

Abstract
Context:
Lithium carbonate is a psychiatric medication commonly used in the treatment of bipolar disorder. It has been implicated in inducing nephrogenic diabetes inspidus, chronic tubulointerstitial nephropathy, and acute tubular necrosis. We describe a case of lithium-induced minimal change disease (MCD) and acute kidney injury (AKI).
Case Report:
A 32-year-old female with a medical history of bipolar disorder treated with chronic lithium therapy presented with anasarca, fatigue, and tremors. Work-up revealed supra-therapeutic lithium levels, hypoalbuminemia, and significant proteinuria. The patient was treated conservatively with fluids and discontinuation of lithium therapy. Subsequently, she developed significant AKI and persistent proteinuria. She underwent a renal biopsy that demonstrated effacement of podocyte foot processes consistent with lithium-induced MCD. This was treated with corticosteroids, which decreased the proteinuria and resolved all the patient's symptoms.
Conclusion:
Lithium-induced MCD is a rare disease that affects patients of all ages. It is often associated with therapeutic lithium and is typically resolved with discontinuation of lithium. In some cases, concurrent AKI may result due to vascular obstruction from hyperalbuminuria and associated renal interstitial edema. Corticosteroids may be needed to reduce the proteinuria and prevent progression to chronic kidney disease. As such, patients on lithium therapy may benefit from monitoring of glomerular function via urinalysis to prevent the onset of nephrotic syndrome.
Keywords: Lithium, minimal change disease, nephrotic syndrome, proteinuria, renal insufficiency
Introduction
Nephrotic syndrome is characterized by proteinuria of >3 g/day, hypoalbuminemia, edema, and hyperlipidemia/lipiduria.[1] Several subtypes of this syndrome exist such as focal segmental glomerulosclerosis, membranous nephropathy, and minimal change disease (MCD). MCD is the most common presentation of nephrotic syndrome in the preadolescent population and accounts for up to 15% of primary nephrotic syndrome in adults.[1] Although most causes of MCD are idiopathic, extraglomerular processes such as toxic reactions to drugs can also precipitate the disease in susceptible patients.[1] Specifically, several cases of lithium-induced nephrotic syndrome have been reported in literature.[2,3,4,5,6,7,8,9] Historically, lithium is commonly associated with nephrogenic diabetes inspidus, chronic tubulointerstitial nephropathy, and acute tubular necrosis.[2,3] Rarely, an idiosyncratic reaction has been established between lithium use and development of proteinuria and nephrotic syndrome.[2,3,4,5,6,7,8,9] Since Duffot and colleagues first identified the association of this metal to nephrotic syndrome in 1973, there have been 24 cases cited in worldwide literature.[2,3,4,5,6,7,8,9] Most cases have linked nephrotic syndrome to therapeutic serum lithium levels and the majority of cases have demonstrated prompt reversal of proteinuria upon discontinuation of lithium. We describe a case of a young woman with no evidence of preexisting renal disease that developed MCD on supratherapeutic lithium levels in whom the renal injury worsened after discontinuation of lithium. The purpose of this case is to stress the importance of close follow-up of renal and glomerular function of patients on acute as well as chronic lithium therapy.
Case Presentation
A 32-year-old female presented to the emergency room with a chief complaint of marked edema of 5 days' duration. Past medical history included: Bipolar disorder, panic disorder, hypothyroidism, and hidradenitis suppurativa. Her daily home medications included lithium carbonate 900 mg, synthroid 75 μg, cymbalata 60 mg, nortryptiline 30 mg, cipralex 30 mg, gabapentin 600-900 mg, and ibuprofen 200 mg. The patient had been on a stable dose of lithium for approximately 5 years experiencing no side effects. She presented to the hospital with a 5-day history of severe fatigue, migraine-like headaches, lethargy, and significant anasarca. Associated with these symptoms were upper extremity tremors as well as polyuria and polydipsia. She denied chest pain, shortness of breath, constitutional symptoms, and any urinary symptoms. Social history included 10-pack year history of tobacco.
On admission, the patient's vital signs were: Blood pressure 115/61, heart rate 61 beats/min, respiratory rate 20 breaths/min, and oxygen saturation 96% on room air. The patient was afebrile. There was generalized + 2 edema of lower extremities, upper extremities, and periorbital regions. Minimal bibasilar crackles were appreciated. The remainder of the cardiovascular and abdominal examination was unremarkable.
Blood work showed a plasma creatinine level of 119 μmol/L (baseline 69 μmol/L) and a blood urea of 10.4 mmol/L. Urine dipstick showed a specific gravity of 1.016, pH 7.5, and 4 + protein; and was negative for glucose, ketones, and nitrites. Microscopy revealed oval fat bodies and free fat droplets. The urine albumin to creatinine ratio was 973.4 mg/mmol and 24-h urine protein was 15.69 g. On admission, serum lithium level was 2.66 mmol/L (therapeutic 0.50-1.50 mmol/L). Serum sodium was 130 mmol/L, potassium 4.5 mmol/L, chloride 105 mmol/L, and bicarbonate 27 mmol/L. A complete blood count demonstrated hemoglobin 106 g/L, white blood cell count 9.46 × 109 /L, and platelet count 234 × 109 /L. Serum albumin was low at 20 g/L; but all liver enzymes, total bilirubin, and coagulation studies were within normal limits. Thyroid-stimulating hormone was 18.90 mU/L and free T4 8.2 pmol/L. Serology for hepatitis B and C, anti-double stranded deoxyribonucleic acid, antineutrophilic cytoplasmic antibodies, ribonucleoprotein, Sjogren's syndrome, scleroderma, inflammatory myositis, and Smith antibody were all negative. Antinuclear antibody was slightly positive on admission. Chest radiographs revealed nonspecific bibasilar infiltrates. Abdominal ultrasound demonstrated normal kidneys with no renal calculi or hydronephrosis.
Lithium treatment was discontinued immediately and the patient was treated conservatively with fluids. Initially the patient refused renal biopsy. Once stabilized, she was discharged from the hospital and lithium remained discontinued. At 2 months follow-up, the patient experienced worsening anasarca, significant acute kidney injury (AKI; serum creatinine 209 μmol/L), and persistent proteinuria. She was readmitted to the hospital and subsequently underwent a renal biopsy, when at the time her creatinine peaked at 348 μmol/l. On light microscopy, there was no global sclerosis, crescent formation, or necrotic lesions capillary wall thickness was normal. There was evidence of moderate focal interstitial fibrosis [Figure 1a]. Immunofluorescence did not demonstrate any immune deposits. Electron microscopy revealed widespread fusion of podocyte foot processes affecting more than 75% of the surface of glomerular capillary loops [Figure 1b]. No electron dense deposits were evident. A diagnosis of MCD secondary to lithium use was established and the patient was started on high dose corticosteroids, initially intravenously, but changed to oral prednisone 80 mg daily on discharge. Over the next 2 months of follow-up, the patient's creatinine, proteinuria, and edema all decreased significantly and she was tapered to oral prednisone 40 mg. At her 4 months follow-up, her albumin/creatinine ratio had decreased to 220 mg/mmol and she remained on oral prednisone 40 mg daily. Her edema as well as AKI had resolved. The progression of renal and glomerular failure is illustrated in Figure 2.
Figure 1.
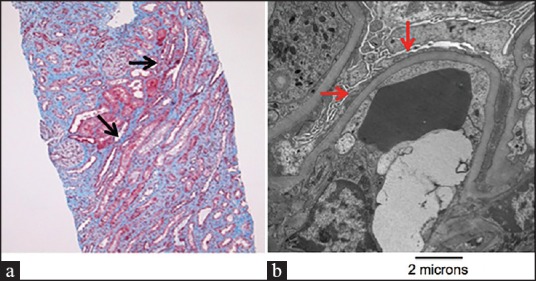
Light and electron microscopic evidence of minimal change disease. (a) Light microscopy identifying focal interstitial fibrosis and cellular mesangial expansion (black arrows). No evidence of ischemic injury to tubular structures. Developed with periodic acid-Schiff stain. (b) Electron microscopic demonstration of widespread podocyte effacement (red arrows). Magnification, ×12,000
Figure 2.
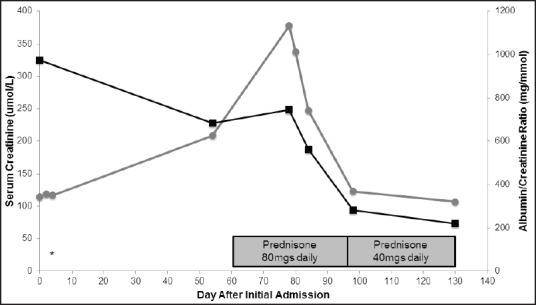
Serum creatinine and albumin/creatinine ratio throughout the hospitalization and post-hospitalization follow-up. Progression of serum creatinine and albumin/creatinine ratio from first day of hospitalization to current follow-up. The start of prednisone is marked. * Indicates the day of lithium discontinuation. ▪ indicates albumin/creatinine levels. • Indicates serum creatinine levels
Discussion
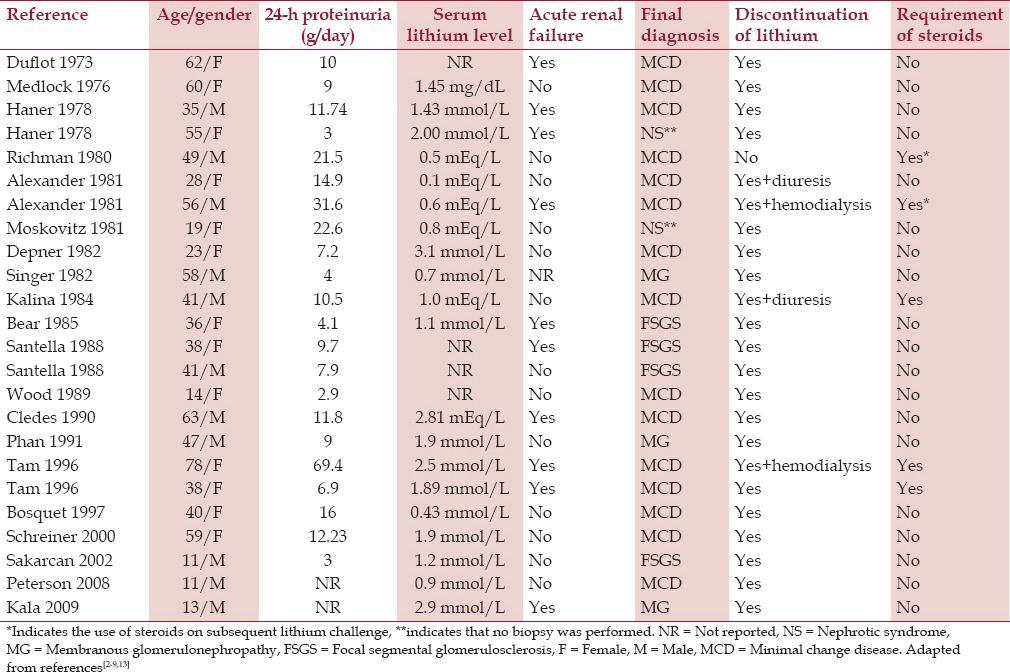
Lithium-induced nephrotic syndrome is an idiosyncratic reaction that has affected the adult and pediatric population as summarized in Table 1.[2,3,4,5,6,7,8,9] Nephrotic syndrome has been associated with therapeutic lithium levels in all but 32% of the cases. A majority of cases have demonstrated complete reversal of proteinuria upon discontinuation of lithium providing evidence for lithium as the potential etiology of the initial renal insult. In cases of persistent proteinuria despite discontinuation of the medication, steroids have been prescribed to prevent chronic renal failure. Additionally, 42% of cases demonstrated concurrent AKI at time of admission. However, to our knowledge, we demonstrate the first case in which the AKI presented at follow-up after discontinuation of lithium use. This may be explained by the hypothesis of nephrosarca. Defined as interstitial edema of the kidney, in association with high degree of albuminuria, nephrosarca may have caused physical obstruction of vasculature and tubular structures resulting in florid AKI.[10,11] Specifically, histological interstitial edema and improvement with diuresis therapy are criteria for the diagnosis of nephrosarca, although highly controversial.[10] Of note there was no tubular injury or marked interstitial nephritis seen on biopsy as is often seen in MCD and AKI associated with extrinsic factors such as nonsteroidal anti-inflammatory use.[12] There was however chronic interstitial fibrosis noted, likely as the result of long-term lithium use in this patient.
Table 1.
All reported cases in worldwide literature of lithium-induced nephrotic syndrome
The pathophysiological mechanism by which lithium induces nephrotic syndrome is not yet delineated. The three-layered permselective barrier consisting of the glomerular capillary, glomerular basement membrane, and podocyte foot processes contains anionic sulfated glycosaminoglycans. It is hypothesized that the cationic lithium may interact with these anionic sites, thereby disrupting the charge barrier and inducing albuminuria.[2,3,4,5,6,7,8,13] Furthermore, toxic circulating lymphokines are associated with damage to the glomerular basement membrane resulting in MCD.[14] It is hypothesized that lithium may induce MCD by activating T lymphocytes and cytokines such as tumor necrosis factor.[13] Lithium is a well-known modulator of the phosphoinositol pathway, which is involved in T cell stimulation.[14] Activating this pathway may result in excessive cytotoxic T cells that may disrupt the selective barrier of the glomerular basement membrane and podocyte foot processes resulting in marked proteinuria and MCD.
Mild proteinuria, presumably from glomerular damage, is a relatively common complication of lithium use. It is estimated up to 42% of patients on lithium may have greater than 1.0 g/day proteinuria.[15] As more evidence suggests lithium-induced proteinuria and nephrotic syndrome, it is inevitable that close follow-up with renal function tests as well as urine dipstick analysis is required to prevent chronic renal failure. Historical data suggests most patients will respond to lithium withdrawal and in rare cases, corticosteroids may be required to reverse the proteinuria. There have been several cases that have demonstrated persistence of proteinuria despite steroid use suggesting alternative hypotheses such as nephrosarca. As such, the importance of early biopsy and early treatment with hemodialysis or aggressive diuresis may prevent progression of AKI. Further research is necessary to better understand the mechanism of concurrent lithium-induced MCD and AKI.
Acknowledgments
We would like to thank Dr. Serge Jothy MD (Pathology, University of Toronto) for providing light microscopy and electron microscopy images.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Waldman M, Crew JR, Valeri A, Busch J, Stokes B, Markowitz G, et al. Adult minimal-change disease: Clinical characteristics, treatment, and outcomes. Clin J Am Soc Nephrol. 2007;2:445–53. doi: 10.2215/CJN.03531006. [DOI] [PubMed] [Google Scholar]
- 2.Bosquet S, Desscombes E, Gauthier T, Fellay G, Regamey C. Nephrotic syndrome during lithium therapy. Nephrol Dial Transplant. 1997;12:2728–31. doi: 10.1093/ndt/12.12.2728. [DOI] [PubMed] [Google Scholar]
- 3.Alexander MP, Farag YM, Mittal BV, Rennke HG, Singh AK. Lithium toxicity: A double-edged sword. Kidney Int. 2008;73:233–7. doi: 10.1038/sj.ki.5002578. [DOI] [PubMed] [Google Scholar]
- 4.Schreiner A, Waldherr R, Rohmeiss P, Hewer W. Focal segmental glomeruloscerlosis and lithium treatment. Am J Psychiatry. 2000;157:834. doi: 10.1176/appi.ajp.157.5.834. [DOI] [PubMed] [Google Scholar]
- 5.Sakarcan A, Thomas DB, O'Reily KP, Richards RW. Lithium-induced nephrotic syndrome in a young pediatric patient. Pediatr Nephrol. 2002;17:290–2. doi: 10.1007/s00467-001-0809-7. [DOI] [PubMed] [Google Scholar]
- 6.Petersen CE, Amaral S, Frosch E. Lithium-induced nephrotic syndrome in a prepubertal boy. J Child Adolesc Psychopharmacol. 2008;18:210–3. doi: 10.1089/cap.2007.0118. [DOI] [PubMed] [Google Scholar]
- 7.Kala GK, Mogri M, Weber-Shrikant E, Springate JE. Lithium-induced membranous glomerulonephropathy in a pediatric patient. Pediatr Nephrol. 2009;24:2267–9. doi: 10.1007/s00467-009-1245-3. [DOI] [PubMed] [Google Scholar]
- 8.Wood IK, Parmelee DX, Foreman JW. Lithium-induced nephrotic syndrome. Am J Psychiatry. 1989;146:84–7. doi: 10.1176/ajp.146.1.84. [DOI] [PubMed] [Google Scholar]
- 9.Richman AV, Masco HL, Rifkin SI, Acharya MK. Minimal-change disease and the nephrotic syndrome associated with lithium therapy. Ann Intern Med. 1980;92:70–2. doi: 10.7326/0003-4819-92-1-70. [DOI] [PubMed] [Google Scholar]
- 10.Lowenstein J, Schacht RG, Baldwin DS. Renal failure in minimal change nephrotic syndrome. Am J Med. 1981;70:227–33. doi: 10.1016/0002-9343(81)90754-3. [DOI] [PubMed] [Google Scholar]
- 11.Furuya R, Kumagai H, Ikegaya N, Kobayashi S, Kimura M, Hishida A, et al. Reversible acute renal failure in idiopathic nephrotic syndrome. Intern Med. 1993;32:31–5. doi: 10.2169/internalmedicine.32.31. [DOI] [PubMed] [Google Scholar]
- 12.Warren GV, Korbet SM, Schwartz MM, Lewis EJ. Minimal change glomerulopathy associated with nonsteroidal antiinflammatory drugs. Am J Kidney Dis. 1989;13:127–30. doi: 10.1016/s0272-6386(89)80130-1. [DOI] [PubMed] [Google Scholar]
- 13.Tam VK, Green J, Schwieger J, Cohen AH. Nephrotic syndrome and renal insufficiency associated with lithium therapy. Am J Kidney Dis. 1996;27:715–20. doi: 10.1016/s0272-6386(96)90108-0. [DOI] [PubMed] [Google Scholar]
- 14.Williams RS, Harwood AJ. Lithium therapy and signal transduction. Trends Pharmacol Sci. 2000;21:61–4. doi: 10.1016/s0165-6147(99)01428-5. [DOI] [PubMed] [Google Scholar]
- 15.Markowtiz GS, Radhakrishnan J, Kambham N, Valeri AM, Hines WH, D'Agati VD. Lithium nephrotoxicity: A progressive combined glomerular and tubulointerstitial nephropathy. J Am Soc Nephrol. 2000;11:1439–48. doi: 10.1681/ASN.V1181439. [DOI] [PubMed] [Google Scholar]