

Abstract
Toll-like receptor-4 (TLR-4) is elevated in skeletal muscle of obese humans, and data from our laboratory have shown that activation of TLR-4 in skeletal muscle via LPS results in decreased fatty acid oxidation (FAO). The purpose of this study was to determine whether overexpression of TLR-4 in skeletal muscle alters mitochondrial function and whole body metabolism in the context of a chow and high-fat diet. C57BL/6J mice (males, 6–8 mo of age) with skeletal muscle-specific overexpression of the TLR-4 (mTLR-4) gene were created and used for this study. Isolated mitochondria and whole muscle homogenates from rodent skeletal muscle (gastrocnemius and quadriceps) were investigated. TLR-4 overexpression resulted in a significant reduction in FAO in muscle homogenates; however, mitochondrial respiration and reactive oxygen species (ROS) production did not appear to be affected on a standard chow diet. To determine the role of TLR-4 overexpression in skeletal muscle in response to high-fat feeding, mTLR-4 mice and WT control mice were fed low- and high-fat diets for 16 wk. The high-fat diet significantly decreased FAO in mTLR-4 mice, which was observed in concert with elevated body weight and fat, greater glucose intolerance, and increase in production of ROS and cellular oxidative damage compared with WT littermates. These findings suggest that TLR-4 plays an important role in the metabolic response in skeletal muscle to high-fat feeding.
Keywords: Toll-like receptor 4, skeletal muscle, high-fat diet, fatty acid oxidation, reactive oxygen species
growing evidence indicates that obesity and metabolic dysregulation are closely associated with chronic low-grade inflammation (1, 20, 21). Our group and others have reported elevated gene expression and protein content of Toll-like receptor 4 (TLR-4) in skeletal muscle samples from obese and/or Type 2 diabetic humans relative to nonobese and/or healthy controls (5, 13). TLR-4, a transmembrane protein, is activated by LPS and plays a critical role in the innate immune system and the induction of inflammatory responses (14). Evidence in both animal and human studies has demonstrated that abnormal TLR-4 expression and signaling may contribute to the pathogenesis of insulin resistance in skeletal muscle (12, 15, 18). Skeletal muscle comprises ∼30% of total body mass and is a major site of postprandial, insulin-mediated glucose disposal, as well as fatty acid oxidation under fasting conditions (16). We recently reported that TLR-4 activation in skeletal muscle by LPS, under both in vitro and in vivo conditions, alters mitochondrial function (6) and results in a shift in substrate metabolism toward glucose utilization with a partitioning of free fatty acids toward neutral lipid synthesis in the fasted state (5). Skeletal muscle is important to the modulation of whole body metabolism; as such, it is important to understand the whole body physiological significance of the modulation of TLR-4 function in skeletal muscle.
Herein, we present studies using a transgenic mouse model with skeletal muscle-specific overexpression of TLR-4 (mTLR-4) under standard chow and high-fat fed conditions. Our hypothesis was that mTLR-4 mice would have a heightened inflammatory state in conjunction with reduced fatty acid oxidative capacity in skeletal muscle, thus leading to greater metabolic derangements during high-fat feeding compared with littermate controls.
MATERIALS AND METHODS
Animal husbandry and generation of muscle-specific overexpression of TLR-4 mouse model.
Animal studies were performed under an approved protocol by the Institutional Animal Care and Use Committee at Virginia Tech. Male C57BL/6J mice with muscle-specific overexpression of the TLR-4 (mTLR-4) gene and their wild-type littermates (WT) were used for all experiments. The mTLR-4 mouse was generated at the Wake Forest University Transgenic Core using the muscle creatine kinase promoter. Mice for all studies were maintained on standard 12:12-h light-dark cycle at a vivarium temperate of 20–22°C. All experiments were performed following an overnight fast (12–16 h), unless stated otherwise.
LPS injection in mTLR-4 mice.
mTLR-4 mice and WT received an intraperitoneal injection with saline or LPS (1 μg/kg body wt) following an overnight fast to determine whether LPS elicited a greater TLR-4 response in mTLR-4 mice. We chose this high LPS dosage because it would increase the activity of the skeletal muscle TLR-4 receptor and allow for adequate comparisons between the two genotypes. LPS from Escherichia coli 0111:B4 (L2630; Sigma-Aldrich, St. Louis, MO) prepared in saline was used for all experiments. The animals were killed and flexor digitorum brevis (FDB) muscle was collected 4 h postinjection to assess IL-6 mRNA expression. The FDB muscle group was used because isolation of single fibers prevents confounding influences of nonmuscle cell types that are present in whole muscle groups (e.g., gastrocnemius and quadriceps).
High-fat feeding studies.
mTLR-4 mice and their WT littermates were fed a control, low-fat diet (Teklad TD.10453, 10% fat, 71% carbohydrate, and 19% protein) or a high-fat diet (Teklad, TD.10505, 45% fat, 37% carbohydrate, and 18% protein) for 16 wk commencing at 6–8 mo of age. Diets were designed and purchased from Harlan (Madison, WI). Following 16 wk of high-fat feeding, fasted mice were killed via carbon dioxide asphyxiation followed by cervical dislocation. Gastrocnemius and quadriceps were harvested, mitochondria were isolated, and measures of mitochondrial respiration, fatty acid oxidation, ROS production, and enzyme activity were performed.
Skeletal muscle whole homogenate preparation.
Approximately 50 mg of fresh muscle samples were immediately placed into 0.2 ml of a modified sucrose EDTA medium on ice containing 250 mM sucrose, 1 mM EDTA, 10 mM Tris·HCl, and 1 mM ATP, at pH 7.4. Muscle samples were then minced with scissors, and buffer was added to a 20-fold diluted (wt/vol) suspension. The minced samples were homogenized in a Potter-Elvehjem glass homogenizer at 12 passes across 30 s at 150 rpm with a motor-driven Teflon pestle, and measures of fatty acid oxidation and maximal enzyme activities were performed.
Mitochondrial isolation from skeletal muscle.
Mitochondria were isolated from red skeletal muscle from the quadriceps femoris and gastrocnemius muscle, as previously described (6). Briefly, tissue samples were collected in buffer containing 67 mM sucrose, 50 mM Tris·HCl, 50 mM KCl, 10 mM EDTA/Tris, and 10% BSA (all from Sigma-Aldrich, St. Louis, MO). Samples were minced and digested in 0.05% trypsin (Invitrogen, Carlsbad, CA) for 30 min. Samples were homogenized and mitochondria were isolated by differential centrifugation.
Real-time quantitative PCR.
Tissue was collected in TRIzol and homogenized, and RNA was extracted. Total mRNA was prepared using TRIzol reagent, according to the manufacturer's protocol (Life Technologies, Carlsbad, CA), treated with DNase I (Ambion, Austin, TX), and extracted using the RNeasy mini kit (Qiagen, Valencia, CA). Total mRNA concentration was quantified using an Agilent Bioanalyzer. Real-time quantitative PCR (RTQ-PCR) was performed using an ABI 7900 Fast HT RTQ-PCR instrument (PE Applied Biosystems, Foster City, CA). IL-6 and TLR-4 gene expression was determined with the TaqMan Universal PCR master mix, according to manufacturer's specifications (Applied Biosystems). Primer and probes sets for all target genes were purchased as prevalidated assays (Applied Biosystems). Relative quantification of target genes was calculated using the 2−ΔCT method. Derivation of the 2−ΔCT equation has been described in Applied Biosystems User Bulletin No. 2 (P/N 4303859). Target gene expression was normalized to β-actin RNA levels.
Western blot analysis.
Western blot analysis was performed using skeletal muscle preparation in 50 mM HEPES, (pH 7.5), 15 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, 2 mM EDTA, 10% glycerol, 1% Triton X-100, 10 mM NaP2O7, 100 mM NaFl, 10 mM PMSF, and 10 g/ml aprotinin. Proteins (∼30 μg per sample) were separated using a 10% Criterion-Tris·HCl gel (Bio-Rad, Hercules, CA) and subsequently transferred to PVDF membrane (Bio-Rad). Blots were probed with primary antibodies against 4-hydroxynonenal (4-HNE; 5 μg/ml; Abcam, Cambridge, MA), TLR-4 (1:250; Santa Cruz Biotechnology, Santa Cruz, CA), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:1,000, Santa Cruz Biotechnology), followed by anti-rabbit (1:10,000; Jackson Immunoresearch, Bar Harbor, ME) or anti-goat secondary antibodies (1:1,000, Santa Cruz Biotechnology), respectively. Proteins were visualized using Super-Signal Chemiluminescent Substrate (Pierce, Rockford, IL) on a ChemiDoc XRS Imaging System (Bio-Rad, Hercules, CA).
Glucose and insulin tolerance testing.
To assess glucose tolerance, mice were fasted overnight and received an intraperitoneal injection of dextrose (1 g/kg body mass). Blood was then collected via tail vein at baseline and then every 30 min for 120 min for the measurement of blood glucose concentrations. To assess insulin tolerance, mice were fasted for 4 h and then received an intraperitoneal injection of insulin (0.5 U/kg body mass; Humulin R). Blood was collected, via the tail vein, at baseline and then every 15 min for 60 min for the measurement of blood glucose concentrations.
Body composition and whole body calorimetry.
Body composition was assessed via the Bruker mini spec LF90. Energy expenditure and respiratory exchange ratio (RER) were measured over 48 h using indirect calorimetry (TSE Systems, Chesterfield, MO). Energy expenditure data were expressed relative to fat-free mass.
Measures of respiration in isolated mitochondria.
Respirometry of isolated mitochondria was performed using an XF24 extracellular flux analyzer (Seahorse Bioscience, North Billerica, MA), as described previously (8). Immediately following isolation and protein quantification, mitochondria were plated on Seahorse cell culture plates at a concentration of 5 μg/well in the presence of pyruvate (10 mM) and malate (5 mM). Experiments were conducted in a sucrose/mannitol solution to maintain the integrity of the mitochondria. Experiments involved 25 s of mixing and 4–7-min measurement cycles, unless otherwise stated. Oxygen consumption was measured under basal conditions (state 2), ADP (5 mM)-stimulated state 3 respiration, oligomycin (2 μM)-induced state 4 respiration, and uncoupled respiration in the presence of the mitochondrial uncoupler FCCP (3 μM) to assess maximal oxidative capacity. All experiments were performed at 37°C.
Fatty acid oxidation.
Palmitate oxidation was assessed in isolated mitochondria and muscle homogenates by measuring and summing 14CO2 production and 14C-labeled acid-soluble metabolites from the oxidation of [1-14C]-palmitic acid (Perkin Elmer, Waltham, MA), respectively, as previously described (5).
Enzyme activity.
Enzyme activities were assessed in isolated mitochondria and muscle homogenates and were determined spectrophotometrically, as previously described (5). Citrate synthase activity was determined by the rate of DNTB reduction upon exposure to acetyl CoA at 412 nm. β-3-hydroxyacyl coenzyme A dehydrogenase (BHAD) and malate dehydrogenase (MDH) activity was determined by the rate of NADH oxidation in the presence of acetoacetyl coA or oxaloacetate, respectively at 340 nm.
Reactive oxygen species production in isolated mitochondria.
Amplex Red (Invitrogen, Grand Island, NY) was used for measures of reactive oxygen species (ROS) production. To measure ROS production from complex 1, complex 3, and reverse electron transfer into complex 1 (REV), isolated mitochondria were plated on a 96-well black plate at a concentration of 5 μg/well under three different conditions, respectively. The three conditions were pyruvate (20 mM), malate (10 mM), oligomycin (2 μM), rotenone (200 nM) for complex 1, pyruvate (20 mM), malate (10 mM), oligomycin (2 μM), SOD (400 U/ml), and antimycin A (2 μM) for complex 3, and succinate (20 mM); oligomycin (2 μM) for REV. Experiments were conducted in sucrose/mannitol solution to maintain the integrity of the mitochondria. Experiments consisted of a 1-min delay and 1-min reading cycles, followed by a 5-s mixing cycle performed every third reading. All experiments were performed at 37°C. Measures for ROS levels were conducted on a microplate reader (BioTek Synergy 2; Winooski, VT). Fluorescence of Amplex Red was measured using a 530-nm excitation filter and a 560-nm emission filter.
Statistical analysis.
Results were analyzed with Student's t-tests or two-way ANOVA with Tukey's post hoc analysis. Results are expressed as means ± SE. The level of significance was set a priori at P < 0.05.
RESULTS
Muscle-specific TLR-4 transgenic mice.
The mTLR-4 mice exhibited an approximately four-fold increase in skeletal muscle TLR-4 mRNA expression compared with WT (Fig. 1A). Protein content of TLR-4 was approximately six-fold higher in FDB muscle fibers from mTLR-4 mice compared with WT (Fig. 1B). To determine whether there was a heightened downstream TLR-4 response in mTLR-4 mice, both mTLR-4 mice and their WT littermates were injected with either saline or LPS (1 μg/kg body wt), and mRNA levels of IL-6, a downstream target of TLR-4 signaling, were assessed. A significant increase in IL-6 mRNA level was induced in response to LPS in both mTLR-4 and WT mice (Fig. 1C), with a more robust response in the mTLR-4 mice. A significant genotype × treatment interaction between mTLR-4 and WT mice and LPS-induced IL-6 mRNA expression was observed (P = 0.003).
Fig. 1.

TLR4 mRNA expression and protein content are increased in muscle-specific (m)TLR4 mice. mTLR4 (n = 4) and WT (n = 4) littermates received an intraperitoneal injection of either saline or LPS at 1 μg/kg body wt to assess the local inflammatory response to endotoxin. Flexor digitorum brevis (FDB) muscle fibers were collected 4 h postinjection, and mRNA levels of IL-6 and TLR-4 were assessed along with TLR-4 protein abundance. mRNA levels (A) and protein content (B) of TLR-4 are significantly elevated in the FDB muscle from mTLR-4 compared with WT mice. FDB IL-6 mRNA is increased to a greater extent in response to an LPS injection in FDB muscle fibers in mTLR-4 mice (C). Data are expressed as means ± SE. *P < 0.05.
Whole body metabolic phenotype of mTLR-4 mice on chow diet.
Mice were maintained on a standard chow diet, and mTLR-4 mice demonstrated a trend toward a greater body weight (Fig. 2A; P = 0.08) and body fat percentage (Fig. 2B; P = 0.12), although these changes did not reach statistical significance. Additionally, muscle-specific TLR-4 overexpression did not alter RER or energy expenditure (Fig. 2, C and D) on this diet. Despite anthropometric similarities, WT mice responded slightly better to a glucose challenge compared with mTLR-4 mice. Glucose tolerance testing revealed significantly elevated blood glucose levels at 90 min following glucose injection in mTLR-4 mice compared with WT (Fig. 2E: 154.2 ± 10.8 vs. 135.7 ± 4.2 mg/dl; P < 0.05). The area under the glucose tolerance curve was also significantly (P < 0.05) higher in mTLR-4 mice compared with WT mice (Fig. 2F).
Fig. 2.

Skeletal muscle TLR-4 over expression induces mild glucose intolerance compared with WT littermates but does not alter body weight, body fat, or energy expenditure on a standard chow diet. mTLR-4 (n = 16) and WT (n = 15) mice were maintained on 12 h light-dark cycles and fed a standard chow diet until 6–8 mo of age. Body mass (A) and body fat (B) were assessed via the Bruker mini spec LF90. Respiratory exchange ratio (C) and energy expenditure (D) were measured over 48-h using indirect calorimetry (TSE Systems, Chesterfield, MO). Following an overnight fast, mice were injected with 1 g/kg dextrose, and blood was measured via the tail vein every 30 min for 2 h to assess glucose tolerance (E and F). Data are expressed as means ± SE. *P < 0.05.
Skeletal muscle substrate metabolism and mitochondrial oxygen consumption in mTLR-4 mice on a standard chow diet.
To determine whether muscle-specific TLR-4 overexpression altered substrate metabolism, we assessed fatty acid oxidation (FAO) in whole muscle homogenates and in isolated mitochondria from red skeletal muscle of mTLR-4 and WT mice. In mice that were fed a standard chow diet, mTLR-4 mice had decreased FAO compared with their WT littermates in red muscle homogenates (Fig. 3A) and a trend toward decreased FAO in isolated mitochondria (P = 0.07; Fig. 3B). Oxidative enzyme activities (Fig. 3, C and D) were also assessed, and a significant decrease in mitochondrial citrate synthase (CS) activity was observed between mTLR-4 and WT mice.
Fig. 3.

Fatty acid oxidation and citrate synthase activity are suppressed in mTLR-4 mice on a standard chow diet. mTLR-4 (n = 9) and their WT (n = 10) littermates were fasted overnight, and mitochondria were isolated from excised red skeletal muscle. Fatty acid oxidation was measured in whole muscle homogenates (A) and isolated mitochondria (B) by assessing [1-14C]-palmitic acid metabolism to 14-CO2 and acid-soluble metabolites. Citrate synthase enzyme activity was assessed in whole muscle (C) and isolated mitochondria (D). Data are expressed as means ± SE. *P < 0.05.
Mitochondria were isolated from red skeletal muscle of mTLR-4 and WT mice for measures of mitochondrial respiration and ROS production. There were no significant differences between mTLR-4 and WT mice in basal (state 2) respiration, ADP-stimulated state 3 respiration, FCCP-stimulated maximal respiration rate, or respiratory control ratio (state 3/state 4 respiration; data not shown). ROS production was similar in complex 1, complex 3, and REV between mTLR-4 and WT mice on the chow diet (data not shown).
The effects of high-fat feeding on the whole body metabolic phenotype of mTLR-4 mice.
High-fat (HF) feeding significantly increased total body mass and body fat in both groups of mice (Fig. 4A) compared with low-fat controls. After 16 wk on the HF diet, mTLR-4 mice gained significantly more body mass and had a greater amount of body fat gain compared with WT high-fat-fed mice (P = 0.01, Fig. 4, B and C). Furthermore, there was a significant genotype × diet interaction between mTLR-4 and WT mice in total body mass and body fat mass gain, indicating that mTLR-4 mice were more susceptible to body weight and fat gain on the high-fat diet compared with WT littermates. Fat-free mass was not different between the two genotypes (data not shown).
Fig. 4.

mTLR-4 mice gain more mass and body fat on a high-fat diet compared with WT littermates. mTLR-4 (n = 15) and WT (n = 15) mice were maintained on 12:12-h light-dark cycles and fed either a 45% high fat (Teklad TD.10505) or control (Teklad TD.10453) diet for 16 wk. Body mass was recorded weekly (A). The effect of the high-fat diet on body mass (B) and body fat (C) gain was genotype-dependent. Data are expressed as means ± SE. *P < 0.05.
At baseline and at 16 wk of HF feeding, mice were placed in calorimetry cages and monitored for 48 h for the measures of energy expenditure (Fig. 5, A and B) and RER (Fig. 5C). The high-fat diet similarly reduced RER in both groups of mice (P < 0.05). Energy expenditure was increased in WT mice in response to high-fat feeding; alternatively, energy expenditure was suppressed in mTLR-4 under HF conditions (P < 0.05). Energy expenditure was expressed relative to fat-free mass. Expressing energy expenditure relative to total body mass and/or total fat mass did not alter the diet × genotype interaction that was observed. There was no significant interaction between diet and genotype in cage activity monitored over 48 h (data not shown).
Fig. 5.
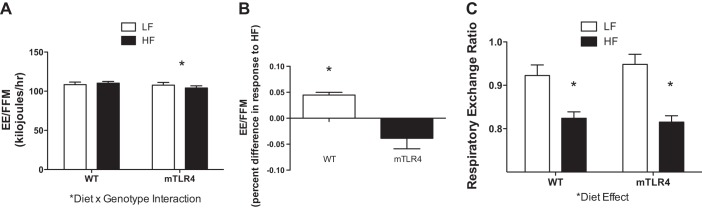
Energy expenditure is increased in WT mice in response to a high-fat diet, whereas mTLR-4 mice exhibit a decrease in energy expenditure following high-fat feeding. Energy expenditure, WT (n = 15) and mTLR4 (n = 15) was measured over 48 h and was expressed per fat free mass, in response to the high-fat diet was dependent on genotype (A and B). Respiratory exchange ratio was determined via indirect calorimetry after 15 wk of high-fat feeding (C) and responded similarly in both genotypes to diet. Data are expressed as means ± SE. *P < 0.05.
The HF diet induced glucose intolerance in both groups of mice; however, the degree of intolerance was dependent on genotype. mTLR-4 mice had significantly greater intolerance compared with the WT mice (Fig. 6, A and B). Furthermore, within the high-fat feeding groups, the mTLR-4 mice experienced greater blood glucose values at 60 and 90 min postinjection compared with WT mice. Sixteen weeks of high-fat feeding did not affect insulin tolerance in WT mice (Fig. 6, C and D); however, the high-fat diet induced mild, yet significant, insulin intolerance in the mTLR-4 mice (P < 0.05).
Fig. 6.

mTLR-4 mice become significantly more glucose intolerant on a high-fat diet compared with WT mice. To assess glucose tolerance mTLR-4 (n = 15) and WT (n = 15), mice were fasted overnight and injected with 1 g/kg body mass of dextrose, and blood glucose was measured via the tail vein at baseline and every 30 min for 2 h (A and B). To assess insulin tolerance, mice were fasted for 4 h and injected with 0.5 U per kg body mass of insulin (Humulin R), and blood glucose was measured via the tail vein at baseline and every 15 min for 1 h (C and D). Data are expressed as means ± SE. *P < 0.05.
The incidence of Type 2 diabetes was defined as two consecutive blood glucose levels above 300 mg/dl (17). On the basis of this definition, the high-fat diet resulted in Type 2 diabetes in all mTLR-4 mice (11/11) but only half of the WT mice (6/12).
The effects of high-fat feeding on skeletal muscle metabolic phenotype of mTLR-4 mice.
When challenged with a high-fat diet for 16 wk, WT mice have a significant increase in FAO in red skeletal muscle homogenates (17.2% higher) and isolated mitochondria (9.7% higher) compared with WT mice on the low-fat diet. Conversely, mTLR-4 mice on the high-fat diet had lower rates of FAO in muscle homogenates (−9.3%) and mitochondria (−8%) compared with the low-fat diet (Fig. 7, A–D). The effect of diet on FAO was significantly dependent on genotype (diet × genotype interaction, P < 0.05).
Fig. 7.

WT mice adapt to high-fat feeding by increasing fatty acid oxidation and oxidative enzyme activity in skeletal muscle, whereas mTLR-4 mice fail to adapt. Fatty acid oxidation was measured in muscle homogenates (A and B) and isolated mitochondria (C and D) from mTLR-4 (n = 15) and WT (n = 15) mice by assessing [1-14C]-palmitic acid metabolism to 14CO2 and acid-soluble metabolites. Maximal activities of oxidative enzymes were measured in isolated mitochondria (E–G). Data are expressed as means ± SE. *P < 0.05.
In addition to FAO, we also sought to examine the response of individual mitochondrial oxidative enzymes to high-fat feeding in mTLR-4 and WT mice (Fig. 7, E–G). MDH activity was significantly increased (P < 0.05) in WT mice, while this adaptation to high-fat feeding was not evident in mTLR-4 mice. We observed that BHAD was lower in mTLR-4 mice on both the high-fat and low-fat diets. This effect of genotype was significant (P = 0.013), but diet composition had no effect on this relationship. No differences were observed with CS activity.
Mitochondrial function from mTLR-4 mice skeletal muscle after HF diet.
Following the HF diet, both WT and mTLR-4 mice responded by increasing basal respiration in isolated mitochondria (data not shown). No differences in genotype or in response to diet were observed in either state 3 or FCCP-stimulated oxygen consumption rate. Respiratory control ratio (state III/state IV), an index of the functional integrity of prepared mitochondria, did not change in response to HF diet in either WT mice or mTLR-4 mice (data not shown).
ROS production experiments showed that the HF diet resulted in a significant increase in ROS production from both complex 1 (34.4% increase; Fig. 8A) and complex 3 (30.0% increase, Fig. 8B) in mTLR-4 mice compared with the low-fat diet; however, this change was not evident in WT mice. Furthermore, there was a significant genotype × diet interaction between mTLR-4 and WT mice, where the mTLR-4 mice on the HF diet had a significantly greater increase in ROS production at complex 3. To determine whether alterations in mitochondrial ROS production have a discernible effect on cellular oxidative stress, we used Western blot analysis to probe for the presence of by-products of lipid peroxidation. Red skeletal muscle preparations were probed with antibodies against 4-HNE, a marker of cellular oxidative stress. Chemiluminescence detection revealed prominent bands at 15, 55, and 60 kDa (Fig. 8C). The abundance and intensity of these bands are indicative of greater lipid peroxidation and were higher in the mTLR-4 mice, which is exacerbated under the high-fat fed condition.
Fig. 8.

ROS production and lipid peroxidation in skeletal muscle are increased in mTLR-4 mice following a high-fat diet. ROS production from complex 1 (A) and complex 3 (B) in isolated mitochondria from mTLR4 (n = 15) and WT (n = 15) mice was assessed via an Amplex Red hydrogen peroxide/peroxidase assay kit. Thirty micrograms of whole muscle homogenates were loaded onto a SDS-PAGE gel and transferred to a PVDF membrane to be probed for 4-HNE as a marker of cellular lipid peroxidation. GAPDH was utilized as a loading control (C). Data are expressed as means ± SE. *P < 0.05.
DISCUSSION
The primary objectives of these studies were to determine whether elevated TLR-4 levels in mouse skeletal muscle influence systemic and skeletal muscle metabolism in the context of chow-fed and high-fat feeding conditions. While on a chow diet, mTLR-4 mice displayed blunted FAO and mild glucose intolerance, compared with WT littermates, despite similarities in whole body metabolism (RER and energy expenditure) and anthropometric indices (body weight and body fat). Importantly, when placed on a high-fat diet, mTLR-4 mice are characterized by 1) a greater propensity to gain weight and body fat, 2) significantly worsened glucose tolerance, 3) an inability to increase red muscle fatty acid oxidative capacity, and 4) increased ROS production and lipid peroxidation (a marker of oxidative stress) compared with their WT littermates. We have previously reported that TLR-4 activation in skeletal muscle induces a metabolic phenotype reminiscent to that observed in obese humans, where fatty acids are partitioned to storage pathways rather than oxidation under fasting conditions (5). The current study is the first to specifically target, and overexpress, TLR-4 levels in red skeletal muscle and corroborates the role for TLR-4 in mediating the detrimental effects of high-fat feeding on skeletal muscle metabolism.
Davis et al. (4) have previously reported that mice lacking TLR-4 are protected from the obesogenic effects of a high-fat diet, but only when the diet is specifically high in saturated fat (SFA). When fed a diet high in unsaturated fat, the WT and TLR4-deficient mice responded similarly. Notably, the diet used in our study (45% fat) is similar to the high SFA diet used in Davis's studies, suggesting that the effects that we observed in the mTLR-4 mice may be more dependent on the high-saturated nature of the diet rather than high calorie consumption alone. In support of this notion, the literature is quite extensive in regard to the effects of SFA on TLR-4 (2, 3, 9, 10). The utilization of the diet chosen in our study was due to the fact that it adequately represents the Western diet that has gained much attention due its potent obesogenic and prodiabetic effects. Although we did not assess food consumption directly in these studies, we observed that the high-fat diet had a profoundly differential effect on glucose tolerance in mTLR-4 mice (all became diabetic) compared with their WT littermates (half became diabetic). In accordance with this finding, Shi et al. (15) have previously shown that TLR-4 links excess fatty acid exposure to insulin resistance. The overall relevance of our findings are important since overconsumption often occurs in concert with increased SFA consumption, relative to other macronutrients, and it is best suited to understand how muscle TLR-4 drives Western diet-associated metabolic syndrome.
The phenotypic differences observed between mTLR-4 and WT mice were only moderate unless challenged with a high-fat diet. Body mass, fat mass, energy expenditure, skeletal muscle inflammatory gene expression (IL-6 and monocyte chemoattractant protein-1) and FAO in isolated mitochondria were all similar between the two groups on a chow diet, with the exception of CS activity, which we found to be lower in isolated mitochondria but not different in the whole muscle homogenates. No differences in CS activity in the whole muscle would be suggestive of no differences in mitochondrial content; however, the fact that CS activities are lower in isolated mitochondria in mTLR-4 mice suggest that TCA flux in isolated mitochondria of these mice may be impaired. The decrements in CS activity may have laid the foundation for the metabolic maladaptation in mTLR-4 mice when administered a chronic dietary intervention. Indeed, when subjected to a high-fat diet, we observed significant phenotypic differences in many of our outcome measures, including body weight and fat, glucose tolerance, red skeletal muscle fatty acid oxidation, and oxidative damage. Glucose tolerance was significantly, but only modestly, different on chow, but was notably significantly different between the genotypes under high-fat feeding conditions. The observation that mTLR-4 mice differ from WT predominantly on a high-fat diet suggests that elevated muscle TLR-4 alone is not enough to produce whole body or local metabolic dysregulation; instead, an interaction must exist between TLR-4 and elevated fat consumption and delivery of fatty acids to peripheral tissues to drive these whole body and/or local metabolic dysregulations.
To determine whether the observed increases in ROS in the mTLR-4 HF mice translated into oxidative damage, we measured the presence of 4-HNE in red skeletal muscle. 4-HNE is formed during lipid peroxidation, and its levels are known to be elevated in response to oxidative stress (19). Indeed, we found that high-fat feeding in WT mice had little to no effect on 4-HNE production, but 4-HNE production increased significantly in the mTLR-4 mice in response to the HFD. The increase in ROS production and subsequent oxidative damage that we observed in the mTLR-4 mice may be a mechanism linking excessive fat consumption to the resultant metabolic dysregulation. In accordance with this hypothesis, it has been shown that ROS production has profound effects on cellular function (7, 11), and TLR-4 has been implicated in the production of excess mitochondrial ROS production. In addition, using antioxidants to prevent oxidative damage has been shown to prevent TLR-4-mediated effects on substrate metabolism (6). Thus, the role of TLR-4 in promoting metabolic disease in the context of excessive fat consumption may lie in its production of excessive ROS. More work specifically manipulating ROS production in our model would need to be conducted to definitively make this conclusion, and future studies will target this mechanism.
In conclusion, we report that overexpression of TLR-4 in skeletal muscle results in an impaired capacity to adequately respond to a high-fat diet. When challenged with excessive fat consumption, WT mice “increase” FAO and energy expenditure, whereas mTLR-4 mice were not able to increase FAO and had “decreased” energy expenditure. These metabolic adaptations, or lack thereof, resulted in significantly greater weight gain, fat gain, and glucose intolerance in mTLR-4 mice.
Perspectives and Significance
Herein, we introduce for the first time a novel mouse model that selectively overexpresses TLR-4 in skeletal muscle. Our goal was to characterize the whole body and skeletal muscle metabolic phenotype that exists within this model in the context of both chow and high-fat feeding. The results from this study lend credence to TLR-4 as a potential mediator of high-fat diet-induced metabolic dysfunction. Specifically, TLR-4-mediated ROS production and resultant oxidative damage may be a highly important contributing factor to symptoms of metabolic syndrome that commonly accompany the consumption of a Western diet. Future studies should be undertaken to determine whether interventions to curb high-fat diet-induced ROS production are sufficient to rescue the maladaptive effects of overfeeding in mTLR-4 mice.
GRANTS
This work was funded by grants from the American Diabetes Association (1-JF-05-24, to M. Hulver) and the National Institutes of Health-NIDDK (2RO1 DK-078765, to M. Hulver).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: R.P.M., R.L.M., M.I.F., and M.W.H. conception and design of research; R.P.M., Y.W., K.V., G.F., J.K., J.R.S., E.S., M.A., M.H., and N.E.B. performed experiments; R.P.M., Y.W., K.V., G.F., J.K., and M.W.H. analyzed data; R.P.M., M.I.F., and M.W.H. interpreted results of experiments; R.P.M. prepared figures; R.P.M. drafted manuscript; R.P.M., Y.W., K.V., A.S.A., N.E.B., M.I.F., and M.W.H. edited and revised manuscript; M.W.H. approved final version of manuscript.
REFERENCES
- 1.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmee E, Cousin B, Sulpice T, Chamontin B, Ferrieres J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56: 1761–1772, 2007. [DOI] [PubMed] [Google Scholar]
- 2.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57: 1470–1481, 2008. [DOI] [PubMed] [Google Scholar]
- 3.Chait A, Kim F. Saturated fatty acids and inflammation: who pays the toll? Arterioscler Thromb Vasc Biol 30: 692–693, 2010. [DOI] [PubMed] [Google Scholar]
- 4.Davis JE, Gabler NK, Walker-Daniels J, Spurlock ME. Tlr-4 deficiency selectively protects against obesity induced by diets high in saturated fat. Obesity (Silver Spring) 16: 1248–1255, 2008. [DOI] [PubMed] [Google Scholar]
- 5.Frisard MI, McMillan RP, Marchand J, Wahlberg KA, Wu Y, Voelker KA, Heilbronn L, Haynie K, Muoio B, Li L, Hulver MW. Toll-like receptor 4 modulates skeletal muscle substrate metabolism. Am J Physiol Endocrinol Metab 298: E988–E998, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frisard MI, Wu Y, McMillan RP, Voelker KA, Wahlberg KA, Anderson AS, Boutagy N, Resendes K, Ravussin E, Hulver MW. Low levels of lipopolysaccharide modulate mitochondrial oxygen consumption in skeletal muscle. Metabolism 64: 416–427, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 114: 1752–1761, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerencser AA, Neilson A, Choi SW, Edman U, Yadava N, Oh RJ, Ferrick DA, Nicholls DG, Brand MD. Quantitative microplate-based respirometry with correction for oxygen diffusion. Anal Chem 81: 6868–6878, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang S, Rutkowsky JM, Snodgrass RG, Ono-Moore KD, Schneider DA, Newman JW, Adams SH, Hwang DH. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J Lipid Res 53: 2002–2013, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim F, Pham M, Luttrell I, Bannerman DD, Tupper J, Thaler J, Hawn TR, Raines EW, Schwartz MW. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ Res 100: 1589–1596, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Kirkinezos IG, Moraes CT. Reactive oxygen species and mitochondrial diseases. Sem Cell Dev Biol 12: 449–457, 2001. [DOI] [PubMed] [Google Scholar]
- 12.Kolek MJ, Carlquist JF, Muhlestein JB, Whiting BM, Horne BD, Bair TL, Anderson JL. Toll-like receptor 4 gene Asp299Gly polymorphism is associated with reductions in vascular inflammation, angiographic coronary artery disease, and clinical diabetes. Am Heart J 148: 1034–1040, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Reyna SM, Ghosh S, Tantiwong P, Meka CS, Eagan P, Jenkinson CP, Cersosimo E, Defronzo RA, Coletta DK, Sriwijitkamol A, Musi N. Elevated toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes 57: 2595–2602, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saitoh S. Chaperones and transport proteins regulate TLR4 trafficking and activation. Immunobiology 214: 594–600, 2009. [DOI] [PubMed] [Google Scholar]
- 15.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116: 3015–3025, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stump CS, Henriksen EJ, Wei Y, Sowers JR. The metabolic syndrome: role of skeletal muscle metabolism. Ann Med 38: 389–402, 2006. [DOI] [PubMed] [Google Scholar]
- 17.Tersey SA, Nishiki Y, Templin AT, Cabrera SM, Stull ND, Colvin SC, Evans-Molina C, Rickus JL, Maier B, Mirmira RG. Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 61: 818–827, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, Prada PO, Hirabara SM, Schenka AA, Araujo EP, Vassallo J, Curi R, Velloso LA, Saad MJ. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 56: 1986–1998, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res 42: 318–343, 2003. [DOI] [PubMed] [Google Scholar]
- 20.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest 115: 1111–1119, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest 112: 1785–1788, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]