

Abstract
Several gastrointestinal proteins have been identified to have insulinotropic effects, including glucose-dependent insulinotropic polypeptide (GIP); however, the direct effects of incretins on skeletal muscle glucose transport remain largely unknown. Therefore, the purpose of the current study was to examine the role of GIP on skeletal muscle glucose transport and insulin signaling in rats. Relative to a glucose challenge, a mixed glucose+lipid oral challenge increased circulating GIP concentrations, skeletal muscle Akt phosphorylation, and improved glucose clearance by ∼35% (P < 0.05). These responses occurred without alterations in serum insulin concentrations. In an incubated soleus muscle preparation, GIP directly stimulated glucose transport and increased GLUT4 accumulation on the plasma membrane in the absence of insulin. Moreover, the ability of GIP to stimulate glucose transport was mitigated by the addition of the PI 3-kinase (PI3K) inhibitor wortmannin, suggesting that signaling through PI3K is required for these responses. We also provide evidence that the combined stimulatory effects of GIP and insulin on soleus muscle glucose transport are additive. However, the specific GIP receptor antagonist (Pro3)GIP did not attenuate GIP-stimulated glucose transport, suggesting that GIP is not signaling through its classical receptor. Together, the current data provide evidence that GIP regulates skeletal muscle glucose transport; however, the exact signaling mechanism(s) remain unknown.
Keywords: glucose transport, incretins, gavage, skeletal muscle
many studies over the past few decades have contributed to the growing understanding of insulin-stimulated glucose transport into skeletal muscle (reviewed in Ref. 9). Glucose disposal is highly regulated and is facilitated by the glucose transporter isoform 4 (GLUT4). In resting muscle, GLUT4 resides predominantly (24, 25, 35) in intracellular vesicular pools that can be stimulated to translocate to the plasma membrane in response to various stimuli, including insulin (7). Binding of insulin to the insulin receptor on the surface of myocytes (41) results in autophosphorylation of the insulin receptor, initiating a signaling cascade involving several intermediates with the end result of GLUT4 translocation to the plasma membrane. The proximal insulin-signaling cascade has been well characterized, and studies have shown that Akt, also known as PKB, is a central mediator of insulin-induced GLUT4 translocation, as inhibition of Akt1 and Akt2 results in a significant decrease in insulin-stimulated glucose uptake (14, 21).
Several gastrointestinal proteins have been identified to have insulinotropic effects and, therefore, influence postprandial glucose homeostasis. The principle mediators of the entero-insular axis involve the gut hormones glucose-dependent insulinotropic polypeptide (GIP; also known as gastric inhibitory polypeptide) and glucagon-like polypeptide (GLP), collectively called the incretins (reviewed in Ref. 4). Although the insulin-stimulating effects of intestinal secretions were postulated in the early 1900s (5, 30), it was not until many decades later that GIP (8) and GLP (22, 29) were characterized. GIP is synthesized and released from K-cells located in the proximal regions of the small intestine (duodenum and jejunum), while L-cells within the ileum primarily secrete GLP (4). Inhibition of GIP signaling, or ablation of the GIP receptor (GIPR), impairs glucose homeostasis (16, 27, 44), and conversely, GIP overexpression improves glucose homeostasis in the face of a high-fat diet challenge (20), highlighting the importance of GIP in regulating postprandial fuel homeostasis. Although believed to work predominantly at the pancreas to stimulate insulin release in response to glucose or lipid ingestion, an extra-pancreatic role for the incretins in stimulating glucose disposal has been suggested. For instance, in 3T3-L1 adipocytes GIP induces the phosphorylation of Akt, GLUT4 accumulation on the plasma membrane, and glucose uptake (40). However, the direct role of the incretins in skeletal muscle glucose uptake remains debated in the literature (1, 3, 32, 37, 38). GLP-1-stimulated glucose uptake has been demonstrated in traditional rodent hind limb muscles (1, 3). However, recently, these studies have been brought into question, suggesting that GLP-1 works predominantly at the microvasculature and does not have a direct effect on skeletal muscle glucose uptake (37, 38). In contrast, the effects of GIP on skeletal muscle glucose uptake are much less studied. To our knowledge, only one study has been conducted. O'Harte et al. (32) demonstrated increased glucose transport in mouse diaphragm in the presence of GIP, although no mechanism of action was investigated.
Therefore, the purpose of the current study was to examine the potential for the incretins to regulate rat skeletal muscle glucose transport and to further our understanding of the possible signaling pathways involved, based on the in vivo response to various oral gavage treatments. We provide evidence that, relative to glucose alone, a mixed glucose+lipid oral challenge increases plasma GIP concentrations, induces skeletal muscle Akt phosphorylation, and improves glucose homeostasis at the whole body level. In addition, using an incubated muscle preparation, we provide evidence that GIP directly stimulates glucose transport and induces skeletal muscle sarcolemmal GLUT4 accumulation in the absence of insulin. Moreover, the ability of GIP to stimulate glucose transport is prevented by the addition of the phosphoinosotide-3 kinase (PI3K) inhibitor wortmannin, suggesting that the activation of the PI3K/Akt pathway is required for these responses. We further demonstrate that GIP and insulin are additive in stimulating skeletal muscle glucose transport. Finally, we show that the specific GIP receptor antagonist, (Pro3)GIP, does not prevent GIP-stimulated glucose transport, suggesting that GIP is not signaling through its classical receptor. Together, the current data provide evidence for an important physiological role for GIP in mediating skeletal muscle glucose transport; however, the exact signaling mechanism remains unknown.
METHODS
Animals
Sprague-Dawley rats [male, Charles River, St. Constant, QC; 9–11 wk of age (∼300 g) for in vivo gavage experiments, ∼5 wk of age (∼100 g) for in vitro incubated soleus strip experiments, and ∼20 wk of age (∼450 g) for quantitative PCR (qPCR) experiments; 150 rats in total] were group housed in a temperature-controlled room with a reverse 12:12-h light-dark cycle (lights on at 8 PM) and were provided with standard rat chow and water ad libitum. All protocols followed the Canadian Council on Animal Care guidelines and were approved by the Animal Care Committee at the University of Guelph.
Gavage Treatment and Blood Collection
On the experimental day, animals were fasted for 4 h prior to treatment (8 AM, the start of the dark cycle). While conscious, animals were given one of three treatments by oral gavage: glucose [GLU, 800 mg: 2 ml of a 40% (wt/vol) solution; BioShop, Burlington, ON, Canada], glucose+lipid [MIX, 800 mg each: 3 ml consisting of 2 ml of glucose solution + 1 ml of generic 100% canola oil + 15 μl Tween-20 (BioShop) to emulsify], or lipid (LIP, 800 mg: 1 ml of canola oil). GLU was used as an insulin-stimulating treatment, while LIP was used as a non-insulin-stimulating control. We also chose to examine the combination of glucose+lipid (MIX). Because the amount of glucose was kept constant between the two glucose-containing treatments, we hypothesized that there would be no difference in circulating insulin between GLU and MIX, but that the inclusion of lipid could affect glucose handling. For blood analyses, the rats served as their own control (at time = 0); however, for the Akt phosphorylation Western blot experiments, we required a nontreated control group as the experiments were terminal. In this case, the animals were provided a gavage treatment of normal saline (2 ml; 0.9% NaCl; SAL; BioShop).
Blood glucose was measured by glucometer (Freestyle Lite, Abbott Laboratories, St. Laurent, QC, Canada) and serum (Microvette300; Sarstedt, Montreal, QC, Canada), and plasma [EDTA Microvette300; Sarstedt; containing 10 μl/ml DPP IV inhibitor (cat. no. DDP4, Millipore, Billerica, MA)] and 15 μl/ml aprotinin (cat. no. A6279; Sigma, Oakville, ON, Canada) were collected from the tail vein at various time points. Blood was centrifuged (3,000 g, 10 min, 4°C) within 20 min of collection, and the serum or plasma was removed and stored at −20°C.
Circulating Hormone Analyses
Insulin ELISA.
Serum insulin was measured in duplicate using a commercially available ELISA kit (cat. no. EZRMI-13K; Millipore), as per the manufacturer's instructions.
Incretin BioPlex.
Plasma GIP and GLP-1 (active form) were measured in duplicate using a commercially available BioPlex kit (cat. no. RMHMAG-84K; Millipore), as per the manufacturer's instructions.
Tissue Sample Collection and Western Blot Analysis
Rats were anesthetized (pentobarbital sodium, 60 mg/kg ip injection) for all tissue collection and killed by removal of the heart or cervical dislocation. Tibialis anterior (red compartment) for Western blot analysis of total and phosphorylated Akt was collected 15 min postgavage and flash frozen in liquid nitrogen and stored at −80°C. Giant sarcolemmal vesicles for plasma GLUT4 determination were obtained as described below. Wild-type (WT) and GIP receptor knockout (GIPR KO) mouse gastrocnemius tissue was a kind gift from Dr. Daniel Drucker (Lunenfeld Tanenbaum Research Institute, Mt. Sinai Hospital, Toronto, ON, Canada). Skeletal muscle (25–50 mg) was homogenized as previously reported (17), and 30 μg of muscle homogenate or 7 μg of plasma membrane protein was loaded for SDS-PAGE. For the GIPR blots, increasing amounts of protein (5–30 μg) were loaded. Proteins were separated on a 7.5% or 10% resolving gel and transferred to PVDF membrane (Roche, Laval, QC, Canada). Membranes were blocked in 7.5% BSA in Tris-buffered saline containing 0.1% Tween-20 (TBS-T) for 1 h at room temperature. Membranes were incubated in primary antibody diluted in blocking solution [anti-GLUT4 (1:4,000), Millipore; anti-tAkt (1:1,000), anti-pAkt (Ser-473) (1:1,000), and anti-pAkt (Thr-308) (1:1,000), Cell Signaling, Danvers, MA; and anti-GIPR (1:1,000), Novus Biologicals, Oakville, ON, Canada] overnight at 4°C. After three washes in TBS-T, membranes were incubated in the appropriate horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology, Dallas, TX), detected using enhanced chemiluminescence (Perkin Elmer, Woodbridge, ON, Canada), and quantified by densitometry (Alpha Innotech Fluorchem HD2, Fisher Scientific, Ottawa, ON, Canada).
Real-Time Quantitative PCR
Rat soleus, extensor digitorum longus, and pancreas (as a positive control) were obtained from anesthetized Sprague-Dawley rats (pentobarbital sodium, 60 mg/kg ip injection), and RNA was extracted using a combined QiaZol and RNeasy tissue kit protocol (Qiagen, Toronto, ON, Canada) followed by DNase treatment (Life Technologies, Burlington, ON, Canada), according to the manufacturer's instructions. Total RNA (0.5 μg) was used to synthesize complementary DNA using SuperScript II Reverse Transcriptase, random primers, and dNTPs (Life Technologies). Gene expression was quantified in duplicate by qPCR (AB 7500; Applied Biosystems, Foster City, CA) using 1 μl of cDNA template and the specific TaqMan gene expression assay for GIPR (Life Technologies). As different tissues were examined, there is no appropriate housekeeping gene with which to compare the expression of the GIPR. Additionally, qPCR was used to indicate the presence or absence of the GIPR; therefore, average raw threshold cycle (Ct) values ± SE are reported.
Skeletal Muscle Glucose Transport
Soleus strips were obtained from anesthetized rats (pentobarbital sodium, 60 mg/kg ip injection) and were equilibrated for 30 min in 2 ml of pregassed (95% O2-5% CO2) modified Krebs-Henseleit buffer [containing 16% vol/vol sodium bicarbonate and 0.1% BSA; (KHB) warmed to 30°C] supplemented with 8 mM glucose and 32 mM mannitol (M1), in a gentle shaking bath (30 rpm). The soleus strips were washed (2 × 10 min) with glucose-free KHB supplemented with 4 mM of pyruvate and 36 mM mannitol (M2) in the presence or absence of 0.5 μg/ml GIP [human full length, dissolved in water; Abcam, Cambridge, MA; cf. (32)]. Soleus strips were then incubated for 20 min (treatment) or 40 min (basal) in KHB supplemented with 4 mM pyruvate, 8 mM 3-O-methyl-d-glucose (including 0.5 μCi/ml 3-O-[3H]methyl-d-glucose; American Radiolabeled Chemicals, St. Louis, MO), and 28 mM mannitol (including 0.2 μCi/ml [14C]mannitol; GE Healthcare; M3). After incubation, the tendon was removed, and muscles were blotted, weighed, and then digested in 1 ml of 1 M NaOH (95°C, 10 min). Glucose transport was calculated from the average of two 200-μl aliquots of muscle digest to quantify intracellular 3-O-[3H]methyl-d-glucose, as described previously (39). In separate experiments, both 500 ng/ml GIP and a 200 times lower dose (2.5 ng/ml) were used to determine the dose responsiveness of GIP. Additionally, basal glucose transport was determined in the presence of GIP (0.5 μg/ml) ± wortmannin [coincubated; 1 μM dissolved in DMSO; Sigma; cf. (23)] to determine the role of the PI3K in mediating GIP stimulation of glucose transport. To control for any nonspecific effects of DMSO on glucose transport, an equal volume of DMSO was added to all reaction vials. In further experiments, glucose transport was determined in basal and insulin-stimulated (500 μU/ml) conditions in the presence and absence of GIP (500 ng/ml). Finally, glucose transport was determined in basal or GIP-stimulated conditions (500 ng/ml) ± the specific GIPR antagonist (Pro3)GIP (0.5 μg/ml; compare Ref. 13) to determine whether GIP signals through its classical receptor in skeletal muscle. As (Pro3)GIP is a competitive inhibitor of the GIPR, we included an additional 30 min of preincubation (in buffer M1) in the presence of (Pro3)GIP, but not GIP.
Isolation of Giant Sarcolemmal Vesicles From Incubated Soleus Strips
To measure the direct effect of GIP, in the absence of insulin, on GLUT4 translocation, soleus muscles were incubated under the same conditions as the glucose transport experiments ± GIP (without radiolabel), and giant sarcolemmal vesicles were prepared, as previously described (19). Briefly, muscle was cut into thin strips and incubated (1 h, 34°C, 100 rpm) in 140 mM KCl/10 mM MOPS (pH 7.4; BioShop) containing collagenase type VII (150 units/ml; Sigma) and aprotinin (10 mg/ml; BioShop). The muscle was washed with additional volumes of KCl/MOPS containing 10 mM Na2EDTA (pH 7.4; BioShop), and the supernatants were combined (7 ml total). Percoll (3.5%; GE Healthcare, Baie d'Urfé, QC, Canada), KCl (28 mM), and aprotinin (10 μg/ml) were added to the supernatant. This solution was layered under 3 ml of 4% Nycodenz (Sigma) and 1 ml KCl/MOPS, and centrifuged (60 g, 45 min, 25°C). The vesicles were harvested from the interface of Nycodenz and KCl/MOPS and pelleted by centrifugation (9,000 g, 5 min, 25°C), and the resulting pellet was resuspended in KCl/MOPS and stored at −80°C for Western blot analysis (as described above).
Statistical Analyses
Data were analyzed using a two-tailed unpaired Student's t-test or a 1-way ANOVA with a Fisher LSD post hoc test where appropriate using the GraphPad Prism v6.0 software package. A value of P < 0.05 was considered significant. All data are reported as means ± SE.
RESULTS
Blood Glucose and Serum Insulin Are Modulated Differently in Response to Different Macronutrient Challenges
To determine the efficacy of our gavage treatments, we measured changes in blood glucose following glucose, lipid, or mixed treatments. Following a glucose gavage (GLU), glucose levels peaked at 9.7 ± 0.1 mM (+63%, P < 0.05) and had returned to baseline by 90 min (Fig. 1A). Similarly, after a mixed glucose+lipid gavage (MIX), glucose levels peaked at 8.2 ± 0.3 mM (+37%, P < 0.05) and had returned to baseline by 90 min (Fig. 1A). Blood glucose was significantly higher in the GLU group compared with the MIX group at both 15 (+42%, P < 0.05) and 30 min (+20%, P < 0.05) postgavage. Additionally, there was a marked (−35%, P < 0.05) decrease in the total area under the curve (AUC) after the MIX treatment compared with GLU alone (Fig. 1B). Blood glucose levels at baseline were the same between groups. Blood glucose did not change following lipid gavage (LIP; Fig. 1A), and LIP was different from GLU and MIX at all time points from 5 to 90 min (for clarity, significance symbols have been omitted from graph).
Fig. 1.

Blood glucose and serum insulin levels postgavage. Blood glucose following glucose (●), mixed (▼), or lipid (■) gavage (A; n = 8) and total glucose area under the curve (AUC; B). Serum insulin following glucose (●), mixed (▼), or lipid (■) gavage (C; n = 8) and total insulin area under the curve (D). Values are expressed as means ± SE. *P < 0.05 different from glucose. †P < 0.05 different from mixed. In both A and C, LIP was different from GLU and MIX at all time points from 5 to 90 min, but for clarity, significance symbols were omitted from the graphs.
Serum insulin was measured to determine whether the change in blood glucose following the gavage treatment was sufficient to cause an increase in circulating insulin. Serum insulin peaked at 10 (MIX; +390%, P < 0.05) and 15 min (GLU; +360%, P < 0.05) postgavage and was not different between the glucose and mixed treatment groups at any time point (Fig. 1C) or by total area under the curve (Fig. 1D). Serum insulin levels at baseline were the same between groups. Serum insulin levels did not change following lipid gavage (Fig. 1C), and LIP was different from GLU and MIX at all time points from 5 to 90 min (for clarity, significance symbols have been omitted from graph).
Akt Phosphorylation Is Increased to a Greater Extent When Lipid Is Included in a Glucose Oral Challenge
Despite similar serum insulin levels, improved glucose clearance following the mixed treatment suggested enhanced insulin signaling, so we examined Akt phosphorylation at Ser-473 and Thr-308 activation sites in skeletal muscle by Western blot analysis. The MIX treatment increased Akt phosphorylation at Thr-308 2.5-fold over saline and significantly more than GLU alone (trend to a 1.7-fold increase over basal, P = 0.10; Fig. 2, A and C). The MIX treatment also increased Akt phosphorylation at Ser-473 greater than any other treatment (P < 0.05; Fig. 2, B and C). There were no changes in either Akt phosphorylation site following LIP treatment compared with saline (Fig. 2, A and B), nor did total Akt change following any treatment (Fig. 2C).
Fig. 2.
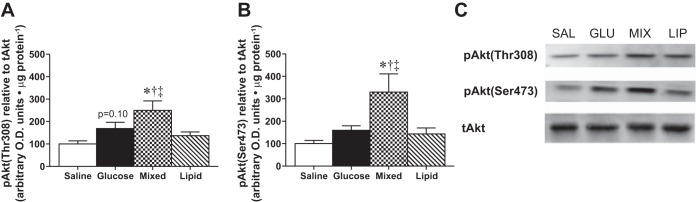
Skeletal muscle Akt phosphorylation postgavage. Whole muscle Akt phosphorylation at Thr-308 (A) and Ser-473 (B) following saline (open bars), glucose (solid bars), mixed (checkered bars), or lipid (striped bars) gavage (n = 8). Representative blots are shown in C. Data are expressed relative to total Akt and as a percentage change over saline. Values are expressed as means ± SE. *P < 0.05 different from saline. †P < 0.05 different from glucose. ‡P < 0.05 different from lipid.
Plasma GIP Levels Are Increased to a Greater Extent in Response to a Glucose+Lipid Gavage Compared with Glucose Alone
To further elucidate what factor(s) could be increasing insulin signaling and improving glucose handling following the mixed gavage, despite circulating insulin levels similar to the glucose gavage, we measured plasma GLP-1 (active form) and GIP. Following all three treatments, plasma GLP-1 was increased by 5 min and remained elevated for 60 min postgavage (Fig. 3A). However, at no time point was there any significant difference between treatments. Changes in GIP levels, however, varied by treatment. Following a glucose gavage, GIP was increased (+75%; P < 0.05) by 10 min, remained elevated, but unchanged through 30 min and had returned to baseline by 60 min (Fig. 3B). Following a mixed gavage, GIP was elevated by 10 min (+50%, P < 0.05), continued to increase through 30 min (+158%, P < 0.05 vs. 0 min), and was still elevated at 60 min (+125%, P < 0.05 vs. 0 min, but not different from 30 min). By 5 min, the increases in GIP following mixed gavage treatment was greater than that caused by glucose alone (P < 0.05; Fig. 3B), and occurred earlier than the increases in Akt phosphorylation. Lipid treatment did not significantly increase plasma GIP (Fig. 3B). Although the plasma concentration of GLP-1 was elevated following all three gavage treatments, it was not different between treatments, and, therefore, unlikely to be responsible for the improved glucose handling following the MIX treatment. For this reason, we focused solely on GIP as a possible mediator of skeletal muscle glucose transport in the following in vitro experiments.
Fig. 3.
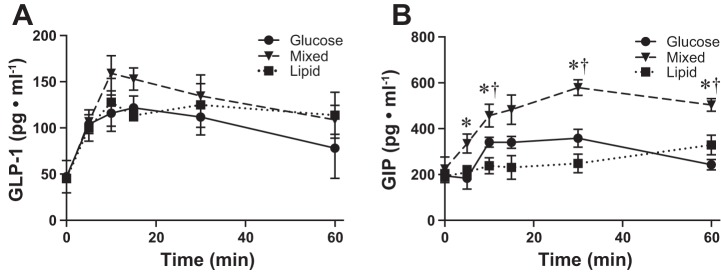
Plasma incretin levels postgavage. Plasma glucagon-like polypeptide (GLP-1) active (A) and glucose-dependent insulinotropic polypeptide (GIP; B) following glucose (●), mixed (▼) or lipid (■) gavage (n = 4). Values are expressed as means ± SE. *P < 0.05 different from glucose. †P < 0.05 different from lipid.
GIP Increases Skeletal Muscle Glucose Transport in the Absence and Presence of Insulin
We next aimed to determine the ability of GIP to directly affect skeletal muscle glucose handling. Ideally, a muscle-specific GIPR knockout model would be utilized to directly determine the in vivo postprandial effects of ablating GIP-mediated signaling in the muscle. Unfortunately, this model does not exist, and using whole body GIPR knockout or pharmacological approaches to ablate GIP signaling would be confounded by the expected decrease in insulin concentrations (26, 31). Therefore, we utilized an isolated skeletal muscle approach to directly determine the ability of GIP to stimulate glucose transport and plasma membrane GLUT4 accumulation in the absence of insulin. Using this approach, we found that GIP increased glucose transport (+25%; P < 0.05; Fig. 4A) and plasma membrane GLUT4 protein content (+50%; P < 0.05; Fig. 4B), suggesting a direct effect of GIP on increasing glucose transport into skeletal muscle.
Fig. 4.
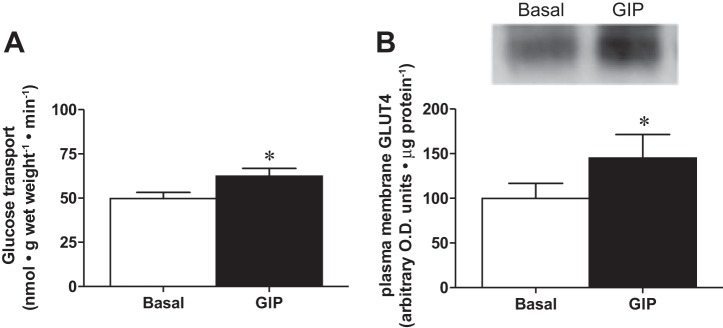
GIP-stimulated glucose transport and GLUT4 plasma membrane content. Basal glucose transport (A) and plasma membrane GLUT4 content (B) in presence (solid bar) and absence (open bar) of GIP. Seven independent glucose transport experiments were performed (n = 7), while the soleus muscle strips of 10 animals were combined for each independent plasma membrane isolation (n = 3). Values are expressed as means ± SE. *P < 0.05 different from basal.
Given the previous observations of enhanced GIP concentrations and muscle Akt phosphorylation following a mixed meal, we next aimed to demonstrate a link between GIP and the canonical insulin-signaling cascade. To accomplish this, we coincubated soleus strips with GIP in the presence or absence of wortmannin. Wortmannin is a well-characterized inhibitor of PI3K, an upstream regulator of Akt (23). Once again, GIP increased glucose transport by +45% (P < 0.05; Fig. 5), and this was prevented by treatment with wortmannin (Fig. 5), suggesting that, in skeletal muscle, GIP directly activates the PI3K/Akt signaling cascade.
Fig. 5.
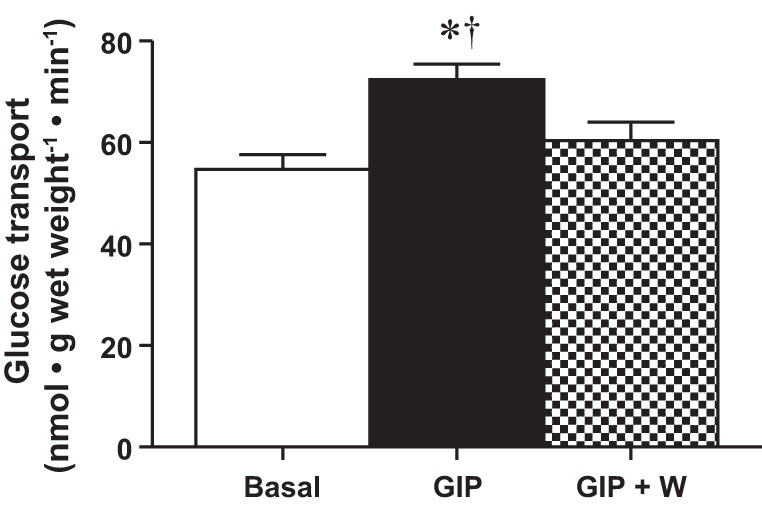
Wortmannin inhibition of GIP-stimulated glucose transport. Basal (open bar), GIP-stimulated (solid bar), and wortmannin-inhibited GIP-stimulated (checkered bar) glucose transport (n = 10). To control for any nonspecific effects of DMSO on glucose transport, an equal volume of DMSO was added to all reaction vials. Values are expressed as means ± SE. *P < 0.05 different from basal. †P < 0.05 different from wortmannin-inhibited.
To further support the link between GIP and the postprandial responses observed following a mixed macronutrient oral challenge, it was also necessary to determine whether GIP increased glucose transport in the presence of submaximal insulin concentrations. Therefore, we determined GIP-stimulated glucose transport in the presence and absence of insulin. Both GIP and insulin increased glucose transport above basal levels (Fig. 6A); however, the coincubation of insulin and GIP further stimulated skeletal muscle glucose transport (Fig. 6A). In addition, the increase in glucose transport in the presence of either GIP or insulin predicted an additive increase in glucose transport that was comparable to the actual response observed (Fig. 6B). Altogether, the current data support an insulin-independent effect of GIP on regulating skeletal muscle glucose transport.
Fig. 6.
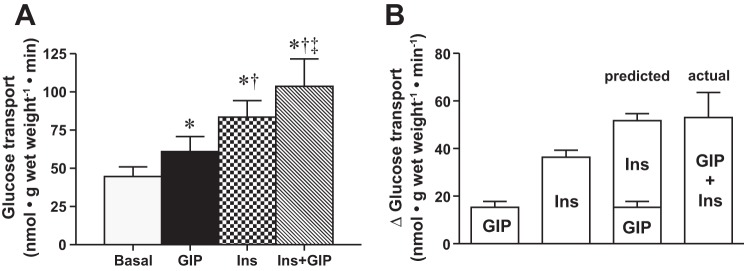
GIP and insulin costimulation of glucose transport. Basal and insulin-stimulated glucose transport (A) in the presence and absence of GIP (n = 7). Comparison of predicted vs. actual combined insulin + GIP-stimulated change in glucose transport (B). Values are expressed as means ± SE. *P < 0.05 different from basal. †P < 0.05 different from GIP stimulated. ‡P < 0.05 different from insulin-stimulated.
GIP Does not Signal Thorough its Traditional Receptor To Increase Glucose Transport in Skeletal Muscle
We next aimed to determine the presence of the GIPR in skeletal muscle. Although the expression of the GIPR has been demonstrated in multiple tissues (42), we are unaware of any published reports of GIPR mRNA levels in skeletal muscle. Using GIPR-specific TaqMan primers and probe set, we demonstrated that the presence of the GIPR in skeletal muscle is around 1% of that found in the pancreas (Table 1). Therefore, to determine whether GIP was stimulating glucose transport in skeletal muscle via signaling through its traditional receptor, we used a specific antagonist of the GIPR, (Pro3)GIP (13). However, GIP-stimulated glucose transport was not attenuated by (Pro3)GIP (Fig. 7), and (Pro3)GIP alone similarly stimulated glucose transport (Fig. 7). (Pro3)GIP differs from GIP in a single GLU to PRO amino acid substitution at position three. Since the sequence identity is 100% at all other positions, it is plausible that both GIP and (Pro3)GIP are interacting with an unknown receptor through another binding motif. This is not surprising as the copy number of the GIPR in skeletal muscle is low (Table 1). Ideally, the low expression of GIPR could be verified at the protein level; however, GIPR antibodies do not appear to be specific, as we detected the same banding pattern in both WT and GIPR knockout muscles (Fig. 8A).
Table 1.
GIP receptor RNA content in various rat tissues
| GIPR | |
|---|---|
| Soleus | 34.7 ± 0.6 |
| EDL | 34.7 ± 0.3 |
| Pancreas | 28.8 ± 0.6 |
Values are raw Ct ± SE (n = 3 or 4). Glucose-dependent insulinotropic polypeptide (GIP) receptor RNA measured by TaqMan quantitative PCR in rat soleus, extensor digitorum longus (EDL), and pancreas.
Fig. 7.
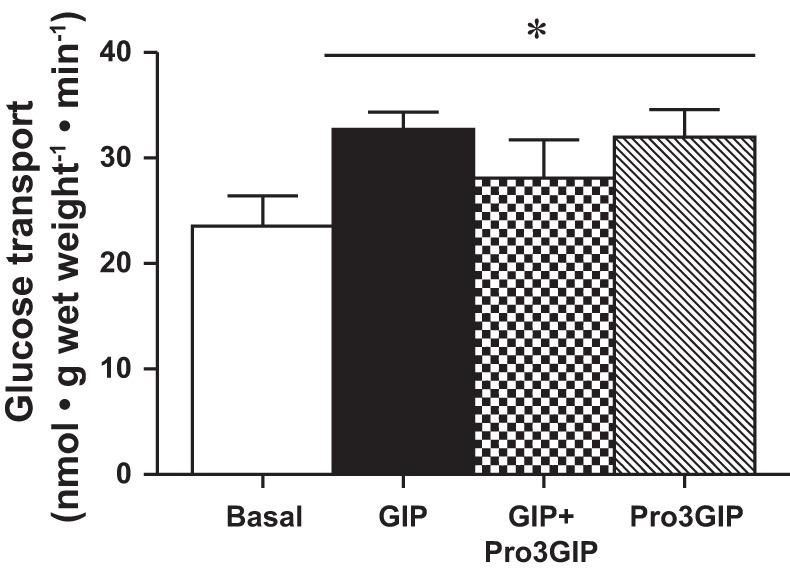
(Pro3)GIP inhibition of GIP-stimulated glucose transport. Basal and GIP-stimulated glucose transport in the presence and absence of the GIPR antagonist (Pro3)GIP (n = 11–14). Values are expressed as means ± SE. *P < 0.05 different from basal.
Fig. 8.
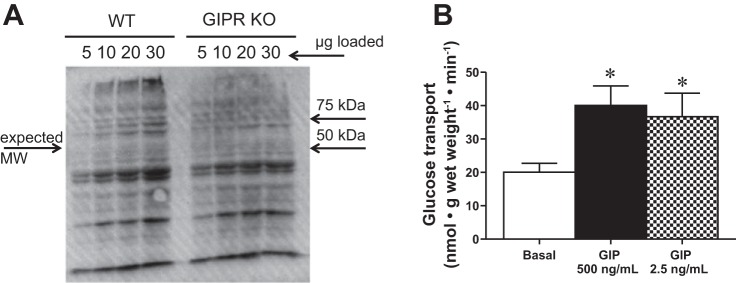
GIP stimulates glucose transport at a near-physiological dose. Western blot (A) of the GIP receptor in WT and GIPR knockout mouse skeletal muscle tissue demonstrating the nonspecific nature of the antibody. Basal and GIP-stimulated glucose transport (B; n = 4–6). Values are expressed as means ± SE. *P < 0.05 different from basal.
Given these unexpected findings, we repeated the GIP-stimulated skeletal muscle glucose transport experiments with a 200 times lower concentration of GIP (2.5 ng/ml) to verify the dose responsiveness of GIP. At this concentration, much closer to the levels seen in vivo, GIP was still able to stimulate glucose transport to levels similar to that observed at the higher concentration (Fig. 8B), suggesting that activation of a novel G protein-coupled receptor is not an artifact of pharmacological concentrations of the hormone.
DISCUSSION
The current study provides evidence that a glucose+lipid oral challenge increases serum GIP concentrations, induces skeletal muscle Akt phosphorylation and, therefore, could be related to improved glucose homeostasis relative to the administration of glucose alone. These responses occurred in the absence of changes in insulin concentrations, suggesting that GIP directly activates skeletal muscle glucose transport. In support of this, in an incubated muscle preparation, we provide evidence that GIP directly stimulates glucose transport in a PI3K-dependent manner and stimulates sarcolemmal GLUT4 accumulation. However, it appears that GIP-stimulated glucose transport does not occur via signaling through its classic receptor. Taken together, the current data provide evidence for an important physiological role for GIP in mediating skeletal muscle glucose transport that is independent of insulin, although the exact mechanism remains to be elucidated.
Improved Glucose Handling Following a Glucose+Lipid Oral Challenge
The initial characterization of postprandial glucose responses following various macronutrient gavage treatments suggested improved glucose clearance when glucose and lipids were combined. Specifically, we found that although both the glucose and mixed treatments caused moderate increases in glucose, peak blood glucose concentrations and the glucose AUC during the subsequent 3 h were attenuated following the glucose+lipid treatment, despite equivalent amounts of glucose in the two challenges. In addition, serum insulin concentrations were not different between these two treatment groups, and therefore, increased insulin cannot account for the observed improvement in glucose homeostasis following a mixed oral challenge. The current data support previous findings in humans of improved glucose clearance despite similar insulin concentrations following a mixed meal compared with the independent ingestion of an identical amount of glucose (18).
In the present study we also observed increased skeletal muscle Akt Ser-473 and Thr-308 phosphorylation following the mixed treatment greater than that seen in the glucose-only group, a response that was independent of circulating insulin concentrations. These data suggest enhanced signaling following the mixed treatment, which could be mediating the observed improvement in glucose homeostasis. To elucidate a potential mechanism, we focused on GIP and GLP-1, as they are secreted by the gut in response to both carbohydrate and lipid ingestion and have been implicated in the activation of various kinases, including Akt (1, 3, 32, 40). GLP-1 similarly accumulated within the circulation in response to all three oral challenges (GLU, MIX, and LIP), and, therefore, cannot account for alterations in glucose homeostasis. In contrast, while GIP responded to both GLU and MIX, GIP was significantly greater in response to the mixed treatment compared with glucose alone. In addition, lipid alone did not alter GIP concentrations. Combined, these data suggest that glucose, and not lipid, is required to initiate the signaling cascade leading to GIP secretion; however, in the presence of glucose, lipids augment this response. These data are in contrast to a recent report showing that GIP secretion is activated by lipids independently, while coingestion with glucose attenuates the stimulatory effects of lipids (36). This discrepancy could be related to the lipid source that was used (lard vs. canola oil) or the dose of lipid, which was three times higher than in the present study. Regardless of these differences, the current study strongly implies that consuming a mixture of lipids and carbohydrates improves glucose clearance beyond a potential insulinotropic effect of incretins. Importantly, plasma GIP accumulated earlier than the time points at which we observed increased skeletal muscle Akt phosphorylation and a reduction in blood glucose. Although this does not demonstrate cause and effect, these data suggest GIP may mediate the observed insulin-independent improvement in glucose homeostasis following a mixed meal.
GIP Enhances Basal and Insulin-Stimulated Glucose Transport in Isolated Soleus Muscle Strips
Given the association between glucose handling, circulating GIP and skeletal muscle Akt activation, we performed experiments in an isolated soleus muscle preparation to elucidate the direct effects of GIP on skeletal muscle glucose transport and insulin signaling. Unlike whole body measurements (34, 37), using isolated soleus muscle strips allowed us to examine the direct effect of GIP on muscle tissue without confounding factors of blood flow or other circulating neuroendocrine factors (6). Using this approach, we found that GIP increased both basal and insulin-stimulated glucose transport. In addition, when we compared the increase in glucose transport in the presence of both GIP and insulin, the predicted transport from the sum of each stimulus alone was identical to what was observed with the combined treatments, further suggesting GIP induces skeletal muscle glucose transport in an insulin-independent manner.
In the current study, we cannot rule out the possibility that GLP-1 has a direct effect on skeletal muscle glucose transport. Several reports have shown that, both in vivo and ex vivo, GLP-1 can increase glucose uptake in rodent skeletal muscle (1, 3, 15, 28); in contrast, a recent study (37) has suggested that GLP-1 increases microvascular recruitment but not glucose uptake directly in both rats and humans. These authors further demonstrate that GLP-1 can reverse impairments in insulin-stimulated glucose uptake caused by short-term high-fat feeding, but that it does not directly stimulate glucose uptake in skeletal muscle (38). Therefore, a direct role for GLP-1 in mediating skeletal muscle glucose uptake remains controversial. In addition, since the circulating levels of GLP-1 were not different between our treatment groups, GLP-1 is unlikely to be contributing to the improved glucose handling following the MIX treatment observed in this study, and, therefore, we focused on GIP. In contrast to GLP-1, in addition to the current study, demonstrating a direct effect of GIP on glucose transport, GIP has been shown to increase glucose uptake in adipocytes (40), and in mice, GIP alone increases glucose transport into isolated abdominal muscle to the same degree as insulin (32). Therefore, although the role of GLP-1 in mediating skeletal muscle glucose uptake remains controversial, evidence is mounting to suggest a direct effect of GIP in skeletal muscle glucose handling.
Wortmannin, but not (Pro3)GIP Inhibits GIP-Induced Increases in Glucose Transport
In the current study, the additivity observed with the coincubation of GIP and insulin suggests that either GIP and insulin are working through separate pathways or that under submaximal insulin stimulation, GIP is able to further stimulate members of the insulin signaling pathway. To determine the necessity of insulin-like signaling in mediating the effects of GIP, we demonstrated that GIP-stimulated glucose transport is inhibited by wortmannin, a PI3K inhibitor. Previous work in adipocytes has suggested GIP induces the phosphorylation of Akt (40), supporting the current interpretations in muscle. In further support of this, we isolated sarcolemmal vesicles from basal and GIP-incubated soleus muscles and found that GIP induced GLUT4 accumulation on the plasma membrane, a PI3K/Akt-mediated process. Combined, these data suggest that intact peripheral signaling through PI3K is required for the beneficial effects of GIP-mediated improvements in skeletal muscle glucose transport.
Surprisingly, when we attempted to inhibit GIP-stimulated glucose transport with the specific GIPR antagonist, (Pro3)GIP, we found that glucose transport was not blunted, but that (Pro3)GIP was also able to stimulate glucose uptake to the same extent as GIP. This suggests to us that GIP is not signaling through its traditional receptor in skeletal muscle.
The GIPR is a member of the G protein-coupled receptor (GPCR) superfamily, of which there are still hundreds of orphans, or receptors with no known ligand (reviewed in Ref. 10); therefore, it is possible that given their sequence homology, GIP and (Pro3)GIP are binding to another GPCR in skeletal muscle to stimulate glucose transport and GLUT4 accumulation at the plasma membrane. In support of this hypothesis, Wang and colleagues (43) demonstrated that both GLUT4 translocation and glucose uptake could be stimulated, in cell culture, by activation of Giβγ, independent of insulin. Additionally, they demonstrated that this was dependent on the phosphorylation of Akt and inhibited by wortmannin, also in agreement with our study. Given the low copy number of the GIPR transcript in skeletal muscle and the technical difficulties with antibodies for the detection of GIPR (and other GPCRs) at the level of expressed protein (this study and Refs. 12 and 33), it is difficult to confirm the presence of the GIPR in rodent skeletal muscle. However, traditional GIP-GIPR signaling does not appear to be required for the glucose transport-stimulating effects of GIP in skeletal muscle.
Perspectives and Significance
Taken together, our data suggest that independent of insulin, GIP can stimulate skeletal muscle glucose uptake in a PI3K-dependent manner, possibly through an unknown GPCR. Given that defects in signal transduction at insulin receptor substrate-1, upstream of PI3K, is believed to be one of the main contributors to insulin resistance (reviewed in Ref. 11), circumventing this portion of the insulin-signaling cascade could lead to improved glucose handling, independent of insulin. This is of clinical relevance, as the incretins are seen as a possible target for the treatment of Type 2 diabetes (reviewed in Ref. 2), a disease state associated with peripheral insulin resistance and reductions in insulin-induced Akt phosphorylation.
Summary
The data presented in this study provide evidence that an oral glucose challenge containing lipids results in lower peak blood glucose, as well as improved glucose clearance, but no difference in circulating insulin compared with glucose alone. This improvement in glucose handling is temporally matched by increased phosphorylation of Akt at Ser-473 and Thr-308, as well as circulating GIP. Additionally, the data demonstrate that GIP increases both basal and insulin-stimulated glucose transport in isolated soleus muscle, a response inhibited by the PI3K inhibitor, wortmannin, but not the GIPR-specific antagonist (Pro3)GIP. Taken together, these data suggest that GIP can improve glucose homeostasis by directly stimulating skeletal muscle glucose; however, the exact mechanism remains to be elucidated.
GRANTS
This work was funded by The Natural Sciences and Engineering Research Council of Canada (D. C. Wright, Grant 341158 and G. P. Holloway, Grant 03656), and infrastructure was purchased with the assistance of the Canadian Foundation for Innovation/Ontario Research Fund (D. C. Wright, Grant 218463 and G. P. Holloway, Grant 25136). L. A. Snook was supported by a Canadian Graduate Scholarship from the Natural Science and Engineering Research Council of Canada. D. C. Wright is a Tier II Canada Research Chair.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: L.A.S., D.C.W., and G.P.H. conception and design of research; L.A.S., E.M.N., and D.J.D. performed experiments; L.A.S., E.M.N., D.C.W., and G.P.H. analyzed data; L.A.S., D.C.W., and G.P.H. interpreted results of experiments; L.A.S. and G.P.H. prepared figures; L.A.S., D.C.W., and G.P.H. drafted manuscript; L.A.S., E.M.N., D.J.D., D.C.W., and G.P.H. edited and revised manuscript; L.A.S., E.M.N., D.J.D., D.C.W., and G.P.H. approved final version of manuscript.
REFERENCES
- 1.Acitores A, Gonzalez N, Sancho V, Arnes L, Valverde I, Malaisse WJ, and Villanueva-Penacarrillo ML. Participation of protein kinases in the stimulant action of GLP-1 on 2-deoxy-d-glucose uptake by normal rat skeletal muscle. Horm Metab Res 37: 275–280, 2005. [DOI] [PubMed] [Google Scholar]
- 2.Ahmadieh H, Azar ST. The role of incretin-based therapies in prediabetes: A review. Primary Care Diabetes 8: 286–294, 2014. [DOI] [PubMed] [Google Scholar]
- 3.Ayala JE, Bracy DP, James FD, Julien BM, Wasserman DH, Drucker DJ. The glucagon-like peptide-1 receptor regulates endogenous glucose production and muscle glucose uptake independent of its incretin action. Endocrinology 150: 1155–1164, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology 132: 2131–2157, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Bayliss WMS, EH. On the causation of the so-called ‘peripheral reflex secretion’ of the pancreas. Proc R Soc Lond Biol 69: 352–353, 1902. [Google Scholar]
- 6.Bonen A, Clark MG, Henriksen EJ. Experimental approaches in muscle metabolism: hindlimb perfusion and isolated muscle incubations. Am J Physiol Endocrinol Metab 266: E1–E16, 1994. [DOI] [PubMed] [Google Scholar]
- 7.Bornemann A, Ploug T, Schmalbruch H. Subcellular localization of GLUT4 in nonstimulated and insulin-stimulated soleus muscle of rat. Diabetes 41: 215–221, 1992. [DOI] [PubMed] [Google Scholar]
- 8.Brown JC, Dryburgh JR, Ross SA, Dupre J. Identification and actions of gastric inhibitory polypeptide. Recent Prog Horm Res 31: 487–532, 1975. [DOI] [PubMed] [Google Scholar]
- 9.Cartee GD, Wojtaszewski JF. Role of Akt substrate of 160 kDa in insulin-stimulated and contraction-stimulated glucose transport. Appl Physiol Nutr Metab 32: 557–566, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Civelli O, Reinscheid RK, Zhang Y, Wang Z, Fredriksson R, Schioth HB. G protein-coupled receptor deorphanizations. Annu Rev Pharmacol Toxicol 53: 127–146, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Draznin B. Molecular mechanisms of insulin resistance: serine phosphorylation of insulin receptor substrate-1 and increased expression of p85alpha: the two sides of a coin. Diabetes 55: 2392–2397, 2006. [DOI] [PubMed] [Google Scholar]
- 12.Drucker DJ, Yusta B. Physiology and pharmacology of the enteroendocrine hormone glucagon-like peptide-2. Annu Rev Physiol 76: 561–583, 2014. [DOI] [PubMed] [Google Scholar]
- 13.Gault VA, O'Harte FP, Harriott P, Mooney MH, Green BD, Flatt PR. Effects of the novel (Pro3)GIP antagonist and exendin(9–39)amide on GIP- and GLP-1-induced cyclic AMP generation, insulin secretion and postprandial insulin release in obese diabetic (ob/ob) mice: evidence that GIP is the major physiological incretin. Diabetologia 46: 222–230, 2003. [DOI] [PubMed] [Google Scholar]
- 14.Gonzalez E, McGraw TE. Insulin signaling diverges into Akt-dependent and -independent signals to regulate the recruitment/docking and the fusion of GLUT4 vesicles to the plasma membrane. Mol Biol Cell 17: 4484–4493, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hansen BF, Jensen P, Nepper-Christensen E, Skjolstrup B. Effects of glucagon-like peptide-1 (7–36)amide on insulin stimulated rat skeletal muscle glucose transport. Acta Diabetol 35: 101–103, 1998. [DOI] [PubMed] [Google Scholar]
- 16.Hansotia T, Drucker DJ. GIP and GLP-1 as incretin hormones: lessons from single and double incretin receptor knockout mice. Regul Pept 128: 125–134, 2005. [DOI] [PubMed] [Google Scholar]
- 17.Herbst EA, Paglialunga S, Gerling C, Whitfield J, Mukai K, Chabowski A, Heigenhauser GJ, Spriet LL, Holloway GP. Omega-3 supplementation alters mitochondrial membrane composition and respiration kinetics in human skeletal muscle. J Physiol 592: 1341–1352, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackson RA, Blix PM, Matthews JA, Morgan LM, Rubenstein AH, Nabarro JD. Comparison of peripheral glucose uptake after oral glucose loading and a mixed meal. Metab Clin Exp 32: 706–710, 1983. [DOI] [PubMed] [Google Scholar]
- 19.Juel C. Muscle lactate transport studied in sarcolemmal giant vesicles. Biochim Biophys Acta 1065: 15–20, 1991. [DOI] [PubMed] [Google Scholar]
- 20.Kim SJ, Nian C, Karunakaran S, Clee SM, Isales CM, McIntosh CH. GIP-overexpressing mice demonstrate reduced diet-induced obesity and steatosis, and improved glucose homeostasis. PloS One 7: e40156, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kramer HF, Witczak CA, Taylor EB, Fujii N, Hirshman MF, Goodyear LJ. AS160 regulates insulin- and contraction-stimulated glucose uptake in mouse skeletal muscle. J Biol Chem 281: 31,478–31,485, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7–36: a physiological incretin in man. Lancet 2: 1300–1304, 1987. [DOI] [PubMed] [Google Scholar]
- 23.Lee AD, Hansen PA, Holloszy JO. Wortmannin inhibits insulin-stimulated but not contraction-stimulated glucose transport activity in skeletal muscle. FEBS Lett 361: 51–54, 1995. [DOI] [PubMed] [Google Scholar]
- 24.Lemieux K, Han XX, Dombrowski L, Bonen A, Marette A. The transferrin receptor defines two distinct contraction-responsive GLUT4 vesicle populations in skeletal muscle. Diabetes 49: 183–189, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Marette A, Richardson JM, Ramlal T, Balon TW, Vranic M, Pessin JE, Klip A. Abundance, localization, and insulin-induced translocation of glucose transporters in red and white muscle. Am J Physiol Cell Physiol 263: C443–C452, 1992. [DOI] [PubMed] [Google Scholar]
- 26.Miyawaki K, Yamada Y, Ban N, Ihara Y, Tsukiyama K, Zhou H, Fujimoto S, Oku A, Tsuda K, Toyokuni S, Hiai H, Mizunoya W, Fushiki T, Holst JJ, Makino M, Tashita A, Kobara Y, Tsubamoto Y, Jinnouchi T, Jomori T, Seino Y. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nature Med 8: 738–742, 2002. [DOI] [PubMed] [Google Scholar]
- 27.Miyawaki K, Yamada Y, Yano H, Niwa H, Ban N, Ihara Y, Kubota A, Fujimoto S, Kajikawa M, Kuroe A, Tsuda K, Hashimoto H, Yamashita T, Jomori T, Tashiro F, Miyazaki J, Seino Y. Glucose intolerance caused by a defect in the entero-insular axis: a study in gastric inhibitory polypeptide receptor knockout mice. Proc Natl Acad Sci USA 96: 14,843–14,847, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizuno A, Kuwajima M, Ishida K, Noma Y, Murakami T, Tateishi K, Sato I, Shima K. Extrapancreatic action of truncated glucagon-like peptide-I in Otsuka Long-Evans Tokushima Fatty rats, an animal model for non-insulin-dependent diabetes mellitus. Metab Clin Exp 46: 745–749, 1997. [DOI] [PubMed] [Google Scholar]
- 29.Mojsov S, Weir GC, Habener JF. Insulinotropin: glucagon-like peptide I (7–37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J Clin Invest 79: 616–619, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moore B. On the treatment of Diabetus mellitus by acid extract of duodenal mucous membrane. Biochem J 1: 28–38, 1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7–36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest 91: 301–307, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O'Harte FP, Gray AM, Flatt PR. Gastric inhibitory polypeptide and effects of glycation on glucose transport and metabolism in isolated mouse abdominal muscle. J Endocrinol 156: 237–243, 1998. [DOI] [PubMed] [Google Scholar]
- 33.Panjwani N, Mulvihill EE, Longuet C, Yusta B, Campbell JE, Brown TJ, Streutker C, Holland D, Cao X, Baggio LL, Drucker DJ. GLP-1 receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate development of atherosclerosis in diabetic male ApoE(-/-) mice. Endocrinology 154: 127–139, 2013. [DOI] [PubMed] [Google Scholar]
- 34.Rose MT, Itoh F, Takahashi Y. Effect of glucose-dependent insulinotropic polypeptide on whole-body glucose utilization in sheep. Exp Physiol 83: 783–792, 1998. [DOI] [PubMed] [Google Scholar]
- 35.Schertzer JD, Antonescu CN, Bilan PJ, Jain S, Huang X, Liu Z, Bonen A, Klip A. A transgenic mouse model to study glucose transporter 4myc regulation in skeletal muscle. Endocrinology 150: 1935–1940, 2009. [DOI] [PubMed] [Google Scholar]
- 36.Shibue K, Yamane S, Harada N, Hamasaki A, Suzuki K, Joo E, Iwasaki K, Nasteska D, Harada T, Hayashi Y, Adachi Y, Owada Y, Takayanagi R, Inagaki N. Fatty acid-binding protein 5 regulates diet-induced obesity via GIP secretion from enteroendocrine K cells in response to fat ingestion. Am J Physiol Endocrinol Metab 308: E583–E591, 2015. [DOI] [PubMed] [Google Scholar]
- 37.Sjoberg KA, Holst JJ, Rattigan S, Richter EA, Kiens B. GLP-1 increases microvascular recruitment but not glucose uptake in human and rat skeletal muscle. Am J Physiol Endocrinol Metab 306: E355–E362, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sjoberg KA, Rattigan S, Jeppesen JF, Lundsgaard AM, Holst JJ, Kiens B. Differential effects of glucagon-like peptide-1 on microvascular recruitment and glucose metabolism in short- and long-term insulin resistance. J Physiol 593: 2185–2198, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith AC, Mullen KL, Junkin KA, Nickerson J, Chabowski A, Bonen A, Dyck DJ. Metformin and exercise reduce muscle FAT/CD36 and lipid accumulation and blunt the progression of high-fat diet-induced hyperglycemia. Am J Physiol Endocrinol Metab 293: E172–E181, 2007. [DOI] [PubMed] [Google Scholar]
- 40.Song DH, Getty-Kaushik L, Tseng E, Simon J, Corkey BE, Wolfe MM. Glucose-dependent insulinotropic polypeptide enhances adipocyte development and glucose uptake in part through Akt activation. Gastroenterology 133: 1796–1805, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sweeney G, Garg RR, Ceddia RB, Li D, Ishiki M, Somwar R, Foster LJ, Neilsen PO, Prestwich GD, Rudich A, Klip A. Intracellular delivery of phosphatidylinositol (3,4,5)-trisphosphate causes incorporation of glucose transporter 4 into the plasma membrane of muscle and fat cells without increasing glucose uptake. J Biol Chem 279: 32,233–32,242, 2004. [DOI] [PubMed] [Google Scholar]
- 42.Usdin TB, Mezey E, Button DC, Brownstein MJ, Bonner TI. Gastric inhibitory polypeptide receptor, a member of the secretin-vasoactive intestinal peptide receptor family, is widely distributed in peripheral organs and the brain. Endocrinology 133: 2861–2870, 1993. [DOI] [PubMed] [Google Scholar]
- 43.Wang L, Hayashi H, Kishi K, Huang L, Hagi A, Tamaoka K, Hawkins PT, Ebina Y. Gi-mediated translocation of GLUT4 is independent of p85/p110alpha and p110gamma phosphoinositide 3-kinases but might involve the activation of Akt kinase. Biochem J 345: 543–555, 2000. [PMC free article] [PubMed] [Google Scholar]
- 44.Yamada C, Yamada Y, Tsukiyama K, Yamada K, Yamane S, Harada N, Miyawaki K, Seino Y, Inagaki N. Genetic inactivation of GIP signaling reverses aging-associated insulin resistance through body composition changes. Biochem Biophys Res Commun 364: 175–180, 2007. [DOI] [PubMed] [Google Scholar]