

Abstract
Whole animal physiological performance is highly polygenic and highly plastic, and the same is generally true for the many subordinate traits that underlie performance capacities. Quantitative genetics, therefore, provides an appropriate framework for the analysis of physiological phenotypes and can be used to infer the microevolutionary processes that have shaped patterns of trait variation within and among species. In cases where specific genes are known to contribute to variation in physiological traits, analyses of intraspecific polymorphism and interspecific divergence can reveal molecular mechanisms of functional evolution and can provide insights into the possible adaptive significance of observed sequence changes. In this review, we explain how the tools and theory of quantitative genetics, population genetics, and molecular evolution can inform our understanding of mechanism and process in physiological evolution. For example, lab-based studies of polygenic inheritance can be integrated with field-based studies of trait variation and survivorship to measure selection in the wild, thereby providing direct insights into the adaptive significance of physiological variation. Analyses of quantitative genetic variation in selection experiments can be used to probe interrelationships among traits and the genetic basis of physiological trade-offs and constraints. We review approaches for characterizing the genetic architecture of physiological traits, including linkage mapping and association mapping, and systems approaches for dissecting intermediary steps in the chain of causation between genotype and phenotype. We also discuss the promise and limitations of population genomic approaches for inferring adaptation at specific loci. We end by highlighting the role of organismal physiology in the functional synthesis of evolutionary biology.
Keywords: complex traits, physiological genomics, QTL mapping, selection experiments, systems genetics
within-species variation in whole animal physiological performance is typically attributable to the combined effects of allelic variation at many loci and environmentally induced variation occurring across all stages of development and even prior to conception. That is, physiological performance can generally be regarded as a classic quantitative trait. The same is generally true for the many subordinate traits that underlie whole animal performance capacities. For example, aerobic exercise performance in rodents is a function of the pulmonary, cardiovascular, muscular, and neural systems, each of which comprises numerous components. For these reasons, quantitative genetics is well suited to the analysis of physiological phenotypes and can be used to infer the microevolutionary processes that have shaped patterns of trait variation within and among species.
In cases where it is possible to identify allelic polymorphisms at specific loci that contribute to variation in physiological traits, the tools and theory of population genetics can provide insights into the evolutionary processes that have shaped observed patterns of genetic variation within species (48, 185). In comparisons of homologous genes among species, analysis of sequence divergence and reconstruction of ancestral sequences permit inferences about historical pathways of molecular evolution (46). Moreover, ancestral sequence reconstructions provide a starting point for genetic engineering experiments that can provide detailed insights into the biochemical and biophysical mechanisms that are responsible for evolved changes in molecular function (76, 77).
Here, we review how genetic approaches can inform our understanding of mechanisms and processes in physiological evolution. We also review approaches for identifying genetic loci that contribute to variation in physiological traits, including both forward and reverse genetics approaches. We do not discuss evolutionary analyses in general, such as the use of phylogenetic comparative methods or ancestral state reconstructions of phenotypes (63, 121, 154); instead, we focus more specifically on how the tools and theory of quantitative genetics, population genetics, and molecular evolution can be used to shed light on the evolution of physiological and biochemical phenotypes. Throughout, we maintain a primary focus on whole organism physiological performance because it plays a pivotal role in both functional biology and evolution.
Insights Into Evolutionary Pattern and Process
Quantitative genetics.
Physiology is an attractive field for quantitative genetic work for a number of reasons. First, the functional significance of many traits is well understood. The traits are not mere markers of an underlying genetic system, but play specific, known roles in adaptive processes. Second, comparative work is often available and can reveal which traits are evolutionarily labile. Third, many physiologists work with animals sampled directly from nature, so that specific natural reference populations can be established for genetic work.
—Arnold (4, p. 207).
Quantitative genetics was developed as a means of estimating the genetic and environmental components of variation in phenotypes that are characterized by more-or-less continuous distributions of trait values, such as body weight or basal metabolic rate (51, 125). Quantitative genetics makes use of variances and covariances in measured trait values among individuals with known pedigree relationships. The additive genetic component of trait variation (the fraction of variation that can be passed on from parents to offspring and that is used to calculate narrow-sense heritability) is of special relevance because it is the primary determinant of a trait's potential for evolutionary change.
As stated by Lynch (124) (p. 497), “For the physiological ecologist, [narrow-sense] heritability is the most useful piece of genetic information since it is both descriptive and predictive.” It is predictive in the sense that resemblance between parents and offspring for a given trait enables us to forecast the short-term response to selection on the trait. At the phenotypic level, selection describes statistical change in the means of traits within the parental generation. Inheritance then determines how the effects of selection are transmitted from the parental generation to the offspring generation, i.e., the genetic response to selection. Both narrow-sense heritability and selection have now been measured for many physiological traits (84, 200), even in human populations (175).
The genetic property of parents that accounts for phenotypic resemblance with their offspring is “additive genetic value”: the sum across loci of the average effects of alleles on a given trait. For a trait that is under polygenic control, an individual's phenotypic value has an additive genetic component (which is transmitted from parents to offspring), a nonadditive genetic component due to dominance and epistasis (which is not transmitted to offspring because the allelic combinations found in parents are broken up when gametes are produced), and an environmental component.
The narrow-sense heritability of a trait is the ratio of variance in additive genetic values (additive genetic variance) divided by variance in phenotypic values (phenotypic variance). This ratio can be calculated as the slope of a regression of additive genetic values (the average trait values for adult offspring) on the measured phenotypic values of parents (assuming the absence of nongenetic parental effects, as might occur through maternal care). The central axiom of quantitative genetics states that—from one generation to the next—the response to selection on a single trait is equal to the product of the narrow-sense heritability of the trait and the directional selection differential, a measure of the strength of selection. The selection differential is the difference in mean trait values between individuals who successfully reproduce in a given generation (actual parents) and the larger, more inclusive set of all potential parents. In the context of a selection experiment, the heritability of a trait is the fraction of the selection differential that is transmitted to the next generation, manifest as a change in the average value of the trait (123).
Multivariate quantitative genetics.
Artificial selection as practiced by animal breeders is often applied to measurements of a single trait (e.g., growth rate, milk yield), but selection in nature inevitably acts on multiple traits. The desire to predict responses to selection that act simultaneously on multiple traits motivated the development of a multivariate theory of quantitative inheritance and selection (3, 6, 14, 109). To predict responses to selection that act simultaneously on multiple traits, we need to measure additive genetic covariances between traits. The measured covariances (standardized as genetic correlations) describe the extent to which pairs of traits may be genetically coupled due to pleiotropy (effects of a single gene on multiple traits) and/or linkage disequilibrium (LD, the nonrandom association between alleles at different loci in the gametic phase). In this multivariate theory, heritability is replaced by the additive genetic variance-covariance matrix (the “G-matrix”) as a descriptor of polygenic inheritance. Likewise, to predict the evolutionary response to selection acting on multiple traits, the single-trait selection differential is replaced by the “directional selection gradient”, which measures the direct force of selection on a given trait, controlling statistically for its correlations with other traits in the analysis. The selection gradient can be measured as the partial regression of relative fitness on the trait of interest, holding all other traits constant. It predicts the change in relative fitness that would be produced by a unit change in the trait in the absence of changes in genetically correlated traits (111). In addition to predicting short-term responses to selection, quantitative genetic models of long-term phenotypic evolution are also potentially applicable to the analysis of physiological traits (7–9, 50, 82, 91, 107), although these models are premised on controversial assumptions about the constancy of genetic parameters over evolutionary time (14).
Quantitative genetic analysis in a multivariate framework can reveal genetic correlations between traits, which can determine whether directional selection on one trait will produce a correlated response in an unselected trait (3, 92, 111). Genetic correlations between traits can provide insights into the causes of physiological trade-offs and constraints that have always been of interest to both physiologists and evolutionary biologists (4, 28, 61, 64, 159). Documentation of genetic correlations between traits within populations can also suggest hypotheses about the nature of trait variation and covariation among species. Information about the sign and magnitude of genetic correlations among traits suggests that certain trajectories of phenotypic change are more likely than others as species diverge by random genetic drift or in response to natural or sexual selection. In other words, genetic correlations may bias pathways of evolution along “genetic lines of least resistance” (166, 167).
An important point to emphasize is that—as with narrow-sense heritabilities—elements of the G-matrix are not static and will change as a result of changes in allele frequencies that are brought about by selection or drift (91, 92). Moreover, correlated selection on multiple traits may favor particular combinations of alleles at multiple loci, thereby permitting the evolution of coadapted gene complexes. Thus, traits may become genetically integrated as a partial step toward the evolution of modularity (131). At any given point in time, the form of the G matrix may reflect not only functional relations among traits but also their past history of correlated selection.
Quantitative genetic analysis can also help characterize the allowable dimensions of organismal form and function and can potentially shed light on why certain phenotypes do not exist. The nonexistence of a particular trait or trait combination can be explained in two general ways: 1) it was not adaptive in ancestral environments (it did not offer a solution to any pressing problem faced by ancestral organisms), so the trait never evolved; or 2) the requisite genetic variation never existed, so the trait could never evolve no matter how adaptive it might have been. The latter hypothesis can be tested by combining analyses of quantitative inheritance with selection experiments (e.g., 205, 206).
Epigenetic sources of phenotypic variation.
When considering environmentally induced changes in physiological traits, it is important to note that some environmental effects, especially those experienced early in life during “critical periods”, can be passed on to offspring. Early-life effects may occur through variation in maternal care, transmission of hormones and nutrients to offspring, developmental plasticity, fetal adaptation and maladaptation, changes in appetite and dietary preferences, energy availability, and compensatory growth. Certain early-life effects can amplify across successive generations (204). Moreover, they can interact with other sources of environmental and genetic variation, including sex (207), in ways that facilitate or constrain evolutionary change at multiple levels of biological organization (11, 26, 141). Much more work is required to fully elucidate the role of epigenetics in physiology and evolution (45, 56, 80, 87, 88).
Of the various potential mechanisms that could be responsible for the persistent effects of early exposures (203), induced alterations in epigenetic regulation likely play an important role in developmental programming of adult behavioral and physiological traits (202). In particular, the epigenetic mechanism of DNA methylation is a prime candidate; developmental establishment of DNA methylation is affected by the environment (90) and, once established, is maintained with high fidelity (30), enabling the long-lasting stability that is the hallmark of developmental programming.
Analysis of phenotypically plastic traits.
Under natural conditions, most physiological traits are characterized by a sizable environmental component of phenotypic variance, and this has important implications for interpreting patterns of trait variation within and among populations. For many physiological phenotypes in animals, it is generally a safe bet that a substantial fraction of the observed trait variation among conspecific populations will be attributable to reversible acclimatization (phenotypic plasticity) rather than genetic differentiation. As stated by Garland and Adolph (62) (p. 194), “Recognizing problems introduced by rampant acclimatization, physiologists may have been inhibited from searching for relatively small population differences. Consequently, the focus on proximate mechanisms of coping with environmental change (i.e., acclimatization) drew more attention than the possibility of genetic differentiation among populations.” The lability of most physiological traits suggests that common-garden or reciprocal transplant experiments are essential for interpreting patterns of trait variation in an evolutionary context (62). Such experiments can be used to characterize the range of phenotypic variation that is achievable via acclimation/acclimatization and can reveal the extent to which phenotypic plasticity might obviate the need for genetically based, evolutionary changes to cope with a changing environment. For example, a given high-altitude animal species may have a higher hematocrit than closely related lowland species when the trait is measured in wild-caught animals at their native elevation, but common-garden or reciprocal transplant experiments would be required to measure the environmental component of the observed trait variation among species. It may be that the elevated hematocrit in the high-altitude species is purely environmentally induced, reflecting an increased production of erythropoietin in response to hypoxemia.
Common-garden experiments are also critical in multispecies comparative studies because environmentally induced variation can confound the detection and interpretation of phylogenetic signal in trait values (63). If environmentally labile traits are not measured under common-garden conditions, then it may not be possible to determine the extent to which phenotypic similarity between a given pair of species is attributable to shared phylogenetic heritage vs. a shared, plastic response to a common environmental factor. For example, a given pair of high-altitude species may have similarly high hematocrits when measured under the hypoxic conditions of their native environment. One possible explanation for the phenotypic similarity is that the two species inherited a constitutively elevated hematocrit from a common ancestor. But before drawing that conclusion, we would first need to rule out the alternative possibility that the two species have simply responded in similar fashion to the same hypoxic stimulus in their shared environment.
Physiology, performance, behavior, and fitness: measuring selection in the wild.
Several different strategies are used to study the evolutionary adaptation of physiology. . . . The one traditionally used by physiological ecologists involves examining comparative data on contemporaneous organisms (e.g., species in different environments) and then deriving post hoc reconstructions of patterns that have evolved over historical time. . . . An alternative approach involves studying selection on phenotypic traits in nature . . . .
—Bennett and Huey (22, p. 265).
If a given performance trait is variable among individuals and if some fraction of that variation is heritable, then it is possible to examine the direct and indirect effects of selection on the trait in real time and to detect responses to multivariate selection over evolutionary time (5, 22). This can be accomplished by measuring the statistical relationship between Darwinian fitness (or estimated components of fitness, such as survival and fecundity) and multivariate phenotypes in natural populations. The strategy is based on multivariate selection theory, which deals with the effects of selection that acts simultaneously on multiple traits (107, 108, 110, 111). With experimental measurements of inheritance and observational data on selection (shifts in trait means within a generation), multivariate selection theory can be used to predict evolutionary change in trait means from one generation to the next [the directional response to selection (107)]. The key parameter that predicts the directional response to selection on a given trait is the selection gradient described above (Quantitative genetics). Arnold (5) described an approach based on path analysis to factor the selection gradient into two parts: a “performance gradient” (the measured effect of the trait on some ecologically relevant measure of organismal performance) and a “fitness gradient” (the effect of performance on fitness; Fig. 1). This paradigm, which focused attention on the importance of measuring whole organism animal performance, has since been extended to include behavior (28, 68, 106) (Fig. 1).
Fig. 1.
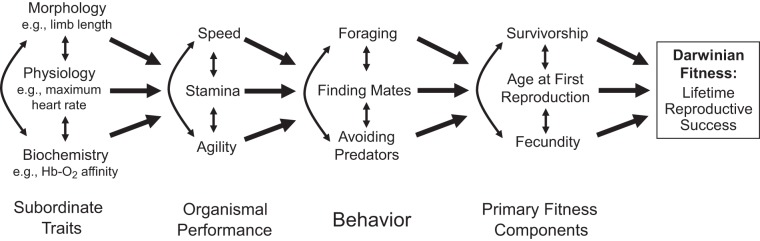
Path diagram of hypothetical relationships among phenotypes at multiple levels of biological organization, ranging from fundamental morphological, physiological, and biochemical traits to Darwinian fitness. Following the standard conventions of path analysis, single-headed arrows indicate putatively causal relationships, whereas double-headed arrows indicate correlations between traits. Not shown are possible direct effects of subordinate traits on behavior or fitness components. For example, individual variation in circulating hormone concentrations might directly affect behavior, growth rates, sexual maturation, and other traits. Arnold (5) first introduced the formalities of path analysis for examining the adaptive significance of interindividual trait variation, and it quickly became a paradigm in ecological and evolutionary physiology. The depiction here builds on a number of subsequent papers that have expanded Arnold's original presentation through both conceptual and empirical approaches (1, 28, 64, 68, 106, 140, 173, 201). Many of these have included aspects of locomotor performance because it is easy to envision how traits such as speed or stamina fit into the scheme.
This approach has been applied successfully in a number of vertebrates (84), including studies of locomotor capacities of garter snakes [Thamnophis sirtalis (89)] and thermogenic capacities of high-altitude deer mice [Peromyscus maniculatus (79)]. These studies followed the same basic formula: physiological performance was measured in wild-caught animals (known-age cohorts in the garter snake study), and rates of survivorship were then estimated using mark-release-recapture protocols. Both studies measured the direction and magnitude of selection on physiological capacities in free-ranging animals, and, therefore, provided insights into the adaptive significance of naturally occurring trait variation. As stated by Bennett and Huey (22) (p. 275): “. . . such studies have the promise of freeing physiological ecology from an implicitly adaptationist program . . . by turning attention to the process of adaptation, rather than its simple assumption. These protocols also open hosts of new and interesting questions and firmly embed studies within the natural environments, demography, and ecology of the organisms investigated.”
Selection experiments and experimental evolution.
Selection experiments may be viewed as the earliest form of “genetic engineering”. Humans were altering the genetic compositions of plant and animal populations through domestication thousands of years before we understood how heredity worked. The impressive nature of these feats was one major line of evidence used by Charles Darwin when he developed the theory of evolution by natural selection. Although he did not recognize them as such (69), intentional and unintentional selection experiments are an experimental way to study “evolution in action”. They are also a way to produce “useful” organisms. The response to selective breeding constitutes the most direct and convincing test of whether a given trait harbors additive genetic variance. More recently, it has been noted that selection experiments are a modern corollary to the August Krogh Principle: if a suitable model does not exist, then create one (21)!
Selection experiments are an excellent way to probe the interrelations among traits, including complex traits, because one can atomize an organism to the desired level, impose selection at that level on a trait of interest, and observe correlated cross-generational changes in other traits (58, 60, 66, 187). They are a good way to test specific hypotheses about putative trade-offs (61) and constraints on the way organisms can evolve (e.g., 205, 206, 211). In particular, they can be a powerful way to demonstrate mechanism, i.e., how organisms work (58, 60). For example, one could impose selection on some measure of whole animal performance and then observe a correlated response in one or more lower-level traits that plausibly cause or permit the change in performance. This hypothesis of causality could then be tested by conducting a second experiment, selecting on the lower-level trait and determining whether performance changes were as predicted. As a hypothetical example, one could breed for high V̇o2 max in rodents (e.g., 72, 73, 211) and potentially observe correlated changes in hematocrit. A second experiment could select for high hematocrit and see whether V̇o2 max increases as predicted. As another example, one might select for basal metabolic rate (BMR) (103, 105), observe a correlated change in circulating thyroid hormone levels, and then conduct another experiment breeding for hormone levels.
Finally, as discussed in a separate section (Linkage mapping/association mapping), selection experiments can aid attempts to find the genes that underlie phenotypic variation. Specifically, genetic crosses between selected lines and nonselected or oppositely selected lines can be used to produce a mapping population with sufficient levels of both genotypic and phenotypic variation for detecting quantitative trait loci (QTL).
As discussed elsewhere, various types of experiments can be considered under the general categories of selection experiments and experimental evolution (60, 69, 93, 189). The key attributes of these experiments include an emphasis on hypothesis testing, tracking of phenotypes (and genotypes) across generations, replication, and potential reproducibility. Beyond those fundamental principles, selection experiments encompass a tremendous range of empirical studies with a wide variety of organisms (69), often including microorganisms and even digital “organisms” (221). Replication is crucial to allow strong inferences regarding both direct and correlated responses, because genetic drift can lead to divergence between selected and control lines for reasons unrelated to functional relationships or pleiotropic gene action (130). Replication also allows the discovery of multiple solutions to adaptive challenges (60, 67, 151, 160, 189).
At one end of the continuum, various organisms have been introduced to new habitats by both intentional and unintentional human activities. English house sparrows and Drosophila species are two familiar examples (86). Sparrows are represented in various museum collections in a way that offers some ability to track phenotypic changes across generations, and these introductions have been “replicated” many times. Physiological ecologists have formulated specific hypotheses about how sparrows should evolve in response to altered environmental conditions, and these can be tested with measurements of individuals sampled from different populations and then raised under common-garden conditions (62, 117, 118, 134).
At the other end of the continuum lie laboratory-based artificial selection experiments, many of which have been conducted with rodents (156, 187, 211). Some of the examples we find most compelling involve replicated selection at the levels of behavior or whole animal physiological performance. For eight generations, Hayes and colleagues (211) selectively bred four replicate lines of laboratory house mice for high mass-independent maximal oxygen consumption (V̇o2 max) during forced treadmill exercise (Fig. 2). The selection criterion included body mass as a covariate, so selection was independent of effects of body mass on metabolic rate. In four other lines, they antagonistically selected for a combination of high mass-independent V̇o2 max and low mass-independent BMR. Four additional lines were maintained as nonselected controls. Compared with controls, V̇o2 max significantly increased by 11.2% in lines bred for V̇o2 max, while BMR did not change significantly (+2.5%). Compared with controls, V̇o2 max significantly increased by 5.3% in antagonistically selected lines, while BMR was not significantly reduced (−4.2%). Overall, analyses indicated a weak positive genetic correlation between V̇o2 max and BMR. These results offer weak support for the idea that selection to increase a mammal's aerobic capacity might be accompanied by an energetic cost in terms of higher resting metabolic rate.
Fig. 2.

Direct and correlated responses to selection for increased mass-independent maximal O2 consumption (V̇o2 max) in replicate lines of house mice after eight generations. Metabolic rates represent means (± SE) of four replicate lines per treatment. Four lines of mice were selectively bred for increased mass-independent metabolic rates (high-MMR). Another four lines were antagonistically selected for a combination of increased mass-independent metabolic rate and low mass-independent basal metabolic rate, BMR (antag-MR). The remaining four lines were maintained as nonselected controls. A: response to directional selection for increased mass-independent maximal metabolic rate (MMR) over eight generations. Directional selection for increased MMR was imposed on high-MMR and antag-MR lines (no selection on control lines). B: changes in mean mass-independent BMR over eight generations. Directional selection for decreased mass-independent BMR was imposed on antag-MR lines (no selection on mass-independent BMR in high-MMR or control lines).
For more than two decades and 70 generations, Garland and colleagues (60, 156, 187, 188) have bred four replicate lines of laboratory house mice for high levels of voluntary wheel running, while also maintaining four nonselected control lines. It is important to emphasize that in these sorts of experiments, the line is the experimental unit. Therefore, statistical comparisons of the selected and control lines use so-called mixed models with line as a random effect nested within line type, and degrees of freedom for testing the effect of selection are (in the wheel-running example) 1 and 6. Even if dozens or hundreds of individual animals are measured for a given comparison, the tests for effects of direct or correlated responses to selection will still be based on 1 and 6 degrees of freedom.
As expected from numerous studies selecting for locomotor behavior or performance in rodents [e.g., see Table 1 in (53)], wheel running responded rapidly to selection in the high runner (HR) lines before reaching apparent limits at generations 16–28, depending on line and sex (29, 67), and wheel running has not increased appreciably in the HR lines for an additional ∼40 generations of selective breeding (T. Garland, Jr., unpublished results). Mice in the HR lines have mainly evolved to run faster, rather than running for more minutes per day (especially in the case of females), perhaps partly because it is more economical (155) or more rewarding (20, 70, 156) to run faster.
Many correlated responses to selection have been documented in the HR mice, including increases in V̇o2 max (101) and treadmill endurance capacity (133). HR mice are also smaller, leaner, reach higher maximum speeds, and run more intermittently during wheel running, and have a reduced incremental cost of transport on a whole-animal basis, although not on a mass-corrected basis (155, 190, 191). Suborganismal traits that have evolved in HR mice include larger hearts (71), increased hindlimb symmetry, larger femoral heads, heavier foot bones, and altered semicircular canal shape (65, 96, 168). Although many of these correlated evolutionary responses would have been anticipated on the basis of physiological knowledge, others might not have been, such as the increase in baseline circulating corticosterone levels or the reduced growth rate and smaller adult body size of HR mice (70, 156, 187). Several lines of evidence indicate altered brain reward systems in HR mice (70), including an increase in overall brain size and the size of the midbrain (102). However, there are a number of evolutionary changes that could have been expected to occur in HR mice (e.g., changes in resting or basal metabolic rate, litter size, maternal care behavior, and general levels of aggression) that did not occur (70). As with any approach in science, well-designed selection experiments may raise more questions than they answer!
Koteja and colleagues (164) used a wild population of bank voles (Myodes glareolus) to begin an ambitious selection experiment (16 total lines) designed to model the adaptive evolution of aerobic metabolism in relation to changes in locomotor activity, predatory behavior, and diet. All three selection treatments were successful: the replicate lines diverged from the nonselected control lines. Several important observations about correlated responses to selection (or lack thereof) have since been published. For example, after 13 or 14 generations, selection for high-activity-related aerobic metabolism did not alter the capacity for nonshivering thermogenesis (174). In the lines selected for increased aerobic exercise capacities, an analysis of variation in tissue-specific transcriptomes from animals sampled at generation 13 revealed that changes in gene expression make a significant contribution to the selection response (104).
Through a glass, darkly: population genetic analysis of single-locus polymorphisms.
Alternative alleles at a single gene may have detectable effects on proximal biochemical phenotypes, but the effects on fitness-related physiological performance may typically fall well below the resolving power of our experimental measurements. From an evolutionary perspective, the key question is whether they also fall below the resolving power of natural selection. Specifically, we want to know whether allelic variation in gene function translates into quantitative trait differences, which, in turn, translate into fitness differences of sufficient magnitude that the deterministic effects of selection are not overwhelmed by the stochastic effects of genetic drift (48, 185) (approximately, when 2Nes>1, where Ne is the effective population size and s is the selection coefficient of the mutant allele). Although it may seldom be possible to measure fitness variation among alternative single-locus genotypes under natural conditions, the effects of selection are expected to leave a historical imprint on levels and patterns of nucleotide variation at causative loci. Because DNA sequence data integrate the effects of fitness variation over long periods of time, neutrality tests based on DNA polymorphism and/or divergence hold the promise for providing indirect, retrospective insights into histories of positive selection at specific loci (185). These indirect inferences can complement direct, experimental measurements of allelic differences in gene function, as well as provide suggestive evidence that the measured differences have adaptive significance. The integration of population-genetic neutrality tests with functional experiments holds the promise of elucidating each step in the so-called “adaptive recursion” (54, 185). As stated by Feder et al. (52): “the general paradigm is that genes encode the phenotype, the phenotype determines the performance of organisms in natural environments in response to ecological or evolutionary stimuli, the performance determines the evolutionary fitness of alternative genotypes, and the fitness determines the frequency of genotypes in the next generation, in recursive fashion . . . .” (p. 320). It is this last step in the recursion that is not generally open to empirical scrutiny in natural populations, as it is typically not possible to quantify how differences in net reproductive rates of alternative genotypes affect the allelic composition of the gamete pool in the following generation. The cumulative effects of fitness variation over many past generations can only be inferred indirectly, and population-genetic neutrality tests—despite their inherent limitations—provide the best available means of making such inferences.
As an example of how population genetic analyses can bolster inferences about physiological adaptation, a multilocus analysis of nucleotide polymorphism in high- and low-altitude populations of deer mice (Peromyscus maniculatus) revealed evidence for a history of spatially varying selection at duplicated genes that encode the subunit chains of the tetrameric α2β2 hemoglobin protein. The population genetic evidence for spatially varying selection on two α-globin gene duplicates and two β-globin gene duplicates, in combination with functional analyses of native and recombinant Hb variants (135, 136, 179–183), corroborated independent lines of experimental evidence, which had demonstrated that allelic variation in hemoglobin-O2 affinity contributes to adaptive variation in whole animal aerobic performance under hypoxia (31, 32).
In addition to providing corroborative support for adaptive hypotheses, the integration of population-genetic neutrality tests and experimental tests of protein function can also serve a useful function by falsifying such hypotheses (38, 161), thereby providing a check on unbridled speculation and adaptive storytelling. As stated by Cheviron et al. (38): “In the absence of experimental validation, analyses of variation in coding sequence can lead to facile inferences about the adaptive significance of observed amino acid replacements. This is especially true in studies of candidate genes in natural populations where there is some plausible a priori expectation about the adaptive significance of changes in protein function.” (p. 2954).
Molecular evolution: analysis of interspecific divergence at specific loci.
In comparisons of homologous protein-coding genes among species, one popular approach for detecting a history of positive selection involves a comparison of relative rates of synonymous and nonsynonymous substitutions at interleaved codon positions (214). This approach has proven useful for identifying episodes of positive selection that involved recurrent amino acid substitutions, as might be expected for proteins involved in coevolutionary arms race dynamics (e.g., immune defense or gamete recognition proteins), but there are good reasons to expect that it is generally less useful for detecting and identifying adaptive modifications of protein function that were caused by small numbers of substitutions (186). Nonetheless, the development of increasingly sophisticated codon-substitution models has spawned a cottage industry of comparative genomic studies that follow a highly formulaic script: 1) compare coding sequences of orthologous genes among multiple species; 2) identify specific genes with a subset of sites in which the ratio of nonsynonymous-to-synonymous substitution rates (dN/dS) is significantly greater than unity (the neutral expectation) in a specific lineage; and 3) tell a “just-so” story about why the observed amino acid substitutions in those genes must have contributed to lineage-specific adaptations. Such analyses can be useful for generating hypotheses about the adaptive significance of observed sequence changes, but such hypotheses ultimately need to be experimentally tested, ideally with protein-engineering experiments that measure the functional effects of specific substitutions on evolutionarily relevant ancestral backgrounds. Studies that integrate evolutionary analyses of sequence divergence with functional analyses of native and/or recombinant proteins can obtain far more detailed and robust evolutionary insights than are possible with analyses of sequence variation alone. Recent examples of this integrative approach include studies of vertebrate hemoglobins (74, 135, 151, 198) and opsins (25, 192, 217–220).
Ancestral protein reconstruction.
In general, conclusive inferences about mechanisms of protein evolution (adaptive or not) require experimental insights into the functional effects of historical mutations on the ancestral genetic background in which they occurred. In recent years, especially refined insights into mechanisms of protein evolution have been obtained by using protein engineering experiments that involved the reconstruction and functional testing of resurrected ancestral proteins (76, 77, 196). Using an alignment of nucleotide sequences (or translated amino acid sequences) from homologous protein-coding genes of extant species, the first step in this approach is to estimate a phylogeny of the sampled sequences (215). Model-based statistical methods are then used to reconstruct ancestral amino acid sequences of proteins at internal nodes in the phylogeny. For each variable site in the alignment, maximum likelihood and/or Bayesian statistical methods are used to infer the most likely character state that existed at a particular node, given the character states in observed (extant) sequences, the estimated phylogeny, and the assumed substitution model. The reconstructed coding sequence of the ancestral gene can then be synthesized and cloned into a plasmid construct, followed by expression and purification of recombinant protein in cultured cells. The “resurrected” ancestral protein can then be used for in vitro functional experiments and structural studies.
This approach has been applied with great success in studies designed to elucidate the molecular mechanisms by which vertebrate steroid receptors evolved to recognize their hormonal ligands. Steroid receptors are transcription factors that have evolved to recognize specific hormonal activators to regulate physiological responses. Following binding of specific steroid ligands, the individual receptors initiate specific downstream signaling cascades. Steroid receptors evolved from an ancestral receptor in the stem lineage of bilaterian animals. Repeated rounds of steroid receptor gene duplication and divergence during vertebrate evolution have produced multiple receptors with affinities for a variety of steroid hormones, including estrogens, androgens, progestogens, corticosteroids, and mineralocorticoids (197). By reconstructing ancestral receptor proteins at key nodes in the phylogeny of steroid receptors, it was possible to identify particular branches of the phylogenetic tree in which important shifts in ligand-specificity evolved (Fig. 3). Protein engineering experiments based on site-directed mutagenesis were then used to introduce historical substitutions into ancestral backgrounds to identify which combinations of substitutions were sufficient to recapitulate the historical shifts in ligand specificities. Integrating the functional tests with crystallographic analyses of ancestral proteins provided insights into the biophysical mechanisms that were responsible for the evolved ligand specificities.
Fig. 3.

Phylogeny of vertebrate steroid receptors and the evolution of hormone specificity. The most ancient steroid receptor, AncSR1, and estrogen receptors (pink) are activated by aromatized steroids, and AncSR2 and its descendants (blue) are activated by nonaromatized hormones. The black box denotes the interval in which two large-effect substitutions occurred, producing an evolutionary shift in hormone recognition.
The most ancient steroid receptor, ancestral to all family members (AncSR1) and the ancestor of all keto-steroid (progestogens, androgens, and corticosteroids) receptors (AncSR2) were synthesized and functionally characterized using reporter gene assays to determine their hormone affinity (Fig. 3). These experiments led to the discovery that the ancestor of all vertebrate steroid receptors was sensitive to many chemicals, like estrogen, that have aromatic rings, and that this specificity has been retained in the estrogen receptor clade. AncSR2 lost sensitivity to estrogens and gained specificity for nonaromatized steroids (49). Functional testing of ancestral proteins on the phylogeny bracketed the functional switch that occurred on the interval between AncSR1 and AncSR2 (Fig. 3). Structural analysis of the ancestral proteins revealed two amino acid substitutions that were predicted to alter the relative affinities for aromatic vs. nonaromatized steroids. When these two substitutions, Glu41Gln and Leu75Met, were introduced into the ancestral receptor, AncSR1, they decreased affinity for aromatic estrogens and increased affinity for nonaromatized steroids, resulting in the derived phenotype. Subsequent experiments reversing these two large-effect historical substitutions in the AncSR2 background convincingly demonstrated that they were causal determinants of the evolved ligand specificity. Additional analyses of these ancestral proteins determined the biophysical basis for this switch in hormone specificity (75).
By revealing the molecular mechanisms underlying the evolution of hormone specificity in steroid receptors, this work provided insights into why estrogen receptors are susceptible to misregulation by environmental chemicals. The minimal recognition criterion of the oldest ancestor was that ligands must have an aromatized A-ring. Since aromatization is at the end of the enzymatic synthesis pathway leading to estrogens, this specificity prevented the receptors from being activated by biosynthetic intermediates, and this rather simple criterion apparently sufficed. After gene duplication, one duplicate receptor retained the ancestral specificity, while the other evolved a promiscuous response to other steroids in this pathway. This second group further diversified with descendent lineages evolving specificity for different hormonal intermediates (195). The minimal specificity requirement for an aromatized ring, inherited from the ancestral receptor, predicts the capacity for estrogen receptors to be disrupted by exogenous xenobiotic phenolics. The ancestral receptors existed within physiological environments where they evolved to distinguish among the various endogenous and exogenous compounds to which they were exposed. Their minimal specificity meant that when novel hormones or new environmental conditions were encountered, their endocrine systems could be sensitive to misregulation.
Through recognition of the evolutionary importance of an aromatized ring to estrogen receptor specificity, we now understand why certain environmental chemicals— those that contain aromatized ring structures—can disrupt estrogen receptor signaling, leading to severe developmental, behavioral, and physiological disorders (47). By reconstructing and testing ancestral proteins, together with careful dissection of molecular mechanisms by which proteins distinguish among binding partners, this work revealed the historical causes of susceptibility to endocrine disruption in modern-day vertebrates and provided information about which types of exogenous compounds might interfere with physiological endocrine signaling (49). This case study demonstrates how knowledge of historical context and molecular evolutionary mechanisms can shed light on current physiological phenomena (49, 75, 197).
Insights Into the Genetic and Mechanistic Basis of Physiological Traits
Linkage mapping/association mapping.
Commuzie and Allison (40) introduced a review of the search for human obesity genes in 1998 with the following: “One of the greatest challenges in biomedical research today is the elucidation of the underlying genetic architecture of complex phenotypes, such as obesity. At first glance, body weight seems exceptionally simple. It can be defined precisely and measured with great accuracy and reliability. However, recent research on obesity has revealed that body weight is, in fact, a truly complex phenotype.” The same applies to almost any behavioral, physiological or morphological trait, and to this day, efforts to characterize the genetic architecture of such traits face the same challenges. Here, we briefly review mapping approaches for characterizing the genetic architecture of complex physiological traits.
Quantitative trait locus mapping experiments attempt to identify specific chromosomal regions—and, ultimately, specific genes and mutations—that contribute to variation in a trait of interest. In the broader enterprise of evolutionary quantitative genetics, the goal of the QTL program is to explain genetic variation for quantitative traits in terms of the underlying genes, the effects of segregating alleles in the context of different environments and different genetic backgrounds, and the frequencies of causal variants in natural populations (13, 126, 127, 129). QTL are typically detected and localized by measuring statistical associations between molecular markers, such as single nucleotide polymorphisms, and the trait of interest in a “mapping population” [e.g., the F2 progeny of a cross between inbred lines; (125)]. The premise of QTL mapping is that marker alleles that are associated with differences in trait values are in LD with the causal variant(s) (in this context, LD describes the nonrandom association between alleles segregating at linked sites along a recombining chromosome).
In addition to the usual factors that affect statistical power (e.g., sample size, range of the independent variable, and measurement accuracy), the power to detect QTL for a given trait is a function of the effect sizes and frequencies of causative alleles. The effect size of a given quantitative trait locus is measured as the mean difference in phenotype between alternative marker genotypes, scaled by the phenotypic variance of the trait within each genotype class. The power to detect a given quantitative trait locus is maximized when the effect size is large and frequencies of alternative marker alleles are equal (=0.50, as is the case for all segregating alleles in the F1 progeny of a cross between two inbred lines). The power to localize the chromosomal position of a given quantitative trait locus depends on the number of recombination events that have occurred between causative variants and marker alleles at linked loci. QTL can be localized to smaller chromosomal intervals when there have been many cross-overs that uncouple genotype-phenotype associations between all marker loci, except those that are tightly linked to the causative variant(s).
In contrast to traditional line-cross mapping populations, association mapping makes use of historical recombination between QTL and marker loci in natural populations (including genome-wide association studies in humans) or in reference panels of recombinant inbred lines, so overall levels of LD are typically much lower. Consequently, association mapping typically has much higher power to localize QTL, as causal variants will have been randomized against a larger number of genetic backgrounds. Moreover, association studies in natural populations simultaneously provide estimates of the frequencies of causal alleles and can, therefore, reveal whether segregating variation for the trait under study is mainly attributable to rare, deleterious alleles maintained at mutation-selection equilibrium, or intermediate-frequency alleles, which would be consistent with selective neutrality or maintenance of variation by some form of balancing selection (13, 127). For any given population, the utility of association mapping (and the number of marker loci that need to be genotyped) depends on the scale and pattern of LD. The ability to localize QTL to narrow intervals is greatest in species like Drosophila melanogaster, where LD decays over short distances [i.e., a few tens of kilobases or less (27)]. In natural populations of house mice, the rate at which LD decays with distance is similar to that in human populations and is much higher than that in laboratory strains of mice (113), suggesting that wild-derived strains of house mice are well suited to fine-scale association mapping.
High-resolution mapping in model organisms has revealed that single QTL often fractionate into multiple, closely linked QTL, some of which segregate alleles with opposing effects on the focal trait (55). Moreover, allelic effects are often conditional on sex, genetic background (i.e., epistasis, or gene × gene interaction), and/or environment (gene × environment interaction). For these reasons, it has proven extremely difficult to move beyond the delimitation of broad QTL intervals (which may contain hundreds of candidate genes for the trait of interest) to the ultimate goal of positional cloning and identification of causative mutations (i.e., quantitative trait nucleotides, or QTN). As stated by Mackay (127), “[Studies of quantitative genetic variation in Drosophila] … highlight how little we know about ‘candidate genes’ affecting quantitative traits: the majority of the genome is uncharted territory with respect to phenotypic effects of naturally segregating alleles affecting even extensively studied phenotypes in a genetic model organism.” (p. 1233). However, for model organisms like Drosophila and mice, the development of community-mapping resources (83, 119, 128, 132) and systems genetics approaches (see below) should greatly facilitate future advances.
As mentioned above (Selection experiments and experimental evolution), one way to create mapping populations that harbor high levels of variation for the trait of interest is to cross lines derived from artificial selection experiments. In such cases, intercrosses are typically carried out to the second (F2) generation, or backcrosses are performed, yielding a high level of variation in the selected trait (137). However, F2 and backcross populations yield minimal recombination events, and the identified QTL are typically located in broad intervals that may contain numerous candidate genes (44). Through random intercrossing over multiple generations beyond the F2, advanced intercross lines (AILs) accumulate recombination events and provide increased mapping resolution (narrowing confidence intervals) (43). Kelly et al. (99) used one of four replicate lines of house mice that were selectively bred for high voluntary wheel-running (discussed above) and the inbred mouse strain C57BL6/J to create an AIL suitable for mapping voluntary wheel-running traits. Animals were genotyped and phenotyped at generations 4 (G4) (n = 800 mice) and 10 (G10) (n = 473 mice) (99, 115). Analyses of the G4 yielded 32 significant and 13 suggestive QTL for running distance, duration, average speed, and maximum speed. The largest QTL effect accounted for 6.6% of the total phenotypic variation, indicating that variation in the measured trait is mainly attributable to allelic differences at many loci that have individually small effects (99). In an effort to prioritize candidate genes, Kelly et al. (97, 98) examined global gene expression profiles in brain and muscle tissue. These two tissues were chosen for the purpose of investigating the motivational aspects (brain), as well as the physical abilities (muscle) that contribute to exercise performance. Examination of QTL for transcript abundance (expression quantitative trait loci, eQTL) that colocalized with QTL for the performance trait implicated hundreds of potential candidate genes that may be functionally important in the control of voluntary wheel-running behavior. This is because colocalization of QTL for a given trait and the expression level of a particular gene suggest that cis-regulatory changes in the gene may contribute to the observed variation in trait values (cis-regulatory elements are noncoding loci that regulate the transcription of nearby genes). A backcross mapping population derived from a different HR line (compared with the AIL above) was also used to identify a causative mutation (autosomal recessive) in a gene of major effect that causes a major reduction in hindlimb muscle mass (78, 95).
These findings represent an important initial step in understanding the genetic architecture of voluntary wheel running behavior in the artificial selection experiment [see Kelly et al. (99) for a discussion of limitations]. However, this approach is limited by the variation segregating in the two parental strains used to map QTL for voluntary activity. This problem of restricted genetic sampling can be circumvented by using reference panels of recombinant inbred lines, such as the Collaborative Cross or the Diversity Outbred mouse population (119, 132). Finally, we note that it is important to consider the evolutionary “relevance” of the mapping population in addition to considerations of experimental tractability or statistical power. If the goal of a QTL study is to identify genes that can affect a particular trait in a particular species, then choices based purely on statistical power, tractability, and cost may reasonably take precedence. These considerations have generally driven development of community mapping resources. However, if the goal is to determine which loci have contributed to phenotypic changes in particular species or populations, then results obtained from “generalized” mapping populations may not be evolutionarily relevant.
Systems genetics.
Understanding the relationship between DNA sequence variation, transcriptional, protein and metabolite networks, and organismal-level phenotypes is the main challenge for the future and will add the missing biological context to genotype-phenotype associations.
— Mackay (127, p. 1235).
. . .[N]atural genetic variation has the character of an ideal multifactorial perturbation, providing a natural experimental design that is helping researchers to uncover the mechanistic basis of the map that connects genotype to phenotype. As the molecular catalogues of genomics yield to the integrated models of systems biology, natural genetic variation will have an increasingly central role.
— Rockman (158, p. 743)
Mechanistic insights into trait formation require a consideration of intermediate steps in the chain of causation that links genotype to phenotype. The goal of the systems genetics approach (10, 39, 129, 171) is to achieve a mechanistic understanding of the mapping function that relates genotype to phenotype—that is, an understanding of how genetic variation is transduced through the transcriptome, proteome, and metabolome to generate phenotypes. Intermediate molecular phenotypes (“endophenotypes”), such as transcript abundance, are also quantitative traits with genetic and environmental components of variance. The general expectation is that additive genetic variation for transcript abundance and protein activity can be related to variation in protein and metabolite abundance, which, in turn, can be related to organismal phenotypes (39, 116), potentially at many levels of biological organization (Fig. 1).
Using genome-wide gene expression profiles for specific tissues, researchers have found that it is possible to identify trait-specific transcriptional modules of coregulated genes by measuring associations between particular physiological traits and levels of transcript abundance. For example, studies of within-population transcriptomic variation in Drosophila melanogaster have revealed that ∼60% of variable transcripts cluster into two discrete modules; levels of transcript abundance are positively correlated within modules but negatively correlated between them (10). When flies reared under standard growth conditions were subjected to environmental changes, the modular organization of the transcriptome was fragmented due to coregulated changes in sets of environmentally responsive genes comprising ∼15% of the transcriptome (10, 222). Regressions of organismal phenotypes (e.g., resistance to starvation stress, time to recover from a chill-induced coma) on transcript abundance implicated numerous candidate genes that formed statistically defined transcriptional modules (10). Because transcriptional modules are typically enriched for functionally related genes whose products interact in the same pathways, identifying discrete modules of intercorrelated genes provides a means of constructing genetic interaction networks, specifically, networks of causal effects that link genotype to phenotype (158).
A similar approach has been applied to an analysis of evolved population differences in physiological performance capacities between high- and low-altitude deer mice. Relative to deer mice that are native to low-altitude environments, high-altitude deer mice have evolved an enhanced thermogenic capacity under hypoxia (35–37), a complex organismal performance trait that is subject to strong directional selection in the wild (79). The enhanced thermogenic performance of high-altitude mice is associated with upregulation of five main transcriptional modules that influence several hierarchical steps in the O2 transport cascade, including tissue O2 diffusion (angiogenesis) and tissue O2 utilization (metabolic fuel use and cellular oxidative capacity) (35, 37). Additional transcriptional modules are associated with tissue-level traits, such as capillary density and the relative proportion of aerobic and glycolytic fibers in skeletal muscle, which help sustain high rates of aerobic metabolism under hypoxic conditions (122, 169) (Fig. 4). In such studies, transcripts that are significantly associated with a particular phenotype are candidate genes for that phenotype (10, 39, 142) and can, therefore, be targeted for additional experiments to test for causal effects.
Fig. 4.

Transcriptional modules of coregulated genes are associated with muscle phenotype in deer mice. A: histological analyses revealed that the gastrocnemius muscle of high-altitude deer mice has a higher capillary density (top) and a higher areal density of oxidative fibers (bottom) relative to that of low-altitude mice. B: Left: transcripts that are differentially expressed between high- and low-altitude populations of deer mice (n = 68 transcripts) cluster into two discrete modules (P1 and P2). Right: transcripts that are differentially expressed between mice in different rearing environments (wild-caught mice sampled at their native elevations vs. F1 lab-reared mice, n = 658 transcripts) cluster into five modules (T1–T5). C: Left: changes in expression of the P2 module (50 transcripts) are significantly associated with evolved (nonplastic) differences in muscle capillary density between high- and low-altitude deer mice (r2 = 0.46, P = 0.001). Right: changes in expression of the T5 module (224 transcripts) are associated with environmentally induced (plastic) changes in the density of oxidative fibers in the gastrocnemius (r2 = 0.30, P = 0.007). For each transcriptional module, an index of overall expression is provided by PC1 scores, where PC1 is the first axis of a principal components analysis on the correlation matrix of transcript abundance for all genes in a given module. In both panels, data points for high- and low-altitude mice are denoted as solid and open symbols, respectively. [Adapted with permission from Fig. 1 of Ref. 169: Adaptive modifications of muscle phenotype in high-altitude deer mice are associated with evolved changes in gene regulation. Scott GR, et al., Mol Biol Evol doi:10.1093/molbev/msv076].
Population genomics.
In principle, genome-wide scans of DNA polymorphism can be used to identify candidate loci for adaptation based on statistical signatures of positive selection. This population genomics approach is premised on the idea that the locus-specific effects of positive selection can be detected against the genome-wide backdrop of neutral variation. Consider a genome-wide survey of DNA polymorphism in samples of individuals from a pair of populations inhabiting distinct, spatially separated environments—for example, high- and low-altitude environments. If a given trait is subject to opposing selection in the high- and low-altitude populations, then the underlying loci are expected to experience an increase in the between-population variance in allele frequency. In principle, it is possible to identify chromosomal regions that harbor such loci by exploiting theoretical predictions about the effects of spatially varying selection on patterns of DNA polymorphism at linked neutral sites. Specifically, spatially varying selection is expected to produce a peak in the between-population component of nucleotide diversity that is centered on the selected polymorphism(s), and the correlation in levels of differentiation between the selected site and linked neutral markers is expected to dissipate at a rate proportional to the genetic map distance between them (Fig. 5) (33).
Fig. 5.
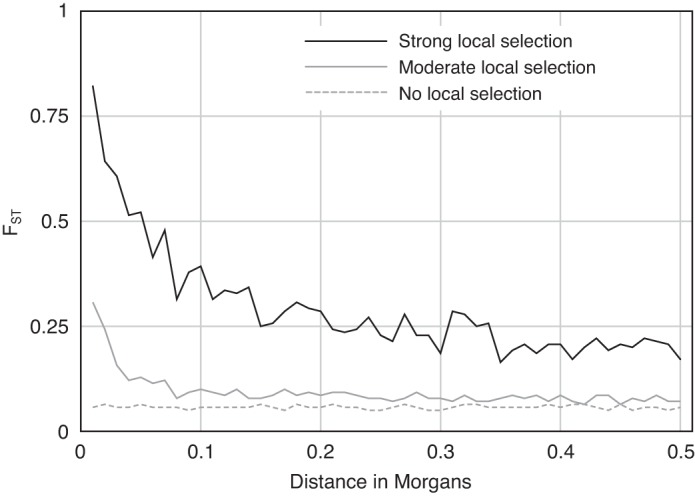
Simulation results showing the effects of spatially varying selection on levels of differentiation at linked sites in a pair of populations at migration selection equilibrium (figure based on data from Ref. 33). The y-axis shows levels of differentiation in site-specific allele frequencies (as measured by FST), and the x-axis shows a measure of genetic map distance (as measured by Morgans, the distance over which the expected average number of cross-over events per generation is 0.1). Local selection was modeled as directional selection that acts in opposite directions in two populations that exchange migrants. Heterozygotes and homozygotes at the selected locus were assigned fitnesses of 1 − (s/2) and 1 − s, respectively (where s is the selection coefficient); s = 0.5 under “strong” selection and s = 0.1 under “moderate” selection. In the absence of local selection (s = 0), FST at linked markers is not correlated with map distance from the selected polymorphism. In the presence of local selection, levels of differentiation are especially high at sites that are closely linked to the selected site, and the rate of decay is inversely proportional to the strength of local selection. In comparisons between populations that are locally adapted to contrasting environments (e.g., high and low altitudes), loci that have contributed to adaptive changes in phenotype would be expected to exhibit especially high FST values relative to the genome-wide average.
In cases in which a given allele has been rapidly driven up to high frequency by positive selection, the surrounding gene region will typically be characterized by a transient increase in LD. Newly arisen mutations will typically exhibit high LD with neighboring nucleotide polymorphisms since insufficient time has elapsed for recombination to randomize associations between alleles at linked sites. By contrast, the neutral expectation is that high-frequency alleles will typically exhibit low LD with neighboring polymorphisms, since the time needed for them to drift to high frequency will generally be sufficient to randomize associations between alleles at linked sites (100). For this reason, high-frequency alleles that exhibit high LD present a combination of features that are not expected under neutrality (162, 163). These features can provide a basis for identifying positively selected loci in genomic survey data, although selection on new mutations and selection on preexisting variants (standing genetic variation) are expected to generate somewhat different patterns of LD and nucleotide variation (41, 81, 85, 139, 145, 146, 152, 193).
Genome scans to detect the effects of positive selection involve two main steps. The first step is to compute a given test statistic across the genome (for individual polymorphisms or chromosomal intervals that harbor multiple polymorphisms). The second step is to calculate whether locus-specific values of the test statistic exceed the critical value for rejecting a neutral model. In practice, locus-specific P values are based on an empirical distribution of the test statistic or a null distribution of the statistic under an assumed probablistic model. Regardless of the choice of test statistic, and regardless of whether parametric or nonparametric methods are used to identify outlier loci, it is important to appreciate that the effects of selection on patterns of variation at or near causative loci depend strongly on the genetic architecture of the selected trait, the intensity of selection on the trait, and the genetic structure of the population under consideration (34, 94, 112, 114, 138, 165, 178, 185). In particular, theoretical and simulation studies have demonstrated that the ability to detect the effects of selection at individual loci is strongly affected by the dominance coefficient of the causative allele (194) and the initial frequency of the causative allele at the onset of selection (81, 85, 139, 145, 146, 152, 193).
In principle, the population genomics approach can be used as a means of “outlier detection” to nominate candidate loci for adaptation, or it can be used to assess evidence for selection on previously identified candidate loci by assessing whether such loci emerge as outliers against a backdrop of genome-wide variation. Both approaches have been used successfully to identify loci involved in evolutionary adaptation to particular environmental conditions. In humans, for example, genome-wide surveys of DNA polymorphism in Tibetan highlanders revealed evidence for strong and recent positive selection on several genes that can be described as upstream regulators of the hypoxia inducible-factor (HIF) oxygen signaling pathway (18, 24, 144, 172, 213, 216). The HIF family of transcription factors plays a key role in regulating oxygen homeostasis by coordinating the transcriptional response to hypoxia. One of the HIF genes that exhibited an especially clear signal of positive selection in the Tibetan population was the EPAS1 gene (endothelial PAS domain protein 1) (Fig. 6), also known as HIF2α, which encodes the oxygen-sensitive α subunit of the HIF-2 transcription factor. The product of EPAS1 is known to play an especially important role in regulating the erythropoietic response to hypoxia (153). It is especially remarkable that evidence for positive selection on EPAS1 in Tibetan highlanders was obtained by several different research groups who used different samples and who employed somewhat different analytical approaches (18, 24, 144, 172, 213, 216). Another HIF-regulatory gene that exhibited strong evidence for selection in both Tibetan and Andean populations was EGLN1 (Egl Nine homolog 1) (24, 144, 172, 212, 213, 216), which encodes the prolyl hydroxylase isozyme (PHD2) that is responsible for hydroxylating the α subunit of the HIF1 transcription factor. Results of these studies provide proof-of-principle that the genome scan approach can successfully identify targets of recent positive selection. The integration of such analyses with association studies and manipulative experiments can provide additional insights into possible phenotypic targets of selection (120).
Fig. 6.

A genome-wide scan of allelic differentiation between population samples of Tibetans (resident at 3,200-3,500 m in Yunnan Province, China) and Han Chinese. The vertical axis of the graph shows the negative log of site-specific P values for allele frequency differences between the Tibetan and Han Chinese population samples (low P values denote allele frequency differences that are too large to explain by genetic drift). The horizontal axis of the graph shows the genomic positions of each assayed nucleotide site, arranged by chromosome number. The red line indicates the threshold for genome-wide statistical significance (P = 5 × 10−7). Values are shown after correction for background population stratification using an intragenomic control. Several noncoding variants flanking the EPAS1 gene are highly significant outliers. [Reprinted from (18) with permission: Natural selection on EPAS1 (HIF2α) associated with low hemoglobin concentration in Tibetan highlanders. Beall CM, et al., Proc Natl Acad Sci USA 107: 11,459-11,464, 2010].
One reason for the popularity of the population genomic approach is that, unlike linkage or association mapping approaches that use phenotypic variation as a starting point, genome scans for selected loci are not biased by preconceived ideas about the specific traits that contribute to adaptive evolution. Thus, when coupled with functional studies of the candidate genes for adaptation that are ultimately identified, the genome scan approach holds the promise of identifying genetic mechanisms of adaptation that were previously unanticipated (178). However, there are many limitations of this approach. One major concern is that the identified candidate loci for positive selection represent a highly biased subset of loci that have actually contributed to past adaptation, so the implicated traits may have an entirely unrepresentative genetic architecture. In particular, loci that have contributed to adaptive changes in highly polygenic traits may be systematically under-represented in genomic maps of positive selection. This is because changes in the mean value of a polygenic trait can be produced by modest shifts in allele frequencies at large numbers of loci that have individually small effects, so the effects of selection at any single locus may not be readily detectable (23, 149, 150, 185). This is an important consideration since many ecologically important (fitness-related) traits typically have a highly polygenic basis (157).
Another important issue concerns the mapping function that relates genotype to phenotype. It is not realistic to expect that the identification of positively selected loci will typically lead to the clear and unambiguous identification of selected phenotypes. For example, the noncoding EPAS1 variants that are present at an elevated frequency among Tibetan highlanders are associated with a reduced hemoglobin concentration (18, 172, 216), which appears to stem from a blunted erythropoietic response to hypoxia (15–17, 19, 147, 210). There are good reasons to think that this blunted response may be physiologically beneficial under conditions of chronic hypoxia at high altitude (147, 148, 177, 184, 199), but that does not mean that hemoglobin concentration was necessarily the trait under direct selection. The reduced hemoglobin concentration may represent a second-order consequence of upstream modifications in O2 chemosensitivity. For example, allelic variation at EPAS1 could affect hemoglobin concentration indirectly through changes in the hypoxic ventilatory response, which modulates erythropoietin production through changes in arterial O2 tension. In such cases, a combination of experimental and analytical methods can be used to determine whether the relationship between genotype and phenotype is causal or consequential (39, 158).
Results of genome scans are useful for generating hypotheses about candidate genes that can be followed up with functional experiments (e.g., 120). All too often, however, results of such analyses are uncritically interpreted in purely adaptive terms without any form of experimental validation. In particular, gene-ontology enrichment analyses of outlier loci often provide a basis for concocting speculative, hand-waving stories about mechanisms of adaptation. The same practice is common in genomic surveys of sequence divergence using codon-substitution models (see Molecular evolution: analysis of interspecific divergence at specific loci). Pavlidis et al. (143) confirmed the suspicions of many skeptics by conducting neutral simulations that demonstrated, firstly, that genome scans for putative targets of positive selection can have soberingly high false-positive rates (see also Ref. 12) and, secondly, that gene ontology annotations of false-positive outliers can be used to construct seemingly plausible narratives about genetic adaptation that are empirically unsubstantiated.
Experimental analyses of physiological traits can play an important role as an adjunct to population genomic analyses by validating selection-nominated candidate genes. However, such experiments have a far more general role to play in integrated “phenotype-first” studies that reveal how variation in regulatory networks gives rise to ecologically relevant trait variation under natural conditions. As described above (Systems genetics), functional genomic analyses that are integrated with experimental analyses of physiological variation can provide the foundation and starting point for investigating the mechanistic and genetic basis of natural variation in whole animal performance (35, 37, 42, 169, 170, 208, 209).
Role of Physiology in the Functional Synthesis of Evolutionary Biology
Dean and Thornton (46) advocated an integration of molecular evolution research with mechanistic studies of genes and gene products to achieve a “functional synthesis of evolutionary biology”. In addition to enriching our basic understanding of the biology of phenotypic evolution, insights into the mechanistic basis of evolutionary changes in molecular function can provide insights into features of causative mutations (dominance coefficients, epistatic interactions, and pleiotropic effects) that influence fixation probabilities and can, therefore, help explain why certain types of mutations make disproportionate contributions to evolutionary change (176, 186). However, insights into mechanism are equally valuable at higher levels of biological organization, suggesting a key role for mechanistic studies of phenotypic evolution that fall squarely within the purview of organismal physiology. For example, an understanding of physiological mechanisms can be used to predict and interpret the causes of genetic correlations between traits (2, 4, 57, 59). As stated by Garland and Carter (64): “. . . an understanding of physiological mechanisms can help in determining whether a particular pattern of phenotypic variation or covariation (e.g., an allometric relationship) represents what could possibly exist or just what selection has allowed.” (p. 586).
Mechanistic studies at different hierarchical levels of biological organization are relevant to different links in the “adaptive recursion” mentioned above. Whereas experimental approaches in molecular biology and biochemistry are needed to establish causal connections between genotype and proximal (biochemical) phenotype, genetically based changes in such phenotypes are visible to selection only to the extent that they impinge on fitness-related aspects of whole organism performance. Thus, experimental studies of whole animal physiology are needed to establish the causal links between proximal phenotypes (subordinate traits such as enzyme activity, and pathway flux) and whole animal physiological performance, and as described above (Physiology, performance, behavior, and fitness: measuring selection in the wild), when experiments on performance are integrated with measures of survivorship and/or reproductive success of free-ranging animals, the measurement of selection gradients can be used to establish the ensuing link between performance and fitness.
The integration of molecular biology, biochemistry, and evolutionary biology has greatly enriched our understanding of the mechanism and process in the evolution of biological macromolecules (46, 77). The marriage of evolutionary biology and organismal physiology has an equally important role to play in the “functional synthesis,” broadly defined.
GRANTS
The authors acknowledge grant support from the National Science Foundation (NSF Grant IOS-1121273 to T. Garland and Grant IOS-1354390 to J. F. Storz) and the National Institutes of Health (Grant GM-104397 to J. T. Bridgham and Grant HL-087216 to J. F. Storz).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: J.F.S., J.T.B., and T.G. prepared figures; J.F.S., J.T.B., S.A.K., and T.G. drafted manuscript; J.F.S., J.T.B., S.A.K., and T.G. edited and revised manuscript; J.F.S., J.T.B., S.A.K., and T.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank G. R. Scott and three anonymous reviewers for helpful suggestions. We thank C. M. Beall, Z. A. Cheviron, and B. W. M. Wone for providing figures.
REFERENCES
- 1.Arnold SJ. Behavior, energy, and fitness. Am Zool 28: 815–827, 1988. [Google Scholar]
- 2.Arnold SJ. Behavioral variation in natural populations. I. Phenotypic, genetic, and environmental correlations between chemoreceptive responses to prey in the garter snake, Thamnophis elegans. Evolution 35: 489–509, 1981. [DOI] [PubMed] [Google Scholar]
- 3.Arnold SJ. Constraints on phenotypic evolution. Am Nat 140: S85–S107, 1992. [DOI] [PubMed] [Google Scholar]
- 4.Arnold SJ. Genetic correlation and the evolution of physiology. In: New Directions in Ecological Physiology, edited by Feder ME, Bennett AF, Burggren WW, and Huey RB, 1987, p. 189–215. [Google Scholar]
- 5.Arnold SJ. Morphology, performance, and fitness. Am Zool 23: 347–361, 1983. [Google Scholar]
- 6.Arnold SJ. Multivariate inheritance and evolution: A review of concepts. In: Quantitative Genetic Studies of Behavioral Evolution, edited by Boake CRB. Chicago, IL: University of Chicago, 1994, p. 17–48. [Google Scholar]
- 7.Arnold SJ. Phenotypic evolution: the ongoing synthesis. Am Nat 183: 729–746, 2014. [DOI] [PubMed] [Google Scholar]
- 8.Arnold SJ, Buerger R, Hohenlohe PA, Ajie BC, Jones AG. Understanding the evolution and stability of the G-matrix. Evolution 62: 2451–2461, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnold SJ, Pfrender ME, Jones AG. The adaptive landscape as a conceptual bridge between micro- and macroevolution. Genetica 112: 9–32, 2001. [PubMed] [Google Scholar]
- 10.Ayroles JF, Carbone MA, Stone EA, Jordan KW, Lyman RF, Magwire MM, Rollmann SM, Duncan LH, Lawrence F, Anholt RRH, Mackay TFC. Systems genetics of complex traits in Drosophila melanogaster. Nat Genet 41: 299–307, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Badyaev AV. Maternal effects as generators of evolutionary change: A reassessment. In: Year in Evolutionary Biology 2008, edited by Schlichting CD and Mousseau TA. New York: Wiley-Blackwell, 2008, p. 151–161. [DOI] [PubMed] [Google Scholar]
- 12.Bank C, Ewing GB, Ferrer-Admettla A, Foll M, Jensen JD. Thinking too positive? Revisiting current methods of population genetic selection inference. Trends Genet 30: 540–546, 2014. [DOI] [PubMed] [Google Scholar]
- 13.Barton NH, Keightley PD. Understanding quantitative genetic variation. Nat Rev Genet 3: 11–21, 2002. [DOI] [PubMed] [Google Scholar]
- 14.Barton NH, Turelli M. Evolutionary quantitative genetics—How little do we know? Ann Rev Genet 23: 337–370, 1989. [DOI] [PubMed] [Google Scholar]
- 15.Beall CM. Two routes to functional adaptation: Tibetan and Andean high-altitude natives. Proc Natl Acad Sci USA 104: 8655–8660, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beall CM, Brittenham GM, Macuaga F, Barragan M. Variation in hemoglobin concentration among samples of high-altitude natives in the Andes and the Himalayas. Am J Hum Biol 2: 639–651, 1990. [DOI] [PubMed] [Google Scholar]
- 17.Beall CM, Brittenham GM, Strohl KP, Blangero J, Williams-Blangero S, Goldstein MC, Decker MJ, Vargas E, Villena M, Soria R, Alarcon AM, Gonzales C. Hemoglobin concentration of high-altitude Tibetans and Bolivian Aymara. Am J Phys Anthropol 106: 385–400, 1998. [DOI] [PubMed] [Google Scholar]
- 18.Beall CM, Cavalleri GL, Deng L, Elston RC, Gao Y, Knight J, Li C, Li JC, Liang Y, McCormack M, Montgomery HE, Pan H, Robbins PA, Shianna KV, Tam SC, Tsering N, Veeramah KR, Wang W, Wangdui P, Weale ME, Xu Y, Xu Z, Yang L, Zaman MJ, Zeng C, Zhang L, Zhang X, Zhaxi P, Zheng YT. Natural selection on EPAS1 (HIF2α) associated with low hemoglobin concentration in Tibetan highlanders. Proc Natl Acad Sci USA 107: 11,459–11,464, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beall CM, Reichsman AB. Hemoglobin levels in a Himalayan high-altitude population. Am J Phys Anthropol 63: 301–306, 1984. [DOI] [PubMed] [Google Scholar]
- 20.Belke TW, Garland T. A brief opportunity to run does not function as a reinforcer for mice selected for high daily wheelrunning rates. J Exp Anal Behav 88: 199–213, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bennett AF. Experimental evolution and the Krogh principle: generating biological novelty for functional and genetic analyses. Physiol Biochem Zool 76: 1–11, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Bennett AF, Huey RB. Studying the evolution of physiological performance. Oxford Surv Evol Biol 7: 251–284, 1990. [Google Scholar]
- 23.Berg JJ, Coop G. A population genetic signal of polygenic adaptation. PLoS Genet 10: 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bigham AW, Mao X, Mei R, Brutsaert T, Wilson MJ, Julian CG, Parra EJ, Akey JM, Moore LG, Shriver MD. Identifying positive selection candidate loci for high-altitude adaptation in Andean populations. Human Genomics 4: 79–90, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Briscoe AD, Bybee SM, Bernard GD, Yuan F, Sison-Mangus MP, Reed RD, Warren AD, Llorente-Bousquets J, Chiao CC. Positive selection of a duplicated UV-sensitive visual pigment coincides with wing pigment evolution in Heliconius butterflies. Proc Natl Acad Sci USA 107: 3628–3633, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burggren WW, Crews D. Epigenetics in comparative biology: Why we should pay attention. Integr Comp Biol 54: 7–20, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carbone MA, Jordan KW, Lyman RF, Harbison ST, Leips J, Morgan TJ, DeLuca M, Awadalla P, Mackay TFC. Phenotypic variation and natural selection at Catsup, a pleiotropic quantitative trait gene in Drosophila. Curr Biol 16: 912–919, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Careau V, Garland T Jr. Performance, personality, and energetics: correlation, causation, and mechanism. Physiol Biochem Zool 85: 543–571, 2012. [DOI] [PubMed] [Google Scholar]
- 29.Careau V, Wolak ME, Carter PA, Garland T Jr. Limits to behavioral evolution: The quantitative genetics of a complex trait under directional selection. Evolution 67: 3102–3119, 2013. [DOI] [PubMed] [Google Scholar]
- 30.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 10: 295–304, 2009. [DOI] [PubMed] [Google Scholar]
- 31.Chappell MA, Hayes JP, Snyder LRG. Hemoglobin polymorphisms in deer mice (Peromyscus maniculatus), physiology of β-globin variants and α-globin recombinants. Evolution 42: 681–688, 1988. [DOI] [PubMed] [Google Scholar]
- 32.Chappell MA, Snyder LRG. Biochemical and physiological correlates of deer mouse α-chain hemoglobin polymorphisms. Proc Natl Acad Sci USA 81: 5484–5488, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Charlesworth B, Nordborg M, Charlesworth D. The effects of local selection, balanced polymorphism and background selection on equilibrium patterns of genetic diversity in subdivided populations. Genet Res 70: 155–174, 1997. [DOI] [PubMed] [Google Scholar]
- 34.Chevin LM, Hospital F. Selective sweep at a quantitative trait locus in the presence of background genetic variation. Genetics 180: 1645–1660, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheviron ZA, Bachman GC, Connaty AD, McClelland GB, Storz JF. Regulatory changes contribute to the adaptive enhancement of thermogenic capacity in high-altitude deer mice. Proc Natl Acad Sci USA 109: 8635–8640, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheviron ZA, Bachman GC, Storz JF. Contributions of phenotypic plasticity to differences in thermogenic performance between highland and lowland deer mice. J Exp Biol 216: 1160–1166, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheviron ZA, Connaty AD, McClelland GB, Storz JF. Functional genomics of adaptation to hypoxic cold-stress in high-altitude deer mice: transcriptomic plasticity and thermogenic performance. Evolution 68: 48–62, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheviron ZA, Natarajan C, Projecto-Garcia J, Eddy DK, Jones J, Carling MD, Witt CC, Moriyama H, Weber RE, Fago A, Storz JF. Integrating evolutionary and functional tests of adaptive hypotheses: a case study of altitudinal differentiation in hemoglobin function in an Andean sparrow, Zonotrichia capensis. Mol Biol Evol 31: 2948–2962, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Civelek M, Lusis AJ. Systems genetics approaches to understand complex traits. Nat Rev Genet 15: 34–48, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Comuzzie AG, Allison DB. The search for human obesity genes. Science 280: 1374–1377, 1998. [DOI] [PubMed] [Google Scholar]
- 41.Coop G, Ralph P. Patterns of neutral diversity under general models of selective sweeps. Genetics 192: 205–U438, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dalziel AC, Rogers SM, Schulte PM. Linking genotypes to phenotypes and fitness: how mechanistic biology can inform molecular ecology. Mol Ecol 18: 4997–5017, 2009. [DOI] [PubMed] [Google Scholar]
- 43.Darvasi A, Soller M. Advanced intercross lines, an experimental population for fine genetic mapping. Genetics 141: 1199–1207, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Darvasi A, Weinreb A, Minke V, Weller JI, Soller M. Detecting marker-QTL linkage and estimating QTL gene effect and map location using a saturated genetic map. Genetics 134: 943–951, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Day T, Bonduriansky R. A unified approach to the evolutionary consequences of genetic and nongenetic inheritance. Am Nat 178: E18–E36, 2011. [DOI] [PubMed] [Google Scholar]
- 46.Dean AM, Thornton JW. Mechanistic approaches to the study of evolution: the functional synthesis. Nat Rev Genet 8: 675–688, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dickerson SM, Gore AC. Estrogenic environmental endocrine-disrupting chemical effects on reproductive neuroendocrine function and dysfunction across the life cycle. Rev Endocr Metab Disord 8: 143–159, 2007. [DOI] [PubMed] [Google Scholar]
- 48.Eanes WF. Analysis of selection on enzyme polymorphisms. Ann Rev Ecol Systemat 30: 301–326, 1999. [Google Scholar]
- 49.Eick GN, Colucci JK, Harms MJ, Ortlund EA, Thornton JW. Evolution of minimal specificity and promiscuity in steroid hormone receptors. PLoS Genet 8: 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Estes S, Arnold SJ. Resolving the paradox of stasis: models with stabilizing selection explain evolutionary divergence on all timescales. Am Nat 169: 227–244, 2007. [DOI] [PubMed] [Google Scholar]
- 51.Falconer DS, Mackay TFC. Introduction to Quantitative Genetics, 4th ed. Harlow, UK: Longman Science and Technology, 1996. [Google Scholar]
- 52.Feder ME, Bennett AF, Huey RB. Evolutionary physiology. Ann Rev Ecol Systemat 31: 315–341, 2000. [Google Scholar]
- 53.Feder ME, Garland T Jr, Marden JH, Zera AJ. Locomotion in response to shifting climate zones: not so fast. Ann Rev Physiol 72: 167–190, 2010. [DOI] [PubMed] [Google Scholar]
- 54.Feder ME, Watt WB. Functional biology of adaptation. In: Genes in Ecology, edited by Berry RJ, Crawford TJ, and Hewitt GM. Oxford, UK: Blackwell Scientific Publications, 1992. [Google Scholar]
- 55.Flint J, Mackay TFC. Genetic architecture of quantitative traits in mice, flies, and humans. Genome Res 19: 723–733, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Furrow RE, Feldman MW. Genetic variation and the evolution of epigenetic regulation. Evolution 68: 673–683, 2014. [DOI] [PubMed] [Google Scholar]
- 57.Garlanda T., Jr Genetic basis of activity metabolism. 1. Inheritance of speed, stamina, and antipredator displays in the garter snake. Thamnophis sirtalis Evolution 42: 335–350, 1988. [DOI] [PubMed] [Google Scholar]
- 58.Garland T., Jr Phylogenetic comparison and artificial selection. Two approaches in evolutionary physiology. In: Hypoxia: From Genes to the Bedside, edited by Roach RC, Wagner PD, and Hackett PH. New York: Kluwer Academic, 2001, p. 107–132. [PubMed] [Google Scholar]
- 59.Garland T., Jr Quantitative genetics of locomotor behavior and physiology in a garter snake. In: Quantitative Genetic Studies of Behavioral Evolution, edited by Boake CRB, 1994, p. 251–277. [Google Scholar]
- 60.Garland T., Jr Selection experiments: an under-utilized tool in biomechanics and organismal biology. In: Vertebrate Biomechanics and Evolution, edited by Bels VL, Gasc JP, and Casinos A. New York: Garland Science, 2003, p. 23–56. [Google Scholar]
- 61.Garland T., Jr Trade-offs. In: Curr Biol 24: R60–R61, 2014. [DOI] [PubMed] [Google Scholar]
- 62.Garland T Jr, and Adolph SC. Physiological differentiation of vertebrate populations. Ann Rev Ecol Systemat 22: 193–228, 1991. [Google Scholar]
- 63.Garland T Jr, Bennett AF, Rezende EL. Phylogenetic approaches in comparative physiology. J Exp Biol 208: 3015–3035, 2005. [DOI] [PubMed] [Google Scholar]
- 64.Garland T Jr, Carter PA. Evolutionary physiology. Ann Rev Physiol 56: 579–621, 1994. [DOI] [PubMed] [Google Scholar]
- 65.Garland T Jr, Freeman PW. Selective breeding for high endurance running increases hindlimb symmetry. Evolution 59: 1851–1854, 2005. [PubMed] [Google Scholar]
- 66.Garland T Jr, Kelly SA. Phenotypic plasticity and experimental evolution. J Exp Biol 209: 2344–2361, 2006. [DOI] [PubMed] [Google Scholar]
- 67.Garland T Jr, Kelly SA, Malisch JL, Kolb EM, Hannon RM, Keeney BK, Van Cleave SL, Middleton KM. How to run far: multiple solutions and sex-specific responses to selective breeding for high voluntary activity levels. Proc R Soc B Biol Sci 278: 574–581, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garland T Jr, Losos JB. Ecological morphology of locomotor performance in squamate reptiles. In: Ecological Morphology: Integrative Organismal Biology, edited by Wainwright PC, and Reilly SM. Chicago, IL: University of Chicago, 1994, p. 240–302. [Google Scholar]
- 69.Garland T Jr, Rose M. Experimental Evolution: Concepts, Methods and Applications of Selection Experiments. Berkeley, CA: University of California, 2009. [Google Scholar]
- 70.Garland T Jr, Schutz H, Chappell MA, Keeney BK, Meek TH, Copes LE, Acosta W, Drenowatz C, Maciel RC, van Dijk G, Kotz CM, Eisenmann JC. The biological control of voluntary exercise, spontaneous physical activity and daily energy expenditure in relation to obesity: human and rodent perspectives. J Exp Biol 214: 206–229, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gebczynski AK, Konarzewski M. Effects of oxygen availability on maximum aerobic performance in Mus musculus selected for basal metabolic rate or aerobic capacity. J Exp Biol 214: 1714–1720, 2011. [DOI] [PubMed] [Google Scholar]
- 72.Gebczynski AK, Konarzewski M. Locomotor activity of mice divergently selected for basal metabolic rate: a test of hypotheses on the evolution of endothermy. J Evol Biol 22: 1212–1220, 2009. [DOI] [PubMed] [Google Scholar]
- 73.Gebczynski AK, Konarzewski M. Metabolic correlates of selection on aerobic capacity in laboratory mice: a test of the model for the evolution of endothermy. J Exp Biol 212: 2872–2878, 2009. [DOI] [PubMed] [Google Scholar]
- 74.Grispo MT, Natarajan C, Projecto-Garcia J, Moriyama H, Weber RE, Storz JF. Gene duplication and the evolution of hemoglobin isoform differentiation in birds. J Biol Chem 287: 12, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harms MJ, Eick GN, Goswami D, Colucci JK, Griffin PR, Ortlund EA, Thornton JW. Biophysical mechanisms for large-effect mutations in the evolution of steroid hormone receptors. Proc Natl Acad Sci USA 110: 11,475–11,480, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Harms MJ, Thornton JW. Analyzing protein structure and function using ancestral gene reconstruction. Curr Opin Struct Biol 20: 360–366, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harms MJ, Thornton JW. Evolutionary biochemistry: revealing the historical and physical causes of protein properties. Nat Rev Genet 14: 559–571, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hartmann J, Garland T Jr, Hannon RM, Kelly SA, Munoz G, Pomp D. Fine mapping of “mini-muscle,” a recessive mutation causing reduced hindlimb muscle mass in mice. J Hered 99: 679–687, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hayes JP, O'Connor CS. Natural selection on thermogenic capacity of high-altitude deer mice. Evolution 53: 1280–1287, 1999. [DOI] [PubMed] [Google Scholar]
- 80.Herman JJ, Spencer HG, Donohue K, Sultan SE. How stable ‘should’ epigenetic modifications be? Insights from adaptive plasticity and bet hedging. Evolution 68: 632–643, 2014. [DOI] [PubMed] [Google Scholar]
- 81.Hermisson J, Pennings PS. Soft sweeps: molecular population genetics of adaptation from standing genetic variation. Genetics 169: 2335–2352, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hohenlohe PA, Arnold SJ. MIPoD: a hypothesis-testing framework for microevolutionary inference from patterns of divergence. Am Nat 171: 366–385, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huang W, Massouras A, Inoue Y, Peiffer J, Ramia M, Tarone AM, Turlapati L, Zichner T, Zhu D, Lyman RF, Magwire MM, Blankenburg K, Carbone MA, Chang K, Ellis LL, Fernandez S, Han Y, Highnam G, Hjelmen CE, Jack JR, Javaid M, Jayaseelan J, Kalra D, Lee S, Lewis L, Munidasa M, Ongeri F, Patel S, Perales L, Perez A, Pu L, Rollmann SM, Ruth R, Saada N, Warner C, Williams A, Wu YQ, Yamamoto A, Zhang Y, Zhu Y, Anholt RRH, Korbel JO, Mittelman D, Muzny DM, Gibbs RA, Barbadilla A, Johnston JS, Stone EA, Richards S, Deplancke B, Mackay TFC. Natural variation in genome architecture among 205 Drosophila melanogaster Genetic Reference Panel lines. Genome Res 24: 1193–1208, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Husak JF. Measuring selection in the wild and manipulating phenotypes (in terrestrial non-human vertebrates). Compr Physiol In press. [DOI] [PubMed] [Google Scholar]
- 85.Innan H, Kim Y. Pattern of polymorphism after strong artificial selection in a domestication event. Proc Natl Acad Sci USA 101: 10,667–10,672, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Irschick DJ, Reznick D. Field experiments, introductions, and experimental evolution: a review and practical guide. In: Experimental Evolution: Concepts, Methods, and Applications of Selection Experiments, edited by Garland T Jr., Rose MR. Berkeley, CA: University of California, 2009, p. 173–193. [Google Scholar]
- 87.Jablonka E. Epigenetic variations in heredity and evolution. Clin Pharmacol Ther 92: 683–688, 2012. [DOI] [PubMed] [Google Scholar]
- 88.Jamniczky HA, Boughner JC, Rolian C, Gonzalez PN, Powell CD, Schmidt EJ, Parsons TE, Bookstein FL, Hallgrimsson B. Rediscovering Waddington in the post-genomic age. Bioessays 32: 553–558, 2010. [DOI] [PubMed] [Google Scholar]
- 89.Jayne BC, Bennett AF. Selection on locomotor performance capacity in a natural population of garter snakes. Evolution 44: 1204–1229, 1990. [DOI] [PubMed] [Google Scholar]
- 90.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet 8: 253–262, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jones AG, Arnold SJ, Borger R. Stability of the G-matrix in a population experiencing pleiotropic mutation, stabilizing selection, and genetic drift. Evolution 57: 1747–1760, 2003. [DOI] [PubMed] [Google Scholar]
- 92.Jones AG, Buerger R, Arnold SJ. Epistasis and natural selection shape the mutational architecture of complex traits. Nat Commun 5: 3709, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kawecki TJ, Lenski RE, Ebert D, Hollis B, Olivieri I, Whitlock MC. Experimental evolution. Trends Ecol Evol 27: 547–560, 2012. [DOI] [PubMed] [Google Scholar]
- 94.Kelly JK. Geographical variation in selection, from phenotypes to molecules. Am Nat 167: 481–495, 2006. [DOI] [PubMed] [Google Scholar]
- 95.Kelly SA, Bell TA, Selitsky SR, Buus RJ, Hua K, Weinstock GM, Garland T Jr, de Villena FPM, Pomp D. A novel intronic single nucleotide polymorphism in the myosin heavy polypeptide 4 gene is responsible for the mini-muscle phenotype characterized by major reduction in hind-limb muscle mass in mice. Genetics 195: 1385–1395, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kelly SA, Czech PP, Wight JT, Blank KM, Garland T Jr. Experimental evolution and phenotypic plasticity of hindlimb bones in high-activity house mice. J Morphol 267: 360–374, 2006. [DOI] [PubMed] [Google Scholar]
- 97.Kelly SA, Nehrenberg DL, Hua K, Garland T Jr, Pomp D. Functional genomic architecture of predisposition to voluntary exercise in mice: expression QTL in the brain. Genetics 191: 643–U546, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kelly SA, Nehrenberg DL, Hua K, Garland T Jr, Pomp D. Quantitative genomics of voluntary exercise in mice: transcriptional analysis and mapping of expression QTL in muscle. Physiol Genomics 46: 593–601, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kelly SA, Nehrenberg DL, Peirce JL, Hua K, Steffy BM, Wiltshire T, de Villena FPM, Garland T Jr, Pomp D. Genetic architecture of voluntary exercise in an advanced intercross line of mice. Physiol Genomics 42: 190–200, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kimura M, Ohta T. Age of a neutral mutant persisting in a finite population. Genetics 75: 199–212, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kolb EM, Kelly SA, Middleton KM, Sermsakdi LS, Chappell MA, Garland T Jr. Erythropoietin elevates V̇o2 max but not voluntary wheel running in mice. J Exp Biol 213: 510–519, 2010. [DOI] [PubMed] [Google Scholar]
- 102.Kolb EM, Rezende EL, Holness L, Radtke A, Lee SK, Obenaus A, Garland T Jr. Mice selectively bred for high voluntary wheel running have larger midbrains: support for the mosaic model of brain evolution. J Exp Biol 216: 515–523, 2013. [DOI] [PubMed] [Google Scholar]
- 103.Konarzewski M, Ksiazek A. Determinants of intra-specific variation in basal metabolic rate. J Comp Physiol B 183: 27–41, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Konczal M, Babik W, Radwan J, Sadowska ET, Pawel K. Initial molecular-level response to artificial selection for increased aerobic metabolism occurs primarily via changes in gene expression. Mol Biol Evol 32: 1461–1473, 2015. [DOI] [PubMed] [Google Scholar]
- 105.Ksiazek A, Czerniecki J, Konarzewski M. Phenotypic flexibility of traits related to energy acquisition in mice divergently selected for basal metabolic rate (BMR). J Exp Biol 212: 808–814, 2009. [DOI] [PubMed] [Google Scholar]
- 106.Lailvaux SP, Husak JF. The life history of whole-organism performance. Q Rev Biol 89: 285–318, 2014. [DOI] [PubMed] [Google Scholar]
- 107.Lande R. Quantitative genetic analysis of multivariate evolution, applied to brain-body size allometry. Evolution 33: 402–416, 1979. [DOI] [PubMed] [Google Scholar]
- 108.Lande R. A quantitative genetic theory of life-history evolution. Ecology 63: 607–615, 1982. [Google Scholar]
- 109.Lande R. Quantitative genetics and evolutionary theory. Ecology 63: 71–84, 1988. [Google Scholar]
- 110.Lande R. Sexual dimorphism, sexual selection, and adaptation in polygenic characters. Evolution 34: 292–305, 1980. [DOI] [PubMed] [Google Scholar]
- 111.Lande R, Arnold SJ. The measurement of selection on correlated characters. Evolution 37: 1210–1226, 1983. [DOI] [PubMed] [Google Scholar]
- 112.Latta RG. Differentiation of allelic frequencies at quantitative trait loci affecting locally adaptive traits. Am Nat 151: 283–292, 1998. [DOI] [PubMed] [Google Scholar]
- 113.Laurie CC, Nickerson DA, Anderson AD, Weir BS, Livingston RJ, Dean MD, Smith KL, Schadt EE, Nachman MW. Linkage disequilibrium in wild mice. PLoS Genet 3: 1487–1495, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Le Corre V, Kremer A. Genetic variability at neutral markers, quantitative trait loci and trait in a subdivided population under selection. Genetics 164: 1205–1219, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Leamy LJ, Kelly SA, Hua K, Pomp D. Exercise and diet affect quantitative trait loci for body weight and composition traits in an advanced intercross population of mice. Physiol Genomics 44: 1141–1153, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lehner B. Genotype to phenotype: lessons from model organisms for human genetics. Nat Rev Genet 14: 168–178, 2013. [DOI] [PubMed] [Google Scholar]
- 117.Liebl AL, Martin LB. Stress hormone receptors change as range expansion progresses in house sparrows. Biol Lett 9: 20130181, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lima MR, Macedo RHF, Martins TLF, Schrey AW, Martin LB, Bensch S. Genetic and morphometric divergence of an invasive bird: the introduced house sparrow (Passer domesticus) in Brazil. PLoS One 7: e53332, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Logan RW, Robledo RF, Recla JM, Philip VM, Bubier JA, Jay JJ, Harwood C, Wilcox T, Gatti DM, Bult CJ, Churchill GA, Chesler EJ. High-precision genetic mapping of behavioral traits in the diversity outbred mouse population. Genes Brain Behav 12: 424–437, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lorenzo FR, Huff C, Myllymaki M, Olenchock B, Swierczek S, Tashi T, Gordeuk V, Wuren T, Ri-Li G, McClain DA, Khan TM, Koul PA, Guchhait P, Salama ME, Xing J, Semenza GL, Liberzon E, Wilson A, Simonson TS, Jorde LB, Kaelin WG Jr, Koivunen P, Prchal JT. A genetic mechanism for Tibetan high-altitude adaptation. Nat Genet 46: 951–956, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Losos JB. Seeing the forest for the trees: The limitations of phylogenies in comparative biology. Am Nat 177: 709–727, 2011. [DOI] [PubMed] [Google Scholar]
- 122.Lui MA, Mahalingam S, Patel P, Connaty AD, Ivy CM, Cheviron ZA, Storz JF, McClelland GB, Scott GR. High-altitude ancestry and hypoxia acclimation have distinct effects on exercise capacity and muscle phenotype in deer mice. Am J Physiol Regul Integr Comp Physiol 308: R779–R791, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lush JL. Animal Breeding Plans, 3rd ed. Ames, IA: Iowa State University Press, 1945. [Google Scholar]
- 124.Lynch CB. Genetic basis of cold adaptation in laboratory and wild mice, Mus domesticus. In: Living in the Cold: Physiological and Biochemical Adaptations, edited by Heller HC, Musacchia XJ, and Wang LCH. New York: Elsevier, 1986, p. 497–540. [Google Scholar]
- 125.Lynch M, Walsh B. Genetics and Analysis of Quantitative Traits. Sunderland, MA: Sinauer Associates, 1998. [Google Scholar]
- 126.Mackay TFC. The genetic architecture of quantitative traits. Ann Rev Genet 35: 303–339, 2001. [DOI] [PubMed] [Google Scholar]
- 127.Mackay TFC. Mutations and quantitative genetic variation: lessons from Drosophila. Philos Trans R Soc B Biol Sci 365: 1229–1239, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mackay TFC, Richards S, Stone EA, Barbadilla A, Ayroles JF, Zhu D, Casillas S, Han Y, Magwire MM, Cridland JM, Richardson MF, Anholt RRH, Barron M, Bess C, Blankenburg KP, Carbone MA, Castellano D, Chaboub L, Duncan L, Harris Z, Javaid M, Jayaseelan JC, Jhangiani SN, Jordan KW, Lara F, Lawrence F, Lee SL, Librado P, Linheiro RS, Lyman RF, Mackey AJ, Munidasa M, Muzny DM, Nazareth L, Newsham I, Perales L, Pu LL, Qu C, Ramia M, Reid JG, Rollmann SM, Rozas J, Saada N, Turlapati L, Worley KC, Wu YQ, Yamamoto A, Zhu Y, Bergman CM, Thornton KR, Mittelman D, Gibbs RA. The Drosophila melanogaster Genetic Reference Panel. Nature 482: 173–178, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mackay TFC, Stone EA, Ayroles JF. The genetics of quantitative traits: challenges and prospects. Nat Rev Genet 10: 565–577, 2009. [DOI] [PubMed] [Google Scholar]
- 130.Majdak P, Bucko PJ, Holloway AL, Bhattacharya TK, DeYoung EK, Kilby CN, Zombeck JA, Rhodes JS. Behavioral and pharmacological evaluation of a selectively bred mouse model of home cage hyperactivity. Behav Genet 44: 516–534, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Marquez EJ. A statistical framework for testing modularity in multidimensional data. Evolution 62: 2688–2708, 2008. [DOI] [PubMed] [Google Scholar]
- 132.Mathes WF, Aylor DL, Miller DR, Churchill GA, Chesler EJ, de Villena FPM, Threadgill DW, Pomp D. Architecture of energy balance traits in emerging lines of the Collaborative Cross. Am J Physiol Endocrinol Metab 300: E1124–E1134, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Meek TH, Lonquich BP, Hannon RM, Garland T Jr. Endurance capacity of mice selectively bred for high voluntary wheel running. J Exp Biol 212: 2908–2917, 2009. [DOI] [PubMed] [Google Scholar]
- 134.Munoz-Garcia A, Ro J, Brown JC, Williams JB. Cutaneous water loss and sphingolipids in the stratum corneum of house sparrows, Passer domesticus L., from desert and mesic environments as determined by reversed phase high-performance liquid chromatography coupled with atmospheric pressure photospray ionization mass spectrometry. J Exp Biol 211: 447–458, 2008. [DOI] [PubMed] [Google Scholar]
- 135.Natarajan C, Hoffman FG, Lanier HC, Wolf CJ, Cheviron ZA, Spangler ML, Weber RE, Fago A, Storz JF. Intraspecific polymorphism, interspecific divergence, and the origins of function-altering mutations in deer mouse hemoglobin. Mol Biol Evol 32: 978–997, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Natarajan C, Inoguchi N, Weber RE, Fago A, Moriyama H, Storz JF. Epistasis among adaptive mutations in deer mouse hemoglobin. Science 340: 1324–1327, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nehrenberg DL, Wang S, Hannon RM, Garland T Jr, Pomp D. QTL underlying voluntary exercise in mice: Interactions with the “mini muscle” locus and sex. J Hered 101: 42–53, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Novembre J, Di Rienzo A. Spatial patterns of variation due to natural selection in humans. Nat Rev Genet 10: 745–755, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Orr HA, Betancourt AJ. Haldane's sieve and adaptation from the standing genetic variation. Genetics 157: 875–884, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Oufiero CE, Garland T Jr. Evaluating performance costs of sexually selected traits. Funct Ecol 21: 676–689, 2007. [Google Scholar]
- 141.Paranjpe DA, Bastiaans E, Patten A, Cooper RD, Sinervo B. Evidence of maternal effects on temperature preference in side-blotched lizards: implications for evolutionary response to climate change. Ecol Evol 3: 1977–1991, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Passador-Gurgel G, Hsieh WP, Hunt P, Deighton N, Gibson G. Quantitative trait transcripts for nicotine resistance in Drosophila melanogaster. Nat Genet 39: 264–268, 2007. [DOI] [PubMed] [Google Scholar]
- 143.Pavlidis P, Jensen JD, Stephan W, Stamatakis A. A critical assessment of storytelling: gene ontology categories and the importance of validating genomic scans. Mol Biol Evol 29: 3237–3248, 2012. [DOI] [PubMed] [Google Scholar]
- 144.Peng Y, Yang Z, Zhang H, Cui C, Qi X, Luo X, Tao X, Wu T, Ouzhuluobu Basang Ciwangsangbu Danzengduojie Chen H, Shi H, Su B. Genetic variations in Tibetan populations and high-altitude adaptation at the Himalayas. Mol Biol Evol 28: 1075–1081, 2011. [DOI] [PubMed] [Google Scholar]
- 145.Pennings PS, Hermisson J. Soft sweeps II-molecular population genetics of adaptation from recurrent mutation or migration. Mol Biol Evol 23: 1076–1084, 2006. [DOI] [PubMed] [Google Scholar]
- 146.Pennings PS, Hermisson J. Soft sweeps III: The signature of positive selection from recurrent mutation. PLoS Genet 2: 1998–2012, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Petousi N, Croft QPP, Cavalleri GL, Cheng HY, Formenti F, Ishida K, Lunn D, McCormack M, Shianna KV, Talbot NP, Ratcliffe PJ, Robbins PA. Tibetans living at sea level have a hyporesponsive hypoxia-inducible factor system and blunted physiological responses to hypoxia. J Appl Physiol 116: 893–904, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Petousi N, Robbins PA. Human adaptation to the hypoxia of high altitude: the Tibetan paradigm from the pregenomic to the postgenomic era. J Appl Physiol 116: 875–884, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pritchard JK, Di Rienzo A. Adaptation: not by sweeps alone. Nat Rev Genet 11: 665–667, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pritchard JK, Pickrell JK, Coop G. The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Curr Biol 20: R208–R215, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Projecto-Garcia J, Natarajan C, Moriyama H, Weber RE, Fago A, Cheviron ZA, Dudley R, McGuire JA, Witt CC, Storz JF. Repeated elevational transitions in hemoglobin function during the evolution of Andean hummingbirds. Proc Natl Acad Sci USA 110: 20,669–20,674, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Przeworski M, Coop G, Wall JD. The signature of positive selection on standing genetic variation. Evolution 59: 2312–2323, 2005. [PubMed] [Google Scholar]
- 153.Rankin EB, Biju MP, Liu Q, Unger TL, Rha J, Johnson RS, Simon MC, Keith B, Haase VH. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest 117: 1068–1077, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rezende EL, Diniz-Filho J. AF Phylogenetic analyses: comparing species to infer adaptations and physiological mechanisms. Compr Physiol 2: 639–674, 2012. [DOI] [PubMed] [Google Scholar]
- 155.Rezende EL, Gomes FR, Chappell MA, Garland T Jr. Running behavior and its energy cost in mice selectively bred for high voluntary locomotor activity. Physiol Biochem Zool 82: 662–679, 2009. [DOI] [PubMed] [Google Scholar]
- 156.Rhodes JS, Kawecki TJ. Behavior and neurobiology. In: Experimental Evolution: Concepts, Methods, and Applications of Selection Experiments, edited by Garland T Jr., and Rose MR. Berkeley, CA: University of California, 2009, p. 263–300. [Google Scholar]
- 157.Rockman MV. The QTN program and the alleles that matter for evolution: all that's gold does not glitter. Evolution 66: 1–17, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Rockman MV. Reverse engineering the genotype-phenotype map with natural genetic variation. Nature 456: 738–744, 2008. [DOI] [PubMed] [Google Scholar]
- 159.Roff DA, Fairbairn DJ. The evolution of trade-offs: where are we? J Evol Biol 20: 433–447, 2007. [DOI] [PubMed] [Google Scholar]
- 160.Rosenblum EB, Roempler H, Schoeneberg T, Hoekstra HE. Molecular and functional basis of phenotypic convergence in white lizards at White Sands. Proc Natl Acad Sci USA 107: 2113–2117, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Runck AM, Weber RE, Fago A, Storz JF. Evolutionary and functional properties of a two-locus β-globin polymorphism in Indian house mice. Genetics 184: 1121–1131, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sabeti PC, Reich DE, Higgins JM, Levine HZP, Richter DJ, Schaffner SF, Gabriel SB, Platko JV, Patterson NJ, McDonald GJ, Ackerman HC, Campbell SJ, Altshuler D, Cooper R, Kwiatkowski D, Ward R, Lander ES. Detecting recent positive selection in the human genome from haplotype structure. Nature 419: 832–837, 2002. [DOI] [PubMed] [Google Scholar]
- 163.Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, Xie X, Byrne EH, McCarroll SA, Gaudet R, Schaffner SF, Lander ES, International HapMap Consortium, Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, Willis TD, Yu F, Yang H, Zeng C, Gao Y, Hu H, Hu W, Li C, Lin W, Liu S, Pan H, Tang X, Wang J, Wang W, Yu J, Zhang B, Zhang Q, Zhao H, Zhao H, Zhou J, Gabriel SB, Barry R, Blumenstiel B, Camargo A, Defelice M, Faggart M, Goyette M, Gupta S, Moore J, Nguyen H, Onofrio RC, Parkin M, Roy J, Stahl E, Winchester E, Ziaugra L, Altshuler D, Shen Y, Yao Z, Huang W, Chu X, He Y, Jin L, Liu Y, Shen Y, Sun W, Wang H, Wang Y, Wang Y, Xiong X, Xu L, Waye MM, Tsui SK, Xue H, Wong JT, Galver LM, Fan JB, Gunderson K, Murray SS, Oliphant AR, Chee MS, Montpetit A, Chagnon F, Ferretti V, Leboeuf M, Olivier JF, Phillips MS, Roumy S, Sallée C, Verner A, Hudson TJ, Kwok PY, Cai D, Koboldt DC, Miller RD, Pawlikowska L, Taillon-Miller P, Xiao M, Tsui LC, Mak W, Song YQ, Tam PK, Nakamura Y, Kawaguchi T, Kitamoto T, Morizono T, Nagashima A, Ohnishi Y, Sekine A, Tanaka T, Tsunoda T, Deloukas P, Bird CP, Delgado M, Dermitzakis ET, Gwilliam R, Hunt S, Morrison J, Powell D, Stranger BE, Whittaker P, Bentley DR, Daly MJ, de Bakker PI, Barrett J, Chretien YR, Maller J, McCarroll S, Patterson N, Pe'er I, Price A, Purcell S, Richter DJ, Sabeti P, Saxena R, Schaffner SF, Sham PC, Varilly P, Altshuler D, Stein LD, Krishnan L, Smith AV, Tello-Ruiz MK, Thorisson GA, Chakravarti A, Chen PE, Cutler DJ, Kashuk CS, Lin S, Abecasis GR, Guan W, Li Y, Munro HM, Qin ZS, Thomas DJ, McVean G, Auton A, Bottolo L, Cardin N, Eyheramendy S, Freeman C, Marchini J, Myers S, Spencer C, Stephens M, Donnelly P, Cardon LR, Clarke G, Evans DM, Morris AP, Weir BS, Tsunoda T, Johnson TA, Mullikin JC, Sherry ST, Feolo M, Skol A, Zhang H, Zeng C, Zhao H, Matsuda I, Fukushima Y, Macer DR, Suda E, Rotimi CN, Adebamowo CA, Ajayi I, Aniagwu T, Marshall PA, Nkwodimmah C, Royal CD, Leppert MF, Dixon M, Peiffer A, Qiu R, Kent A, Kato K, Niikawa N, Adewole IF, Knoppers BM, Foster MW, Clayton EW, Watkin J, Gibbs RA, Belmont JW, Muzny D, Nazareth L, Sodergren E, Weinstock GM, Wheeler DA, Yakub I, Gabriel SB, Onofrio RC, Richter DJ, Ziaugra L, Birren BW, Daly MJ, Altshuler D, Wilson RK, Fulton LL, Rogers J, Burton J, Carter NP, Clee CM, Griffiths M, Jones MC, McLay K, Plumb RW, Ross MT, Sims SK, Willey DL, Chen Z, Han H, Kang L, Godbout M, Wallenburg JC, L'Archevêque P, Bellemare G, Saeki K, Wang H, An D, Fu H, Li Q, Wang Z, Wang R, Holden AL, Brooks LD, McEwen JE, Guyer MS, Wang VO, Peterson JL, Shi M, Spiegel J, Sung LM, Zacharia LF, Collins FS, Kennedy K, Jamieson R, Stewart J. Genome-wide detection and characterization of positive selection in human populations. Nature 449: 913-U912, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sadowska ET, Baliga-Klimczyk K, Chrzascik KM, Koteja P. Laboratory model of adaptive radiation: A selection experiment in the bank vole. Physiol Biochem Zool 81: 627–640, 2008. [DOI] [PubMed] [Google Scholar]
- 165.Savolainen O, Lascoux M, Merila J. Ecological genomics of local adaptation. Nat Rev Genet 14: 807–820, 2013. [DOI] [PubMed] [Google Scholar]
- 166.Schluter D. Adaptive radiation along genetic lines of least resistance. Evolution 50: 1766–1774, 1996. [DOI] [PubMed] [Google Scholar]
- 167.Schluter D. The Ecology of Adaptive Radiation. Oxford, UK: Oxford University Press, 2000. [Google Scholar]
- 168.Schutz H, Jamniczky HA, Hallgrimsson B, Garland T Jr. Shape-shift: semicircular canal morphology responds to selective breeding for increased locomotor activity. Evolution 68: 3184–3198, 2014. [DOI] [PubMed] [Google Scholar]
- 169.Scott GR, Elogio TS, Lui MA, Storz JF, Cheviron ZA. Adaptive modifications of muscle phenotype in high-altitude deer mice are associated with evolved changes in gene regulation. Mol Biol Evol doi: 10.1093/molbev/msv076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Shaw JR, Hampton TH, King BL, Whitehead A, Galvez F, Gross RH, Keith N, Notch E, Jung D, Glaholt SP, Chen CY, Colbourne JK, Stanton BA. Natural selection canalizes expression variation of environmentally induced plasticity-enabling genes. Mol Biol Evol 31: 3002–3015, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Sieberts SK, Schadt EE. Moving toward a system genetics view of disease. Mamm Genome 18: 389–401, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Simonson TS, Yang Y, Huff CD, Yun H, Qin G, Witherspoon DJ, Bai Z, Lorenzo FR, Xing J, Jorde LB, Prchal JT, Ge R. Genetic evidence for high-altitude adaptation in Tibet. Science 329: 72–75, 2010. [DOI] [PubMed] [Google Scholar]
- 173.Sinervo B, Calsbeek R. Physiological epistasis, ontogenetic conflict and natural selection on physiology and life history. Integr Comp Biol 43: 419–430, 2003. [DOI] [PubMed] [Google Scholar]
- 174.Stawski C, Koteja P, Sadowska ET, Jefimow M, Wojciechowski MS. Selection for high activity-related aerobic metabolism does not alter the capacity of non-shivering thermogenesis in bank voles. Comp Biochem Physiol A Mol Integr Physiol 180: 51–56, 2015. [DOI] [PubMed] [Google Scholar]
- 175.Stearns SC, Byars SG, Govindaraju DR, Ewbank D. Measuring selection in contemporary human populations. Nat Rev Genet 11: 611–622, 2010. [DOI] [PubMed] [Google Scholar]
- 176.Stern DL. Evolutionary developmental biology and the problem of variation. Evolution 54: 1079–1091, 2000. [DOI] [PubMed] [Google Scholar]
- 177.Storz JF. Genes for high altitudes. Science 329: 40–41, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Storz JF. Using genome scans of DNA polymorphism to infer adaptive population divergence. Mol Ecol 14: 671–688, 2005. [DOI] [PubMed] [Google Scholar]
- 179.Storz JF, Kelly JK. Effects of spatially varying selection on nucleotide diversity and linkage disequilibrium: Insights from deer mouse globin genes. Genetics 180: 367–379, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Storz JF, Natarajan C, Cheviron ZA, Hoffmann FG, Kelly JK. Altitudinal variation at duplicated β-globin genes in deer mice: effects of selection, recombination, and gene conversion. Genetics 190: 203–216, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Storz JF, Runck AM, Moriyama H, Weber RE, Fago A. Genetic differences in hemoglobin function between highland and lowland deer mice. J Exp Biol 213: 2565–2574, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Storz JF, Runck AM, Sabatino SJ, Kelly JK, Ferrand N, Moriyama H, Weber RE, Fago A. Evolutionary and functional insights into the mechanism underlying high-altitude adaptation of deer mouse hemoglobin. Proc Natl Acad Sci USA 106: 14,450–14,455, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Storz JF, Sabatino SJ, Hoffmann FG, Gering EJ, Moriyama H, Ferrand N, Monteiro B, Nachman MW. The molecular basis of high-altitude adaptation in deer mice. PLoS Genet 3: 448–459, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Storz JF, Scott GR, Cheviron ZA. Phenotypic plasticity and genetic adaptation to high-altitude hypoxia in vertebrates. J Exp Biol 213: 4125–4136, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Storz JF, Wheat CW. Integrating evolutionary and functional approaches to infer adaptation at specific loci. Evolution 64: 2489–2509, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Storz JF, Zera AJ. Experimental approaches to evaluate the contributions of candidate protein-coding mutations to phenotypic evolution. Methods Mol Biol 772: 377–396, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Swallow J, Hayes J, Koteja P, Garland T Jr. Selection experiments and experimental evolution of performance and physiology. In: Experimental Evolution: Concepts, Methods, and Applications of Selection Experiments, edited by Garland T and Rose MR. Berkeley, CA: University of California, 2009, p. 301–351. [Google Scholar]
- 188.Swallow JG, Carter PA, Garland T Jr. Artificial selection for increased wheel-running behavior in house mice. Behav Genet 28: 227–237, 1998. [DOI] [PubMed] [Google Scholar]
- 189.Swallow JG, Garland T Jr. Selection experiments as a tool in evolutionary and comparative physiology: insights into complex traits—an introduction to the symposium. Integr Comp Biol 45: 387–390, 2005. [DOI] [PubMed] [Google Scholar]
- 190.Swallow JG, Koteja P, Carter PA, Garland T Jr. Artificial selection for increased wheel-running activity in house mice results in decreased body mass at maturity. J Exp Biol 202: 2513–2520, 1999. [DOI] [PubMed] [Google Scholar]
- 191.Swallow JG, Koteja P, Carter PA, Garland T Jr. Food consumption and body composition in mice selected for high wheel-running activity. J Comp Physiol B 171: 651–659, 2001. [DOI] [PubMed] [Google Scholar]
- 192.Tada T, Altun A, Yokoyama S. Evolutionary replacement of UV vision by violet vision in fish. Proc Natl Acad Sci USA 106: 17,457–17,462, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Teshima KM, Coop G, Przeworski M. How reliable are empirical genomic scans for selective sweeps? Genome Res 16: 702–712, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Teshima KM, Przeworski M. Directional positive selection on an allele of arbitrary dominance. Genetics 172: 713–718, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Thornton JW. Evolution of vertebrate steroid receptors from an ancestral estrogen receptor by ligand exploitation and serial genome expansions. Proc Natl Acad Sci USA 98: 5671–5676, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Thornton JW. Resurrecting ancient genes: experimental analysis of extinct molecules. Nat Rev Genet 5: 366–375, 2004. [DOI] [PubMed] [Google Scholar]
- 197.Thornton JW, Need E, Crews D. Resurrecting the ancestral steroid receptor: Ancient origin of estrogen signaling. Science 301: 1714–1717, 2003. [DOI] [PubMed] [Google Scholar]
- 198.Tufts DM, Natarajan C, Revsbech IG, Projecto-Garcia J, Hoffman FG, Weber RE, Fago A, Moriyama H, Storz JF. Epistasis constrains mutational pathways of hemoglobin adaptation in high-altitude pikas. Mol Biol Evol 32: 287–298, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Villafuerte FC, Cardenas R, Monge CC. Optimal hemoglobin concentration and high altitude: a theoretical approach for Andean men at rest. J Appl Physiol 96: 1581–1588, 2004. [DOI] [PubMed] [Google Scholar]
- 200.Visscher PM, Hill WG, Wray NR. Heritability in the genomics era. Concepts and misconceptions. Nat Rev Genet 9: 255–266, 2008. [DOI] [PubMed] [Google Scholar]
- 201.Walker JA. A general model of functional constraints on phenotypic evolution. Am Nat 170: 681–689, 2007. [DOI] [PubMed] [Google Scholar]
- 202.Waterland RA. Epigenetic mechanisms affecting regulation of energy balance: many questions, few answers. Ann Rev Nutr 34: 337–355, 2014. [DOI] [PubMed] [Google Scholar]
- 203.Waterland RA, Garza C. Potential mechanisms of metabolic imprinting that lead to chronic disease. Am J Clin Nutr 69: 179–197, 1999. [DOI] [PubMed] [Google Scholar]
- 204.Waterland RA, Travisano M, Tahiliani KG, Rached MT, Mirza S. Methyl donor supplementation prevents transgenerational amplification of obesity. Int J Obes 32: 1373–1379, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Weber KE. How small are the smallest selectable domains of form? Genetics 130: 345–353, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Weber KE. Selection on wing allometry in Drosophila melanogaster. Genetics 126: 975–989, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Whitaker KW, Totoki K, Reyes TM. Metabolic adaptations to early life protein restriction differ by offspring sex and post-weaning diet in the mouse. Nutr Metab Cardiovasc Dis 22: 1067–1074, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Whitehead A. Comparative genomics in ecological physiology: toward a more nuanced understanding of acclimation and adaptation. J Exp Biol 215: 884–891, 2012. [DOI] [PubMed] [Google Scholar]
- 209.Whitehead A, Roach JL, Zhang S, Galvez F. Genomic mechanisms of evolved physiological plasticity in killifish distributed along an environmental salinity gradient. Proc Natl Acad Sci USA 108: 6193–6198, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Winslow RM, Chapman KW, Gibson CC, Samaja M, Monge CC, Goldwasser E, Sherpa M, Blume FD, Santolaya R. Different hematologic responses to hypoxia in sherpas and Quechua indians. J Appl Physiol 66: 1561–1569, 1989. [DOI] [PubMed] [Google Scholar]
- 211.Wone B, Madsen P, Donovan E, Labocha M, Sears M, CJ D, Sorenson D, Hayes J. A strong response to selection on mass-independent maximal metabolic rate without a correlated response in basal metabolic rate. Heredity 114: 419–427, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Xiang K, Ouzhuluobu Peng Y, Yang Z, Zhang X, Cui C, Zhang H, Li M, Zhang Y, Bianba Gonggalanzi Basang Ciwangsangbu Wu T, Chen H, Shi H, Qi X, Su B. Identification of a Tibetan-specific mutation in the hypoxic gene EGLN1 and its contribution to high-altitude adaptation. Mol Biol Evol 30: 1889–1898, 2013. [DOI] [PubMed] [Google Scholar]
- 213.Xu S, Li S, Yang Y, Tan J, Lou H, Jin W, Yang L, Pan X, Wang J, Shen Y, Wu B, Wang H, Jin L. A genome-wide search for signals of high-altitude adaptation in Tibetans. Mol Biol Evol 28: 1003–1011, 2011. [DOI] [PubMed] [Google Scholar]
- 214.Yang ZH, Bielawski JP. Statistical methods for detecting molecular adaptation. Trends Ecol Evol 15: 496–503, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Yang ZH, Kumar S, Nei M. A new method of inference of ancestral nucleotide and amino-acid sequences. Genetics 141: 1641–1650, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZXP, Pool JE, Xu X, Jiang H, Vinckenbosch N, Korneliussen TS, Zheng H, Liu T, He W, Li K, Luo R, Nie X, Wu H, Zhao M, Cao H, Zou J, Shan Y, Li S, Yang Q, Asan Ni P, Tian G, Xu J, Liu X, Jiang T, Wu R, Zhou G, Tang M, Qin J, Wang T, Feng S, Li G, Huasang Luosang J, Wang W, Chen F, Wang Y, Zheng X, Li Z, Bianba Z, Yang G, Wang X, Tang S, Gao G, Chen Y, Luo Z, Gusang L, Cao Z, Zhang Q, Ouyang W, Ren X, Liang H, Zheng H, Huang Y, Li J, Bolund L, Kristiansen K, Li Y, Zhang Y, Zhang X, Li R, Li S, Yang H, Nielsen R, Wang J, Wang J. Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329: 75–78, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Yokoyama S. Synthesis of experimental molecular biology and evolutionary biology: an example from the world of vision. Bioscience 62: 939–948, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Yokoyama S, Tada T, Zhang H, Britt L. Elucidation of phenotypic adaptations: molecular analyses of dim-light vision proteins in vertebrates. Proc Natl Acad Sci USA 105: 13,480–13,485, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Yokoyama S, Yang H, Starmert WT. Molecular basis of spectral tuning in the red- and green-sensitive (M/LWS) pigments in vertebrates. Genetics 179: 2037–2043, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Yuan F, Bernard GD, Le J, Briscoe AD. Contrasting modes of evolution of the visual pigments in Heliconius butterflies. Mol Biol Evol 27: 2392–2405, 2010. [DOI] [PubMed] [Google Scholar]
- 221.Zaman L, Meyer JR, Devangam S, Bryson DM, Lenski RE, Ofria C. Coevolution drives the emergence of complex traits and promotes evolvability. PLoS Biol 12: e1002023, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Zhou S, Campbell TG, Stone EA, Mackay TFC, Anholt RRH. Phenotypic plasticity of the Drosophila transcriptome. PLoS Genet 8: e1002593, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]