ERH. ERH is a small, highly conserved nuclear protein that is believed to function in transcriptional regulation, the cell cycle, mitosis, and optimal cell growth under stress conditions (12, 14–17). Recent studies also implicate ERH in RNA processing. Exogenously expressed ERH colocalizes with exogenously expressed SC35 in nuclear speckles believed to be splicing factor compartments (18). Additionally, ERH interacts with the Sm complex subunit SNRPD3 (which binds spliceosome snRNAs) and is important for the splicing of CENPE mRNA (17). Depletion of ERH results in reduced gene expression of a subset of cell cycle and DNA repair genes. However, other studies indicate a more direct function of ERH in DNA replication due to colocalization with CIZ1, a replication protein found at replication factories (15, 19).
Here we report that ERH is important for the replication stress response. We demonstrate that ERH-deficient cells exhibit reduced recovery from replication stress challenges and slower DNA replication fork elongation even in unchallenged cells. Localization, proteomic, and transcriptomic analyses indicate that the mechanism of ERH function in these processes is by ensuring proper mRNA splicing of ATR and other repair and DNA processing proteins.
MATERIALS AND METHODS
Cell culture.
U2OS and HEK293T cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 7.5% fetal bovine serum (FBS).
RNA interference (RNAi) and plasmids.
Cells were transfected with 10 nM siRNA by using DharmaFECT 1 (Dharmacon) according to the manufacturer's instructions and incubated for 72 h prior to experimental assays. siRNAs used were the All-Stars negative control (Qiagen), ATR siRNA (CCUCCGUGAUGUUGCUUGA) (Dharmacon), a pool of four ERH siRNAs (AGACAUACCAGCCUUAUAA, GGGAAAUAAUUGUGUUGGA, AAGAGAAGAUCUACGUGCU, and UAGCCAAGAUUGACUGUAU), ERH siRNA 1 (CCAACAGGCUGGGAAAUAA), ERH siRNA 2 (GAACUUAUGCUGACUACGA), ERH siRNA 3 (UCAGUCAGUUGUUUGAUUU), and ERH siRNA 4 (GUCCCUCUAUCACAUAUGA) (Dharmacon).
FLAG- and green fluorescent protein (GFP)-tagged ERH expression vectors were generated by using the Gateway Technology system. ERH-GFP lentivirus was generated by transfection of pLL5.0-ERH-GFP, dR7.74psPAX2, and pMD2.G into HEK293T cells. Virus was collected, and U2OS cells were transduced twice to obtain a stable cell line expressing ERH-GFP.
Antibodies.
Antibodies used for immunofluorescence (IF) include Alexa Fluor 488 azide-conjugated, Cy5–goat anti-mouse, Alexa Fluor 594–goat anti-mouse, and Alexa Fluor 568–goat anti-rabbit antibodies (Life Technologies), γH2AX antibody (Millipore), and RPA32 antibody (Abcam). Antibodies used for immunoblotting include antibody to ERH (Fisher), antibodies to pCHK1 Ser317, MRE11, and protein-arginine methyltransferase 1 (PRMT1) (Cell Signaling), antibodies to ATR, CHK1, and RAD9 (Santa Cruz), antibodies to KU70 and RPA32 (Abcam), antibodies to TOPBP1, ATRIP, THRAP3, DGCR8, and CHTOP (Bethyl Labs), antibody to RAD50 (Genetex), antibody to FLAG M2 (Sigma), antibody to ORC2 (BD Pharmingen), and antibody to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Millipore).
Replication recovery/restart assay.
Seventy-two hours after transfection with siRNAs, U2OS cells were treated with 2 mM hydroxyurea (HU) for 24 h and then washed and released into medium containing 10 μM 5-ethynyl-2′-deoxyuridine (EdU) for 4 h. Cells were subsequently fixed with 3.7% paraformaldehyde–phosphate-buffered saline (PBS). Cells were permeabilized for 20 min with 0.5% Triton X-100–PBS, washed with PBS, and incubated for 30 min in click chemistry reaction buffer (Alexa Fluor 488 azide, 2 mg/ml sodium ascorbate, 2 mM copper sulfate in PBS) to conjugate biotin to EdU incorporated into the DNA. Cells were then washed, blocked for 30 min with 10% Image-iT FX signal enhancer (Life Technologies) in PBS, washed, blocked in 10% FBS–PBS, and incubated with γH2AX antibody in 1% bovine serum albumin (BSA)–PBS overnight at 4°C. The following day, cells were washed, incubated with Alexa Fluor 568–goat anti-rabbit secondary antibody in 1% BSA–PBS for 20 min, washed, incubated in 200 ng/ml 4′,6-diamidino-2-phenylindole (DAPI) in PBS for 3 min, washed, and imaged by using an Opera QEHS (quadruple-excitation high-sensitivity) system (PerkinElmer). Nuclei were defined by the DAPI signal, and γH2AX and EdU intensity levels for each nucleus were obtained by using Columbus software (PerkinElmer).
Viability assays.
Seventy-two hours following transfection with siRNAs, U2OS cells were treated with 0.2 mM HU, 1 nM gemcitabine (Gem), or 5 nM camptothecin (CPT) for 72 h, and cell viability was measured with alamarBlue. Statistical differences were determined by using one-way analysis of variance (ANOVA).
Immunoblot analyses.
Cells were lysed in NETN buffer (50 mM Tris [pH 8], 200 mM NaCl, 1% IGEPAL) supplemented with protease and phosphatase inhibitors (5 μg/ml aprotinin, 5 μg/ml leupeptin, 1 mM NaF, 1 mM sodium orthovanadate, 1 mM dithiothreitol [DTT], 1 mM phenylmethylsulfonyl fluoride [PMSF], and 20 mM β-glycerophosphate). Lysates were separated by SDS-PAGE, transferred onto a nitrocellulose membrane, and probed for proteins of interest. Chromatin fractionations were performed as previously described (20).
Immunofluorescence.
To examine levels of γH2AX in ERH-depleted cells, U2OS cells were transfected with nontargeting (NT) and ERH siRNAs in 96-well plates and fixed 72 h later. Cells were permeabilized with 0.5% Triton X-100–PBS for 5 min, blocked with 5% BSA–PBS, incubated in primary antibody diluted in 1% BSA–PBS for 1 h at 37°C, washed, incubated in secondary antibody for 20 min at room temperature, and stained with 200 ng/ml DAPI in PBS, and images were obtained by using an Image Express Micro XL instrument (Molecular Devices). γH2AX intensity levels were determined for each nucleus by using MetaXpress software (Molecular Devices) and analyzed by using the Kruskal-Wallis test. To assess colocalization of ERH with damage sites, U2OS cells stably expressing ERH-GFP were plated onto coverslips and treated with 2 mM HU for 5 h and fixed or with 5 Gy gamma irradiation (IR) and fixed 1 h later. IF was performed as described above, and coverslips were mounted with Prolong Gold containing DAPI. Images were obtained by using a Zeiss AxioPlan2 microscope equipped with a Zeiss camera.
For microirradiation, U2OS cells stably expressing ERH-GFP were sensitized to DNA damage by incubation with 10 μM bromodeoxyuridine (BrdU) for 24 h, and cells were then microirradiated by using a UVA laser as described previously (21). Cells were fixed 10 min later and processed for IF analysis.
Flow cytometry.
The HU recovery assay was performed as previously described (9), except that cells were treated for 20 h with 3 mM HU and harvested 10 h following release.
DNA fiber labeling assay.
U2OS cells were labeled with 20 μM 5-iodo-2′-deoxyuridine (IdU) for 20 min, washed twice with equilibrated HEPES-buffered saline, and then labeled with 100 μM 5-chloro-2′-deoxyuridine (CldU) for 20 min. DNA fiber analysis was then performed as described previously (22).
RNA extraction and quantitative PCR analysis.
U2OS cells were transfected with siRNA, cell pellets were collected at 72 h posttransfection, and total RNA was isolated by using the Qiagen RNeasy minikit. RNA was reverse transcribed to cDNA by using the iScript cDNA synthesis kit (Bio-Rad) for gene expression levels and the High-Capacity cDNA reverse transcription kit (Applied Biosystems) for splicing assays. iQ SYBR green supermix (Bio-Rad) was used to evaluate the expression of ERH, RAD50, ATRIP, CHK1, and ATR mature and prespliced mRNAs. For the detection of mature mRNA, the following interexon-spanning primers were used: ERH forward (F) primer GGCCAGAAGGCAGAACTTATG and reverse (R) primer GGGACTGTTGGGATTCATTCTT, RAD50 F primer TAAGTGTGCAGAAATTGACCGAG and R primer GACGTACCTGCCGAAGTGTT, ATRIP F primer GTGAACGAGCAAATAAACTGGC and R primer GAGGCTTGTATCCTGACTCCG, CHK1 F primer ATATGAAGCGTGCCGTAGACT and R primer TGCCTATGTCTGGCTCTATTCTG, and ATR F primer ACCTCAGCAGTAATAGTGATGGA and R primer GGCCACTGTATTCAAGGGAAAT. For detection of ATR splicing, the following primers were used: exon 5 (E5) F (ACCTCAGCAGTAATAGTGATGGA), intron 5-6 (I5-6) R (TGGCTAAATACAAACTGAATGAAGG), and E6 R (AGTGCACTCCATAATATGCTCTT) for the exon 5/6 junction; E11 F (TGGAAGATCCAGACAAAGATGTTA), I11-12 R (GCTAGGTTGACGTAAACTCAAATG), I11-12 F (GAGTTTACGTCAACCTAGCTCTAA), and E12 R (CAAGGTATCCTTCAGCTCATTATTTC) for the exon 11/12 junction; E24 F (GTTCGGGAAATACTAGAACCTCAT), I24-25 F (GTACTGAGGTAGTCAACTGAAGG), and E25 R (GATGCTGACCATTCTGCAAAG) for the exon 24/25 junction; E35 F (GCTACTAGTGGGCCGATTTATG), I35-36 R (ACGTAGTCAACAGAGTTAACTGAAA), I35-36 F (CCAGCATAGCGTGGGATTT), and E36 R (CCATGGGCATCAATTTGTCATAG) for the exon 35/36 junction; and E43 F (AGTGGTTTCTGAGAACATTCC), I43-44 R (TCCATATACATATGAGGCCAAT), I43-44 F (ACGTTGTTATGGTTGAATGTTTA), and E44 R (TGACATTACTGCAGTGGAAC) for the exon 43/44 junction. The following primers were used for detection of KU70 splicing: E2 F (GATGCCTCCAAGGCTATGTT), I2-3 R (CCTCCCAATGTCCTGAGATTAC), I2-3 F (GAAGGTAGGGATCTTGCCTTATT), and E3 R (CAAGAGATCTCGATCACTGCTTAT) for the exon 2/3 junction; E6 F (GTACAGGCGGTTTGCTTCT), I6-7 R (GGCTGTAGCGAGTCATGTT), I6-7 F (CTCGTAGCCTTCCCATTTGAT), and E7 R (GCCTCAGGTAATGGTGTTTCT) for the exon 6/7 junction; and E11 F (AGCTTGTTTACCCACCAGATTA), I11-12 R (AGGATATTGCCCACTGTGATAAG), I11-12 F (CTGGGATGCCACTTGTATGTA), and E12 R (GCTCCTCTTCTGAATACTCCAC) for the exon 11/12 junction. GAPDH was used as an endogenous control for all experimental samples (F primer GGTGGTCTCCTCTGACTTCAACAGCG and R primer GTTGCTGTAGCCAAATTCGTTGTCAT). Statistical differences of mature mRNA levels in nontargeting control and ERH-depleted cells were determined by using one-way ANOVA.
For mRNA stability assays, U2OS cells were transfected with siRNA and treated with 5 μg/ml actinomycin D for 0, 2, 4, 6, and 24 h to inhibit transcription. Cell pellets were collected, total RNA was isolated, and quantitative PCR (qPCR) for mature ATR mRNA was performed.
Mass spectrometry and immunoprecipitations.
To identify proteins that interact with ERH, HEK293T cells were transfected with a FLAG-ERH expression vector. FLAG-ERH was immunoprecipitated from nuclear extracts with anti-FLAG M2 beads (Sigma) and eluted from the beads with FLAG peptide. Eluted proteins were processed and analyzed by mass spectrometry essentially as described previously (23, 24). FLAG immunoprecipitations from mock-transfected HEK293T cells were processed in parallel to identify nonspecific contaminating proteins from the purification.
To confirm data from the mass spectrometry analysis, the same immunoprecipitation procedure was used, except that the lysate was treated with 10 μg/ml RNase A (Sigma) for 30 min to eliminate interactions bridged by RNA.
RNA sequencing analysis.
U2OS cells were transfected with nontargeting or ERH siRNA 3 for 72 h, and RNA was harvested by using the Qiagen miRNeasy minikit. Two biological replicates were collected per sample. Total RNA samples were verified by quality control analysis, rRNA reduction was performed, cDNA libraries were prepared by using the Illumina Tru-seq RNA sample preparation kit, and paired-end 75-bp high-throughput sequencing was performed by using the Illumina HiSeq2500 system. Quality of the RNA sequencing (RNA-seq) data was thoroughly examined at multiple stages of data processing, as outlined previously (25). Raw data and alignment quality control were performed by using QC3 (26), and expression analysis was carried out by using MultiRankSeq (27). Raw data were aligned with TopHat 2 (28) against the human HG19 reference genome. Gene and intron expression levels were quantified and normalized into fragments per kilobase of transcript per million reads (FPKM) by using Cufflinks (29). Differential gene expression was performed by using Cufflinks with a false discovery rate (FDR) of <0.05 to correct for multiple testing. Intron fold change levels were determined after adjusting for overall expression. Gene ontology analysis was performed by utilizing GORILLA (30, 31).
RESULTS
ERH deficiency results in DNA damage and inefficient restart of replication following replication stress.
A whole-genome siRNA screen to discover proteins that function in the replication stress response identified ERH as a protein of interest. Briefly, U2OS cells transfected with siRNA were treated with HU for 24 h and allowed to recover in the presence of EdU for 4 h (Fig. 1A). The samples were then examined by immunofluorescence to detect γH2AX and EdU intensity levels as measures of DNA damage and replication restart. As a control, cells depleted for ATR exhibited a high level of γH2AX and a low level of EdU incorporation compared to those of nontargeting (NT) controls (Fig. 1B), reflecting fork collapse in ATR-deficient cells (22, 32). A complete data set from the screen will be reported elsewhere. ERH was one of the genes for which four of four siRNAs tested caused increased γH2AX and decreased EdU incorporation compared to those of nontargeting controls, indicating that it may function in the replication stress response (Fig. 1B and C).
FIG 1.

ERH is important for recovery following replication stress. (A) Flowchart of the assay used to identify replication genes. (B and C) Nontargeting (NT), ATR, and ERH (pool of 4 or individual) siRNAs were processed as depicted in panel A, and γH2AX and EdU intensity levels were determined from immunofluorescence images from the replication restart (B) and validation (C) assays. The means and standard errors of the means of data from three biological replicates are reported. (D) Seventy-two hours after siRNA transfection, cells were treated with 2 mM HU for 20 h and then released into medium containing nocodazole for 10 h. Samples were collected, fixed, and stained with propidium iodide, and DNA content was measured by using a FACSCalibur instrument. (E to G) Seventy-two hours following transfection with NT, ATR, and ERH siRNAs, U2OS cells were split and treated with 0.2 mM HU (E), 1 nM gemcitabine (F), or 5 nM camptothecin (G) for 72 h and then examined for viability compared to untreated controls. (The means and standard errors of the means of data from three replicate experiments are shown. **** denotes a P value of <0.0001.) (H) U2OS cells were transfected with nontargeting or ERH siRNAs, and protein levels were examined at 72 h posttransfection by immunoblotting to confirm ERH knockdown.
To further characterize how ERH depletion affected replication recovery following a replication stress challenge, cells were arrested by treatment with HU for 20 h and then released into nocodazole to prevent cells from progressing past mitosis. Cells transfected with nontargeting siRNA progressed completely through S phase 10 h after removal of HU, while cells transfected with ERH siRNAs exhibited reduced progression through S phase (Fig. 1D).
ERH-deficient cells also exhibited hypersensitivity to the replication stress-inducing agents HU, gemcitabine, and camptothecin (Fig. 1E to G). Knockdown of ERH was confirmed by immunoblotting (Fig. 1H). These data confirm that ERH is necessary for recovery from an acute replication stress challenge.
ERH function is important for DNA replication and genome stability even in the absence of exogenous damaging agents.
Silencing of ERH, even in otherwise untreated cells, resulted in increased levels of γH2AX compared to those in nontargeting controls (Fig. 2A), indicating that ERH is also important for the maintenance of genomic stability in the absence of exogenous sources of replication stress.
FIG 2.
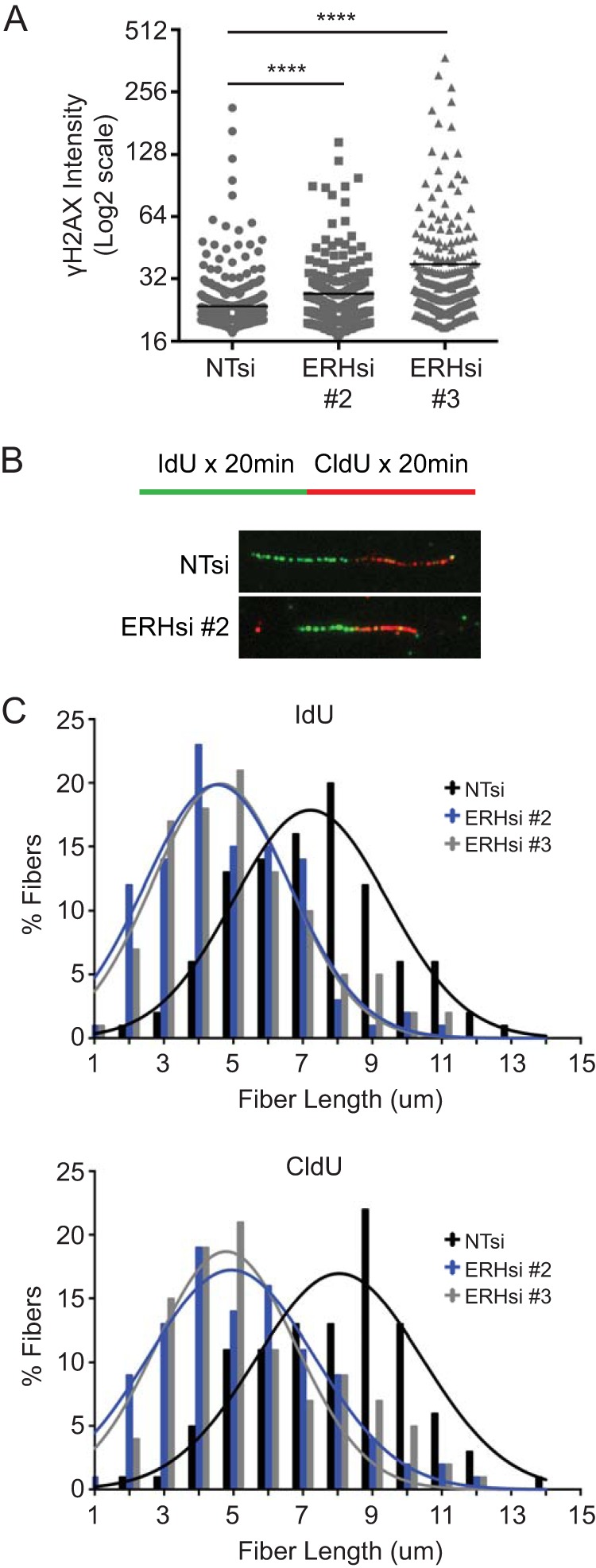
ERH is important for DNA replication and genome stability in the absence of exogenous stress. (A) U2OS cells were transfected with nontargeting and ERH siRNAs and fixed, and γH2AX levels were determined by immunofluorescence. (**** denotes a P value of <0.0001.) (B and C) Cells were transfected as described above for panel A and labeled with IdU for 20 min, followed by labeling with CldU for 20 min. Cells were collected and permeabilized, and DNA was spread onto glass slides. Immunofluorescence images were collected (B), and DNA fiber lengths were quantitated (C). The means of data from three replicate experiments are represented.
To better understand its function in the absence of exogenous DNA-damaging agents, we examined replication in ERH-depleted cells utilizing a DNA fiber labeling assay. Cells were labeled with IdU, followed by a labeling period with CldU, to allow the incorporation of these nucleoside analogs into newly synthesized DNA. DNA fibers were visualized by immunofluorescence, and the lengths of each label were measured to determine the amount of incorporation (Fig. 2B). ERH depletion resulted in reductions in fiber lengths of both IdU and CldU compared to those in nontargeting siRNA controls, indicating a problem in replication elongation (Fig. 2C). Thus, ERH is necessary for proper DNA replication in the absence of exogenous damage in addition to recovery following an acute replication stress challenge.
ERH is important for ATR signaling but is not observed at sites of replication or DNA damage.
The phenotypes associated with a loss of ERH function mimic those caused by an inhibition of the ATR pathway. Thus, we assessed whether the loss of ERH alters ATR signaling. To measure ATR pathway activation, control and ERH-depleted cells were treated with HU, and CHK1 phosphorylation levels were examined. CHK1 phosphorylation was reduced in ERH-deficient cells compared to that in nontargeting controls (Fig. 3A). We also noted a reduction in total CHK1 levels in these cells.
FIG 3.

ERH affects ATR pathway signaling, although it is not observed at sites of replication stress. (A) U2OS cells transfected with nontargeting and ERH siRNAs were treated with 2 mM and 3 mM HU for 2 and 4 h; cells were lysed; and protein levels of pCHK1, total CHK1, ERH, and GAPDH were examined by immunoblot analysis. A representative blot of three replicate experiments is depicted. (B and C) U2OS cells stably expressing GFP-tagged ERH were treated with 2 mM HU for 5 h and fixed (B and C) or were treated with 5 Gy IR and fixed 1 h later (C) prior to immunofluorescence imaging. (D) U2OS cells expressing ERH-GFP were microirradiated to induce DNA damage, incubated for 10 min, and analyzed by immunofluorescence imaging.
Since our data indicate that ERH is necessary for recovery following HU arrest and is important for ATR pathway signaling, we sought to determine whether ERH localized to sites of DNA replication or stalled replication forks. ERH did not colocalize with either RPA or γH2AX foci in untreated cells or cells treated with either HU or ionizing radiation (Fig. 3B and C). We also did not observe ERH recruitment to sites of DNA damage caused by microirradiation (Fig. 3D). Additionally, ERH localization at replication forks was not detected by the isolation of proteins on nascent DNA (iPOND) method in >30 experiments analyzed by both immunoblotting and mass spectrometry (33, 34) (data not shown). Since ERH does not localize to replication forks or sites of damage, ERH may affect replication and replication stress responses indirectly.
ERH interacts with RNA processing proteins.
To better understand the mechanism of action of ERH in the replication stress response, we sought to identify proteins that interact with ERH using mass spectrometry. ERH protein complexes were purified from nuclear extracts of HEK293T cells. Highly abundant nonspecific proteins detected in both nontargeting control and ERH samples, such as tubulin, filamin, and spectrin, were excluded as interacting partners. All ERH-interacting proteins detected with substantially higher peptide counts than those in the controls are depicted in Fig. 4A. Several proteins previously reported to interact with ERH were detected. These proteins include CIZ1 and POLDIP3/SKAR, which were identified by yeast two-hybrid assays (14, 15), and CHTOP, POLDIP3/SKAR, BCLAF1, and PRMT1, which were previously detected in unbiased screens for mitotic protein complexes (35). Our data set also identified interactions with THRAP3, DGCR8, DROSHA, C1QBP, and FAM208B (Fig. 4A). Most of these ERH-interacting proteins are linked to RNA processing. BCLAF1, THRAP3, C1QBP, CHTOP, and POLDIP3 were previously associated with mRNA splicing or processing (36–43), and the microprocessor complex proteins DGCR8 and DROSHA function in microRNA (miRNA) biogenesis (44–48). We confirmed the interactions of ERH with THRAP3, DGCR8, CHTOP, and PRMT1 by standard coimmunoprecipitation (Fig. 4B). Treatment of extracts with RNase A did not disrupt the interaction of ERH with any of the proteins, suggesting direct protein-protein interactions (Fig. 4B).
FIG 4.

ERH interacts with RNA processing proteins. (A) FLAG beads were used to immunoprecipitate FLAG-tagged ERH from HEK293T nuclear extracts. Shown are ERH-interacting proteins identified by mass spectrometry along with the number of peptides and their listed function. For comparison, 797 ERH peptides were observed. (B) FLAG-ERH immunoprecipitates (IP) were obtained as described above for panel A except that the lysates were treated with RNase A, where indicated, prior to immunoblotting.
ATR expression levels are reduced as a result of ERH depletion.
Because ERH interacted with RNA processing proteins, we next examined whether ERH loss altered the expression of ATR pathway proteins. Both ATR protein and mRNA levels were diminished in comparison to those in nontargeting controls (Fig. 5A and B). Additionally, protein levels of several other replication and DNA damage response proteins (ATRIP, TOPBP1, RAD50, and MRE11) and mRNA levels of RAD50 were reduced upon ERH knockdown (Fig. 5A and B). Not all replication and DNA damage proteins are affected by ERH loss, however, as RAD9, RPA32, and KU70 levels remained unchanged. In contrast to the reduction in ATRIP protein levels, ATRIP mRNA levels were increased in ERH-depleted cells. This observation is likely explained by the reduced ATRIP protein stability when ATR is absent (49). No change in CHK1 mRNA levels was observed with ERH depletion (Fig. 5B). We noted that one ERH siRNA (siRNA 3) caused a slight reduction of CHK1 protein levels (Fig. 3A), but since this was not observed with other ERH siRNAs, it may be an off-target effect or due to differences in knockdown efficiency.
FIG 5.

ATR levels are reduced in ERH-deficient cells. (A) U2OS cells expressing nontargeting and ERH siRNAs were collected at 72 h posttransfection and lysed, and immunoblot analysis was performed for the indicated proteins. Data from a representative experiment are shown, but quantitative immunoblotting demonstrated statistically significant decreases in the levels of ATR, RAD50, TOPBP1, ATRIP, and MRE11 in ERH-depleted cells. (B) Cells were transfected with siRNA as indicated, total RNA was extracted and reverse transcribed into cDNA, and qPCR was performed to analyze the fold differences of mRNA levels in ERH-depleted cells compared to those of nontargeting controls. (* denotes a P value of <0.05. ** denotes a P value of <0.01.) (C) Cells were transfected with siRNA as indicated, and ATR levels in nuclear soluble and chromatin fractions were examined by immunoblotting. ORC2 is included as a loading control for chromatin fractions.
To assess whether reduced ATR protein levels resulted in less ATR bound to chromatin, chromatin fractionation samples were analyzed. As expected, ATR levels in nuclear soluble fractions were reduced in both untreated and HU-treated ERH-deficient cells compared to those in control cells (Fig. 5C, left). Similar levels of ATR were observed in the chromatin fractions of untreated control and ERH-deficient cells. While nontargeting siRNA controls exhibited increased ATR levels on chromatin with HU treatment, ATR levels in ERH-depleted samples were not increased (Fig. 5C, right). These data indicate that reduced ATR protein levels in ERH-deficient cells result in less ATR localization to chromatin following replication stress.
ATR mRNA is inefficiently spliced in ERH-depleted cells.
As our mass spectrometry data indicated that ERH interacted with multiple RNA processing enzymes, and we observed reduced protein and mRNA expression of ATR as well as other damage response proteins, we examined specifically how ERH depletion affects ATR mRNA. U2OS cells transfected with nontargeting or ERH siRNA were treated with the transcriptional inhibitor actinomycin D for up to 24 h to examine the effect of ERH loss on the stability of ATR mRNA. ATR mRNA levels were substantially lower in ERH-depleted cells, as expected (Fig. 6A). However, no reduction of ATR mRNA stability was observed compared to that in control cells (Fig. 6A). The remaining ATR mRNA population in ERH-depleted cells actually appeared to be more stable.
FIG 6.

ERH is important for splicing of ATR mRNA. (A) Seventy-two hours following transfection of nontargeting and ERH siRNAs into U2OS cells, cells were treated with actinomycin D (ActD) for the indicated times. Total RNA was extracted and reverse transcribed into cDNA, and qPCR was performed to examine ATR mRNA levels. (B) Schematic of primer design for analysis of prespliced and mature mRNA levels in the absence of ERH. A primer pair crossing exon/exon junctions for analysis of spliced mRNA levels and two primer sets detecting prespliced mRNA at exon/intron and intron/exon junctions were used. (C) Cells were transfected as described above for panel A, total RNA was extracted, and cDNA was obtained by using random and oligo(dT) primers. qPCR was performed to analyze pre- and postspliced mRNA levels of ATR. KU70 mRNA levels were also examined as a control. Mean log2 values and standard errors of the means are depicted.
We next examined the effect of ERH loss on mRNA splicing. Spliced and prespliced ATR mRNA levels were evaluated by qPCR using primers designed for multiple exon junctions throughout the gene. Three sets of primers were utilized per junction: one exon/exon set, to detect spliced mRNA, and one exon/intron set and one intron/exon set, to detect prespliced mRNA (Fig. 6B). Cells with deficient mRNA splicing mechanisms should exhibit higher levels of prespliced RNA detected by primers at exon/intron boundaries and decreased levels of mature mRNA detected by exon/exon boundary primers. At all five ATR exon junctions examined, spliced mRNA levels were reduced in ERH-depleted cells (Fig. 6C). In contrast, prespliced RNA levels were increased at four of the five junctions (Fig. 6C). (The asterisks in Fig. 6C denote junctions where qPCR was not successful. Multiple primer pairs were tested; however, PCR primers that provide quantifiable data across the junctions were not identified.) Three KU70 exon junctions were also examined as a negative control because the KU70 protein expression level was unchanged upon the loss of ERH (Fig. 5A). Both prespliced and spliced KU70 mRNA levels showed no appreciable difference between nontargeting control and ERH samples (Fig. 6C). Collectively, these data indicate that ERH is important for the efficient splicing of ATR mRNA. These results suggest that the defects in ATR signaling, replication, and the replication stress response in ERH-deficient cells may be due primarily to defects in the processing of ATR pathway mRNAs, leading to reductions in the expression levels of multiple ATR pathway proteins.
Global effect of ERH deficiency on gene expression and RNA splicing.
To determine whether ERH specifically affects the splicing of ATR pathway genes or is part of a broader regulatory program, RNA sequencing (RNA-seq) analysis was performed to globally examine the effect of ERH loss on gene expression and mRNA splicing. In ERH-deficient cells, gene expression analysis revealed that the expression levels of 1,611 genes were significantly decreased and that those of 1,990 genes were significantly increased compared to the levels in control cells (see Table S1 in the supplemental material). As anticipated, ATR levels were significantly reduced in ERH-depleted cells (P value of 0.04). Genes exhibiting significantly reduced expression in ERH-depleted cells were enriched for nuclear processes, including DNA replication, DNA metabolism, the cell cycle, chromatin assembly and organization, and chromosome organization and segregation (Table 1; see also Table S1 in the supplemental material). Reduced levels of genes involved in these important nuclear pathways likely account for the decreased replication and repair rates observed following replication stress in ERH-depleted cells. Genes with increased levels in ERH-deficient cells were found to function in signal transduction, proliferation, cell death, development, extracellular matrix organization, cell adhesion and motility, protein processing, proteolysis, and several biosynthesis pathways (Table 1; see also Table S1 in the supplemental material). Some of these transcript level changes may be secondary effects. Nonetheless, these data indicate that ERH contributes to DNA replication and the replication stress response by ensuring proper expression of genes involved in these processes.
TABLE 1.
Gene ontology analysis of biological processes enriched in genes exhibiting decreased and increased expression levels with ERH knockdown in comparison to nontargeting siRNA controls
| Biological process | No. of genes | P value | q value |
|---|---|---|---|
| Decreased expression | |||
| Cell cycle process | 125 | 5.41 × 10−43 | 7.26 × 10−39 |
| Chromosome organization | 49 | 1.49 × 10−23 | 2.22 × 10−20 |
| Cell division | 49 | 7.09 × 10−19 | 9.53 × 10−16 |
| Chromatin assembly or disassembly | 24 | 2.27 × 10−16 | 1.79 × 10−13 |
| DNA metabolic process | 67 | 1.07 × 10−15 | 7.55 × 10−13 |
| Chromatin organization | 62 | 2.68 × 10−15 | 1.71 × 10−12 |
| Microtubule-based process | 50 | 5.58 × 10−15 | 3.4 × 10−12 |
| Nuclear division | 38 | 8.89 × 10−14 | 4.42 × 10−11 |
| Chromosome segregation | 22 | 4.87 × 10−14 | 2.51 × 10−11 |
| Protein complex assembly | 74 | 9.44 × 10−14 | 4.53 × 10−11 |
| Spindle organization | 20 | 4.52 × 10−13 | 1.9 × 10−10 |
| Organelle fission | 38 | 1.46 × 10−12 | 5.92 × 10−10 |
| Macromolecular complex assembly | 78 | 2.07 × 10−11 | 6.19 × 10−9 |
| Cell cycle checkpoint | 23 | 6.25 × 10−10 | 1.38 × 10−7 |
| Sister chromatid segregation | 10 | 1.09 × 10−9 | 2.28 × 10−7 |
| DNA conformation change | 18 | 2.08 × 10−9 | 4.17 × 10−7 |
| Regulation of organelle organization | 67 | 1.14 × 10−8 | 1.94 × 10−6 |
| DNA repair | 36 | 3.34 × 10−8 | 5.34 × 10−6 |
| DNA replication | 16 | 7.7 × 10−5 | 6.08 × 10−3 |
| Increased expression | |||
| Regulation of intracellular signal transduction | 87 | 1.3 × 10−9 | 1.75 × 10−5 |
| Regulation of cell death | 88 | 4.18 × 10−8 | 7.02 × 10−5 |
| Regulation of cell proliferation | 86 | 4.44 × 10−8 | 5.97 × 10−5 |
| Positive regulation of signaling | 83 | 1.73 × 10−7 | 1.66 × 10−4 |
| Extracellular matrix organization | 33 | 2.53 × 10−7 | 2.12 × 10−4 |
| Cell adhesion | 64 | 3.63 × 10−7 | 2.21 × 10−4 |
| Cell motility | 54 | 3.76 × 10−7 | 2.1 × 10−4 |
| Positive regulation of cell communication | 82 | 4.47 × 10−7 | 2.22 × 10−4 |
| Regulation of hydrolase activity | 70 | 2.59 × 10−6 | 9.4 × 10−4 |
| Regulation of protein metabolic process | 115 | 3.35 × 10−6 | 1.07 × 10−3 |
| Glycosaminoglycan biosynthetic process | 14 | 9.11 × 10−6 | 2.45 × 10−3 |
| Aminoglycan biosynthetic process | 14 | 1.02 × 10−5 | 2.69 × 10−3 |
| Positive regulation of response to stimulus | 92 | 1.35 × 10−5 | 3.12 × 10−3 |
| Regulation of proteolysis | 45 | 7.08 × 10−5 | 9.41 × 10−3 |
| Epithelium development | 20 | 8.99 × 10−5 | 1.14 × 10−2 |
| Tissue development | 39 | 1.07 × 10−4 | 1.28 × 10−2 |
| Regulation of cellular component movement | 40 | 1.24 × 10−4 | 1.43 × 10−2 |
| Chemotaxis | 20 | 7.57 × 10−4 | 5.01 × 10−2 |
To determine the effect of ERH loss on mRNA splicing, we calculated the fold changes of intron levels adjusted for overall gene expression between ERH-deficient and control samples. Genes exhibiting a change of 1.5-fold or higher in ERH-depleted cells were selected as being indicative of RNA splicing defects. As expected, this analysis indicated a defect in ATR splicing in ERH-deficient cells (1.56-fold change) (see Table S2 in the supplemental material). CENPE, which is also defectively spliced in ERH-deficient cells (17), exhibited a 1.87-fold change. Of genes with FPKM values greater than 1, 749 (5.4%) exhibited evidence of defective splicing, indicating some level of specificity (see Table S2 in the supplemental material). This relatively small percentage indicates that ERH is not a general splicing factor but regulates only a subset of transcripts. Indeed, gene ontology analysis of genes displaying increased intron levels in ERH-depleted cells showed enrichment in a number of nuclear processes, including nucleosome assembly and organization, DNA replication-dependent nucleosome assembly and organization, DNA modification, gene silencing, and DNA recombination (Table 2). Among replication and replication stress genes, defective splicing was evident for ATR, POLA1, MSH2, and FANCM transcripts.
TABLE 2.
Gene ontology analysis of biological processes enriched in genes exhibiting defective mRNA splicing in ERH-deficient cells in comparison to nontargeting controls
| Biological process | No. of genes | P value | q value |
|---|---|---|---|
| Nucleosome assembly | 20 | 5.4 × 10−9 | 6.57 × 10−5 |
| Protein heterotetramerization | 10 | 2.4 × 10−8 | 1.46 × 10−4 |
| Protein-DNA complex assembly | 21 | 2.96 × 10−8 | 1.2 × 10−4 |
| Nucleosome organization | 21 | 6.05 × 10−8 | 1.84 × 10−4 |
| Chromatin silencing at ribosomal DNA | 10 | 1.16 × 10−7 | 2.82 × 10−4 |
| DNA replication-dependent nucleosome organization | 9 | 1.31 × 10−7 | 2.65 × 10−4 |
| DNA replication-dependent nucleosome assembly | 9 | 1.31 × 10−7 | 2.27 × 10−4 |
| DNA methylation | 12 | 1.02 × 10−6 | 1.24 × 10−3 |
| DNA alkylation | 12 | 1.02 × 10−6 | 1.13 × 10−3 |
| Chromatin silencing | 10 | 1.87 × 10−5 | 1.75 × 10−2 |
| DNA modification | 13 | 4.6 × 10−5 | 3.73 × 10−2 |
| Methylation | 27 | 1.38 × 10−4 | 9.29 × 10−2 |
| Chromatin assembly or disassembly | 12 | 2.29 × 10−4 | 1.46 × 10−1 |
| Gene silencing | 12 | 2.62 × 10−4 | 1.59 × 10−1 |
| Protein heterooligomerization | 12 | 2.99 × 10−4 | 1.65 × 10−1 |
| Negative regulation of hematopoietic progenitor cell differentiation | 5 | 3.13 × 10−4 | 1.66 × 10−1 |
| Regulation of response to external stimulus | 40 | 4.29 × 10−4 | 2.17 × 10−1 |
| Reciprocal meiotic recombination | 6 | 6.42 × 10−4 | 3 × 10−1 |
| Reciprocal DNA recombination | 6 | 6.42 × 10−4 | 2.89 × 10−1 |
| Regulation of megakaryocyte differentiation | 5 | 9.36 × 10−4 | 4.06 × 10−1 |
DISCUSSION
We identified ERH by a whole-genome RNAi screen designed to find genes that function in the replication stress response. ERH deficiency causes reduced DNA replication restart from an acute replication stress challenge and persistent levels of DNA damage. Cells lacking ERH do not complete DNA replication after release from a replication block. They are also hypersensitive to replication stress-inducing agents, including HU, camptothecin, and gemcitabine. These data are consistent with the observations of Krzyzanowski et al. (16), who reported that Schizosaccharomyces pombe auxotrophic erh1Δ mutants are hypersensitive to stress agents, including HU.
ERH loss causes increased DNA damage in both the presence and absence of exogenous stress. Furthermore, ERH-deficient cells exhibit slowed elongation of DNA replication forks. Thus, ERH function is also important to maintain genome integrity in cells even without the addition of an acute replication stress challenge.
In the absence of ERH expression, we observed reduced levels of phosphorylated CHK1, indicating that the ATR pathway is compromised. We did not observe colocalization of ERH with RPA or γH2AX foci following HU and IR treatment, localization to laser-induced DNA damage, or localization at replication forks by iPOND analysis. These data are in contrast to a previously reported observation that overexpressed ERH-enhanced GFP (EGFP) colocalizes with overexpressed Ciz1-mCherry, which was reported to be located at sites of DNA replication (15). While we cannot exclude the possibility that ERH localizes directly to sites of DNA replication under some conditions, our analysis suggests an indirect function of ERH in the replication stress response.
Indeed, we found that ERH complexes with multiple RNA processing proteins. These include proteins involved in RNA splicing and processing, such as THRAP3, BCLAF1, C1QBP, CHTOP, and POLDIP3, and proteins involved in miRNA biogenesis, such as DROSHA and DGCR8 (36–48). Consistent with a function of ERH in RNA processing, we observed defective splicing of ATR mRNA in ERH-deficient cells, leading to decreased mature ATR mRNA and protein levels. These data coincide with a study reported while this article was in revision, which also observed an ATR splicing defect (50). In addition to the effect on ATR, we observed decreases in the levels of the DNA damage response proteins ATRIP, TOPBP1, RAD50, and MRE11. These data are consistent with a previous report that found reduced expression levels of several DNA repair proteins in ERH-depleted cells, including ATR, MRE11, and RAD50 (17).
Importantly, our RNA-seq analysis demonstrated decreased expression of numerous DNA replication and repair genes in ERH-deficient cells, including ATR, MCM2, MCM3, MCM5, MCM6, RFC2, RFC3, ORC1, GINS2, POLA1, POLD3, POLE2, MRE11A, MSH2, BLM, and FANCM. Decreased expression of these important replication and repair genes, along with numerous additional cell cycle and DNA packaging genes, most likely contributes to the reduced replication rates and persistence of DNA damage observed in ERH-deficient cells. ERH loss resulted in splicing defects in genes that function in DNA packaging, processing, and repair. However, genes involved in other cellular processes also displayed insufficient splicing. Therefore, while ERH affects splicing of transcripts important for replication and repair, it also impacts splicing of a broader range of genes.
ERH is a small protein that does not contain a known functional domain. Crystallization studies demonstrated that ERH forms a homodimer, and conserved surface residues indicate that they may function as protein-protein interaction interfaces (51–53). It is possible that ERH functions as a cofactor for the assembly or function of RNA processing complexes. In this regard, it is interesting that ERH interacts with protein-arginine methyltransferase 1 (PRMT1). Arginine methylation is associated with RNA processing, including mRNA splicing (54), and a number of RNA-binding proteins undergo arginine methylation (54–56). PRMT1 methylates arginine-glycine-rich (RG-rich) regions. CHTOP is a known substrate of PRMT1, and several of the ERH-interacting proteins that we identified contain RG-rich regions that may be potential methylation sites. Thus, it is possible that ERH regulates posttranslational modifications in multiple RNA processing proteins to influence their function.
In conclusion, our data are consistent with a model in which ERH is needed for the proper splicing and expression of replication stress proteins, including ATR. Thus, ERH loss of function causes defects in ATR signaling, replication, and replication stress responses. This function likely explains the original observation of a genetic interaction of Drosophila E(r) and rudimentary (r) genes. rudimentary encodes a protein required for the pyrimidine biosynthetic pathway, so its mutation creates replication stress, which requires the ATR pathway. Mutations in Drosophila E(r) impair the replication stress response and cause the enhanced phenotype.
Supplementary Material
ACKNOWLEDGMENTS
The Vanderbilt Institute for Integrative Biosystems Research and Education and the Vanderbilt Institute of Chemical Biology High Throughput Screening Facility were utilized for high-content imaging. The Vanderbilt Mass Spectrometry Research Center Proteomics Laboratory was utilized for mass spectrometry analysis. The Vanderbilt Technologies for Advanced Genomics Facility was utilized for RNA sequencing.
This work was supported by NIH grant CA102792 to D.C. G.K. was supported by NIH grants T32 ES007028 and F32 GM103254, K.N.M. was supported by NIH grant T32CA093240, and M.E.L. was supported by an NSF graduate research fellowship. Support for the RNA-seq analysis was provided by CTSA award no. UL1TR000445 from the National Center for Advancing Translational Sciences. Funding for the Opera instrument was provided by grant 1S10RR027485.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01276-14.
REFERENCES
- 1.Cimprich KA, Cortez D. 2008. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petermann E, Helleday T. 2010. Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol 11:683–687. doi: 10.1038/nrm2974. [DOI] [PubMed] [Google Scholar]
- 3.Branzei D, Foiani M. 2010. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 11:208–219. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- 4.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. 2005. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev 19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zou L, Elledge SJ. 2003. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 6.Ball HL, Ehrhardt MR, Mordes DA, Glick GG, Chazin WJ, Cortez D. 2007. Function of a conserved checkpoint recruitment domain in ATRIP proteins. Mol Cell Biol 27:3367–3377. doi: 10.1128/MCB.02238-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ball HL, Myers JS, Cortez D. 2005. ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Mol Biol Cell 16:2372–2381. doi: 10.1091/mbc.E04-11-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumagai A, Lee J, Yoo HY, Dunphy WG. 2006. TopBP1 activates the ATR-ATRIP complex. Cell 124:943–955. doi: 10.1016/j.cell.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 9.Mordes DA, Glick GG, Zhao R, Cortez D. 2008. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev 22:1478–1489. doi: 10.1101/gad.1666208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nam EA, Cortez D. 2011. ATR signalling: more than meeting at the fork. Biochem J 436:527–536. doi: 10.1042/BJ20102162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeman MK, Cimprich KA. 2014. Causes and consequences of replication stress. Nat Cell Biol 16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wojcik E, Murphy AM, Fares H, Dang-Vu K, Tsubota SI. 1994. Enhancer of rudimentaryp1, e(r)p1, a highly conserved enhancer of the rudimentary gene. Genetics 138:1163–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freund JN, Jarry BP. 1987. The rudimentary gene of Drosophila melanogaster encodes four enzymic functions. J Mol Biol 193:1–13. doi: 10.1016/0022-2836(87)90621-8. [DOI] [PubMed] [Google Scholar]
- 14.Smyk A, Szuminska M, Uniewicz KA, Graves LM, Kozlowski P. 2006. Human enhancer of rudimentary is a molecular partner of PDIP46/SKAR, a protein interacting with DNA polymerase delta and S6K1 and regulating cell growth. FEBS J 273:4728–4741. doi: 10.1111/j.1742-4658.2006.05477.x. [DOI] [PubMed] [Google Scholar]
- 15.Lukasik A, Uniewicz KA, Kulis M, Kozlowski P. 2008. Ciz1, a p21 cip1/Waf1-interacting zinc finger protein and DNA replication factor, is a novel molecular partner for human enhancer of rudimentary homolog. FEBS J 275:332–340. doi: 10.1111/j.1742-4658.2007.06203.x. [DOI] [PubMed] [Google Scholar]
- 16.Krzyzanowski MK, Kozlowska E, Kozlowski P. 2012. Identification and functional analysis of the erh1(+) gene encoding enhancer of rudimentary homolog from the fission yeast Schizosaccharomyces pombe. PLoS One 7:e49059. doi: 10.1371/journal.pone.0049059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weng MT, Lee JH, Wei SC, Li Q, Shahamatdar S, Hsu D, Schetter AJ, Swatkoski S, Mannan P, Garfield S, Gucek M, Kim MK, Annunziata CM, Creighton CJ, Emanuele MJ, Harris CC, Sheu JC, Giaccone G, Luo J. 2012. Evolutionarily conserved protein ERH controls CENP-E mRNA splicing and is required for the survival of KRAS mutant cancer cells. Proc Natl Acad Sci U S A 109:E3659–E3667. doi: 10.1073/pnas.1207673110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banko MI, Krzyzanowski MK, Turcza P, Maniecka Z, Kulis M, Kozlowski P. 2013. Identification of amino acid residues of ERH required for its recruitment to nuclear speckles and replication foci in HeLa cells. PLoS One 8:e74885. doi: 10.1371/journal.pone.0074885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coverley D, Marr J, Ainscough J. 2005. Ciz1 promotes mammalian DNA replication. J Cell Sci 118:101–112. doi: 10.1242/jcs.01599. [DOI] [PubMed] [Google Scholar]
- 20.Mendez J, Stillman B. 2000. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol 20:8602–8612. doi: 10.1128/MCB.20.22.8602-8612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, Lukas J. 2006. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol 173:195–206. doi: 10.1083/jcb.200510130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA, Cortez D. 2013. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev 27:1610–1623. doi: 10.1101/gad.214080.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacCoss MJ, McDonald WH, Saraf A, Sadygov R, Clark JM, Tasto JJ, Gould KL, Wolters D, Washburn M, Weiss A, Clark JI, Yates JR III. 2002. Shotgun identification of protein modifications from protein complexes and lens tissue. Proc Natl Acad Sci U S A 99:7900–7905. doi: 10.1073/pnas.122231399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez MN, Emfinger CH, Overton M, Hill S, Ramaswamy TS, Cappel DA, Wu K, Fazio S, McDonald WH, Hachey DL, Tabb DL, Stafford JM. 2012. Obesity and altered glucose metabolism impact HDL composition in CETP transgenic mice: a role for ovarian hormones. J Lipid Res 53:379–389. doi: 10.1194/jlr.M019752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo Y, Ye F, Sheng Q, Clark T, Samuels DC. 2014. Three-stage quality control strategies for DNA re-sequencing data. Brief Bioinform 15:879–889. doi: 10.1093/bib/bbt069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo Y, Zhao S, Sheng Q, Ye F, Li J, Lehmann B, Pietenpol J, Samuels DC, Shyr Y. 2014. Multi-perspective quality control of Illumina exome sequencing data using QC3. Genomics 103:323–328. doi: 10.1016/j.ygeno.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo Y, Zhao S, Ye F, Sheng Q, Shyr Y. 2014. MultiRankSeq: multiperspective approach for RNAseq differential expression analysis and quality control. Biomed Res Int 2014:248090. doi: 10.1155/2014/248090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. 2009. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eden E, Lipson D, Yogev S, Yakhini Z. 2007. Discovering motifs in ranked lists of DNA sequences. PLoS Comput Biol 3:e39. doi: 10.1371/journal.pcbi.0030039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, Lukas J. 2013. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155:1088–1103. doi: 10.1016/j.cell.2013.10.043. [DOI] [PubMed] [Google Scholar]
- 33.Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. 2011. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 25:1320–1327. doi: 10.1101/gad.2053211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sirbu BM, McDonald WH, Dungrawala H, Badu-Nkansah A, Kavanaugh GM, Chen Y, Tabb DL, Cortez D. 2013. Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J Biol Chem 288:31458–31467. doi: 10.1074/jbc.M113.511337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hutchins JR, Toyoda Y, Hegemann B, Poser I, Heriche JK, Sykora MM, Augsburg M, Hudecz O, Buschhorn BA, Bulkescher J, Conrad C, Comartin D, Schleiffer A, Sarov M, Pozniakovsky A, Slabicki MM, Schloissnig S, Steinmacher I, Leuschner M, Ssykor A, Lawo S, Pelletier L, Stark H, Nasmyth K, Ellenberg J, Durbin R, Buchholz F, Mechtler K, Hyman AA, Peters JM. 2010. Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science 328:593–599. doi: 10.1126/science.1181348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee K-M, Hsu I-W, Tarn W-Y. 2010. TRAP150 activates pre-mRNA splicing and promotes nuclear mRNA degradation. Nucleic Acids Res 38:3340–3350. doi: 10.1093/nar/gkq017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beli P, Lukashchuk N, Wagner SA, Weinert BT, Olsen JV, Baskcomb L, Mann M, Jackson SP, Choudhary C. 2012. Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol Cell 46:212–225. doi: 10.1016/j.molcel.2012.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merz C, Urlaub H, Will CL, Luhrmann R. 2007. Protein composition of human mRNPs spliced in vitro and differential requirements for mRNP protein recruitment. RNA 13:116–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bracken CP, Wall SJ, Barre B, Panov KI, Ajuh PM, Perkins ND. 2008. Regulation of cyclin D1 RNA stability by SNIP1. Cancer Res 68:7621–7628. doi: 10.1158/0008-5472.CAN-08-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petersen-Mahrt SK, Estmer C, Ohrmalm C, Matthews DA, Russell WC, Akusjarvi G. 1999. The splicing factor-associated protein, p32, regulates RNA splicing by inhibiting ASF/SF2 RNA binding and phosphorylation. EMBO J 18:1014–1024. doi: 10.1093/emboj/18.4.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang CT, Hautbergue GM, Walsh MJ, Viphakone N, van Dijk TB, Philipsen S, Wilson SA. 2013. Chtop is a component of the dynamic TREX mRNA export complex. EMBO J 32:473–486. doi: 10.1038/emboj.2012.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma XM, Yoon SO, Richardson CJ, Julich K, Blenis J. 2008. SKAR links pre-mRNA splicing to mTOR/S6K1-mediated enhanced translation efficiency of spliced mRNAs. Cell 133:303–313. doi: 10.1016/j.cell.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 43.Folco EG, Lee CS, Dufu K, Yamazaki T, Reed R. 2012. The proteins PDIP3 and ZC11A associate with the human TREX complex in an ATP-dependent manner and function in mRNA export. PLoS One 7:e43804. doi: 10.1371/journal.pone.0043804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R. 2004. The Microprocessor complex mediates the genesis of microRNAs. Nature 432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 45.Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. 2004. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev 18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heras SR, Macias S, Plass M, Fernandez N, Cano D, Eyras E, Garcia-Perez JL, Caceres JF. 2013. The Microprocessor controls the activity of mammalian retrotransposons. Nat Struct Mol Biol 20:1173–1181. doi: 10.1038/nsmb.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davis-Dusenbery BN, Hata A. 2010. MicroRNA in cancer: the involvement of aberrant microRNA biogenesis regulatory pathways. Genes Cancer 1:1100–1114. doi: 10.1177/1947601910396213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Winter J, Jung S, Keller S, Gregory RI, Diederichs S. 2009. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol 11:228–234. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 49.Cortez D, Guntuku S, Qin J, Elledge SJ. 2001. ATR and ATRIP: partners in checkpoint signaling. Science 294:1713–1716. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- 50.Weng MT, Tung TH, Lee JH, Wei SC, Lin HL, Huang YJ, Wong JM, Luo J, Sheu JC. 2015. Enhancer of rudimentary homolog regulates DNA damage response in hepatocellular carcinoma. Sci Rep 5:9357. doi: 10.1038/srep09357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arai R, Kukimoto-Niino M, Uda-Tochio H, Morita S, Uchikubo-Kamo T, Akasaka R, Etou Y, Hayashizaki Y, Kigawa T, Terada T, Shirouzu M, Yokoyama S. 2005. Crystal structure of an enhancer of rudimentary homolog (ERH) at 2.1 angstroms resolution. Protein Sci 14:1888–1893. doi: 10.1110/ps.051484505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li H, Inoue M, Yabuki T, Aoki M, Seki E, Matsuda T, Nunokawa E, Motoda Y, Kobayashi A, Terada T, Shirouzu M, Koshiba S, Lin YJ, Guntert P, Suzuki H, Hayashizaki Y, Kigawa T, Yokoyama S. 2005. Solution structure of the mouse enhancer of rudimentary protein reveals a novel fold. J Biomol NMR 32:329–334. doi: 10.1007/s10858-005-7959-z. [DOI] [PubMed] [Google Scholar]
- 53.Wan C, Tempel W, Liu ZJ, Wang BC, Rose RB. 2005. Structure of the conserved transcriptional repressor enhancer of rudimentary homolog. Biochemistry 44:5017–5023. doi: 10.1021/bi047785w. [DOI] [PubMed] [Google Scholar]
- 54.Bedford MT, Clarke SG. 2009. Protein arginine methylation in mammals: who, what, and why. Mol Cell 33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boisvert FM, Cote J, Boulanger MC, Richard S. 2003. A proteomic analysis of arginine-methylated protein complexes. Mol Cell Proteomics 2:1319–1330. doi: 10.1074/mcp.M300088-MCP200. [DOI] [PubMed] [Google Scholar]
- 56.Pahlich S, Zakaryan RP, Gehring H. 2006. Protein arginine methylation: cellular functions and methods of analysis. Biochim Biophys Acta 1764:1890–1903. doi: 10.1016/j.bbapap.2006.08.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.