

Abstract
Purpose
Myocilin (MYOC) is a well-established primary open-angle glaucoma (POAG) risk gene, with rare variants known to have high penetrance. The most common clinically relevant risk variant, Gln368Ter, has an allele frequency of 0.1% to 0.3% in populations of European ancestry. Detection of rare MYOC variants has traditionally been conducted using Sanger sequencing. Here we report the use of genotyping arrays and imputation to assess whether rare variants including Gln368Ter can be reliably detected.
Methods
A total of 1155 cases with advanced POAG and 1992 unscreened controls genotyped on common variant arrays participated in this study. Accuracy of imputation of Gln368Ter variants was compared with direct sequencing. A genome-wide association study was performed using additive model adjusted for sex and the first six principal components.
Results
We found that although the arrays we used were designed to tag common variants, we could reliably impute the Gln368Ter variant (rs74315329). When tested in 1155 POAG cases and 1992 controls, rs74315329 was strongly associated with risk (odds ratio = 15.53, P = 1.07 × 10−9). All POAG samples underwent full sequencing of the MYOC gene, and we found a sensitivity of 100%, specificity of 99.91%, positive predictive value of 95.65%, and negative predictive value of 100% between imputation and sequencing. Gln368Ter was also accurately imputed in a further set of 1801 individuals without POAG. Among the total set of 3793 (1992 + 1801) individuals without POAG, six were predicted (probability > 95%) to carry the risk variant.
Conclusions
We demonstrate that some clinically important rare variants can be reliably detected using arrays and imputation. These results have important implications for the detection of clinically relevant incidental findings in ongoing and future studies using arrays.
Keywords: primary open-angle glaucoma, MYOC, Gln368Ter, rare variants, imputation
This study demonstrated that Gln368Ter variant, a rare variant within MYOC gene strongly associated with primary open-angle glaucoma, can be accurately screened by imputation using common single nucleotide polymorphisms (SNPs) directly genotyped on genotyping arrays.
Glaucoma is a major cause of blindness worldwide. Primary open-angle glaucoma (POAG; Online Mendelian Inheritance in Man [OMIM] 137760) is the most common subtype of glaucoma, which is characterized by a progressive loss of peripheral vision, although patients may remain undiagnosed until central vision is affected.1,2 Treatment to lower intraocular pressure delays the progression of visual field loss. Several genetic loci have been associated with POAG in linkage and genome-wide association studies (GWAS).3–9 Mutations in the myocilin (MYOC) gene (OMIM 601652) have been reported in different populations and found to account for 2% to 5% of unselected POAG patients.10 Gln368Ter is the most common mutation in populations of European ancestry and confers a high risk of POAG.3,11,12 The Gln368Ter mutation has been observed across multiple populations,11 and was shown to be associated with an average onset of POAG in the fifth and sixth decades.13 Previous studies in 15 Australian families, a large French Canadian family, and two unrelated French Canadian families suggested that this mutation has derived from a common ancestor, showing a founder effect.14,15 Detection of the Gln368Ter mutation is clinically important as it allows for early diagnosis and intervention. However, the risk allele has a frequency of approximately 0.09% to 0.1% among multiple ethnicities (http://www.ncbi.nlm.nih.gov/clinvar/variation/7949/, http://exac.broadinstitute.org/variant/1-171605478-G-A [both in the public domain]), 0.1% in the European American population (http://evs.gs.washington.edu/EVS/ [in the public domain]), and 0.26% in the 1000 Genomes phase 1 European population. This is remarkably similar to the Gln368Ter frequency of 0.09% found in the Blue Mountains Eye Study consisting predominantly of European Australians.16 Sanger sequencing is traditionally used to detect this mutation, and it is not directly genotyped on commonly used genotyping arrays.
Genome-wide association studies have identified thousands of common variants (i.e., variants with a minor allele frequency [MAF] > 5%) associated with human complex diseases17 (http://www.genome.gov/gwastudies/ [in the public domain]). Together the GWAS hits from common variants explain little genetic variance of complex traits, resulting in the “missing heritability” problem.18–20 The heritability of POAG and its endophenotypes including intraocular pressure and vertical cup-to-disc ratio was estimated at 0.81, 0.42, and 0.66, respectively, in a previous study from our group.21 Since the identified genetic variants contributing to the risk of POAG and its endophenotypes explain a small proportion of the genetic variance, missing heritability is an important issue for POAG as it is for many other complex traits. Part of the missing heritability may be due to excluding the rare variants (MAF < 5%) from the standard GWAS.18–20 Although next-generation sequencing technologies have enabled efficient identification of rare variants,22 the cost of sequencing is high, limiting sample size in many situations and leading to low statistical power to identify rare variants associated with complex traits.23
Genotype imputation is a less expensive approach to impute genotypes of untyped genetic variants. However, one study showed that the proportion of well-imputed single nucleotide polymorphisms (SNPs) (imputation quality score [INFO] > 0.4) was only 69%, 60%, and 49% for SNPs with MAF from 0.3% to 5% for individuals genotyped on Omini1M, HumanHap 610, and Illumina 317k arrays, respectively, where 1000 Genomes pilot was used as the reference panel for imputation.24 However, none of the very rare variants (MAF < 0.3%) were well imputed.24 In addition, given that statistical power is proportional to allele frequency and imputation quality, standard GWAS may be underpowered to test low-frequency imputed variants. Methods for association analysis of imputed rare variants are mainly based on combining information across the rare variants within a gene or pathway while accounting for genotype uncertainty due to the imputation.25,26 Thus, it remains unclear whether variants with MAF < 0.3% can be accurately imputed and used in GWAS.
We previously performed a GWAS for POAG using the variants with MAF > 1% imputed to the 1000 Genomes phase 1,7 and in this present study, we explicitly considered the accuracy of imputing rare variants (MAF < 1%) including the Gln368Ter mutation using common variants captured on genotyping arrays. We then investigated whether it is possible to detect the previously established association of the Gln368Ter mutation with POAG from imputed data using a standard GWAS, and whether we can detect other GWAS hits for POAG using imputed rare variants.
Methods
Study Design
In total, 1155 cases with advanced POAG from the Australian and New Zealand Registry of Advanced Glaucoma (ANZRAG) were available for this study, of whom 618 were genotyped on Illumina Omni1M and 537 were genotyped on Illumina OmniExpress array. Controls included 1992 individuals drawn from the Australian Cancer Study (225 esophageal cancer cases, 317 Barrett's esophagus cases, and 552 controls genotyped on Illumina HumanOmni1-Quad) or from a study of inflammatory bowel diseases (303 cases and 595 controls genotyped on Illumina HumanOmniExpressExome). The cohort detail and diagnostic criteria have been previously published.7,27 The data from cases and controls were merged and cleaned (see details below), and the overlapping SNPs between the arrays were used as the basis of imputation to the 1000 Genomes phase 1 reference panel and subsequent GWAS. The research followed the tenets of the Declaration of Helsinki. All participants provided written informed consent. Approval was obtained from the Human Research Ethics Committees of Southern Adelaide Health Service/Flinders University, University of Tasmania, QIMR Berghofer Institute of Medical Research (Queensland Institute of Medical Research), and the Royal Victorian Eye and Ear Hospital.
Quality Control (QC)
The QC was performed in PLINK28 (http://pngu.mgh.harvard.edu/∼purcell/plink/ [in the public domain]) by removing individuals with more than 3% missing genotypes, SNPs with call rate < 97%, MAF < 1%, and Hardy-Weinberg equilibrium P < 0.0001 in controls and P < 5 × 10−10 in cases. The same QC protocol was used before merging the cases and controls to avoid mismatches between the merged datasets. Following merging, the genotypes for 569,249 SNPs common to the arrays were used for subsequent analyses. The autosomal markers were used to compute identity by descent in PLINK, with one of each pair of individuals with relatedness of >0.2 removed. The smartpca package from EIGENSOFT software (http://www.hsph.harvard.edu/alkes-price/software/ [in the public domain]) was used to compute principal components for all participants and reference samples of known northern/western European ancestry (1000 Genomes British, CEU [Utah Residents with Northern and Western European Ancestry], Finland participants).29,30 Ancestry outliers with PC1 or PC2 values > 6 standard deviations from the known northern/western European ancestry group were excluded.
Imputation
We used IMPUTE231 to perform imputation with the 1000 Genomes phase 132 (March 2012 release) as the reference panel. The worldwide reference panel was used, with SNPs with a MAF < 0.1% in Europeans filtered out. Imputation was performed in 1-Mb sections with the recommended settings for IMPUTE2 including a 250-kb buffer flanking imputation sections and the effective size of the sampled population as 20,000.31 Imputation quality can be objectively assessed by the average concordance between input SNPs genotypes and their “best guess” genotypes imputed from the surrounding SNPs; we achieved a very acceptable ≥0.95 across the genome. Single nucleotide polymorphisms with an INFO < 0.4 were discarded. The imputation maximum posterior probability was used to assign the best guess imputed genotypes for rs74315329 and two SNPs in linkage disequilibrium (LD) with rs74315329 (measurement of the degree to which alleles at two genetic loci are associated, where r2 = 0 indicates independent alleles and r2 = 1 indicates completely correlated alleles), rs187423359, and rs182384379 (r2 = 0.5 with rs74315329), with setting the threshold of calling genotypes to 0.6.
Statistical Analysis
Of the SNPs with INFO > 0.4 from imputation, only very well-imputed SNPs (INFO > 0.8) were carried forward for association analysis. SNPTEST33,34 was used to perform association testing on the imputed data using additive model (-frequentist 1) and full dosage scores (-method expected) adjusting for sex and the first six principal components. Genomic inflation factor lambda (λ) was calculated to investigate the presence of population stratification and inflation. The P values were corrected for genomic inflation factor λ (1.06) by dividing the χ2 values by 1.06.
Sequencing
We screened the POAG cases in this study for the Gln368Ter mutation using direct sequencing as previously described.12,35,36
Genotyping and Imputation for the Twin 610K Study
To assess imputation quality using the HumanHap610 array we examined 1801 unrelated individuals genotyped on the Illumina HumanHap610 array from the Brisbane Adolescent Twin Study.37,38 Following cleaning, 504,071 SNPs were available for imputation. Imputation was performed as for the POAG cohorts above.
Results
Genotype data from 1155 individuals with advanced POAG from the ANZRAG and 1992 controls were combined and cleaned, and 569,249 SNPs were used as the base of imputation to the 1000 Genomes phase 1 reference panel. A panethnicity reference panel was used, with SNPs with a MAF < 0.1% in European 1000 Genomes samples filtered out to exclude the singleton or monomorphic SNPs. In total, 5,537,665 SNPs were imputed with MAF < 1%, of which 3,260,097 (59%) were imputed with an acceptable imputation quality (INFO > 0.4). This ratio of SNPs with INFO > 0.4 drops marginally to 53% (466,199 from 876,619 SNPs) for the SNPs with MAF < 0.1% (allele frequencies reported here are in the imputed samples). The proportion of well-imputed SNPs with INFO > 0.8 was lower at 11% (615,714 SNPs) for SNPs with MAF < 1%, and 4% (35,512 SNPs) for the SNPs with MAF < 0.1%. These data suggest that a high proportion of acceptable quality SNPs were imputed in this study, even for SNPs with MAF < 0.1%.
The Gln368Ter (rs74315329) rare variant (MAF = 0.1% in our controls, MAF = 1.2% in our cases estimated from the imputation data) had a high imputation quality (INFO = 0.93). The imputation maximum posterior probability (posterior probabilities for each of the three genotypes of a SNP in the population, i.e., homozygous for wild-type allele, heterozygous, and homozygous for mutant allele) was used to assign the best guess imputed genotypes for the Gln368Ter variant (the threshold of calling genotypes was set to 0.6). The best guess imputed genotypes were then compared with the genotypes obtained from direct sequencing to investigate the concordance between the genotypes obtained from imputation and sequencing. None of the individuals were homozygous for the mutant allele (A allele) (imputation posterior probability was zero for the A/A genotype in all the individuals). Table 1 shows the imputation probabilities and results of direct sequencing for individuals with posterior probabilities > 0 for the heterozygous genotype (A/G). Of the 37 individuals in Table 1, 30 (all case samples) had also the genotypes available from sequencing. Of the 30, 28 (93.3%) were confirmed by sequencing to be carriers (Table 1). In addition, four POAG unaffected individuals were also carriers of the mutation based on the imputation results, three of them with high confidence (imputation posterior probabilities > 0.95). The other POAG cases in this study who are not included in Table 1 (n = 1124) were not carriers of the mutation as confirmed by both imputation and sequencing. Overall, we found a sensitivity of 96.29%, specificity of 99.91%, positive predictive value of 96.29%, and negative predictive value of 99.91% for imputation of the Gln368Ter variant compared with direct sequencing. When only individuals with high imputation posterior probabilities (>0.9) are included to reduce the uncertainty for the best guess genotypes obtained from imputed data, the accuracy is higher at a sensitivity of 100%, specificity of 99.91%, positive predictive value of 95.65%, and negative predictive value of 100% (Table 2).
Table 1.
Imputation Maximum Posterior Probability, Best Guess Imputed Genotypes, and Genotypes From Direct Sequencing for rs74315329, rs187423359, and rs182384379
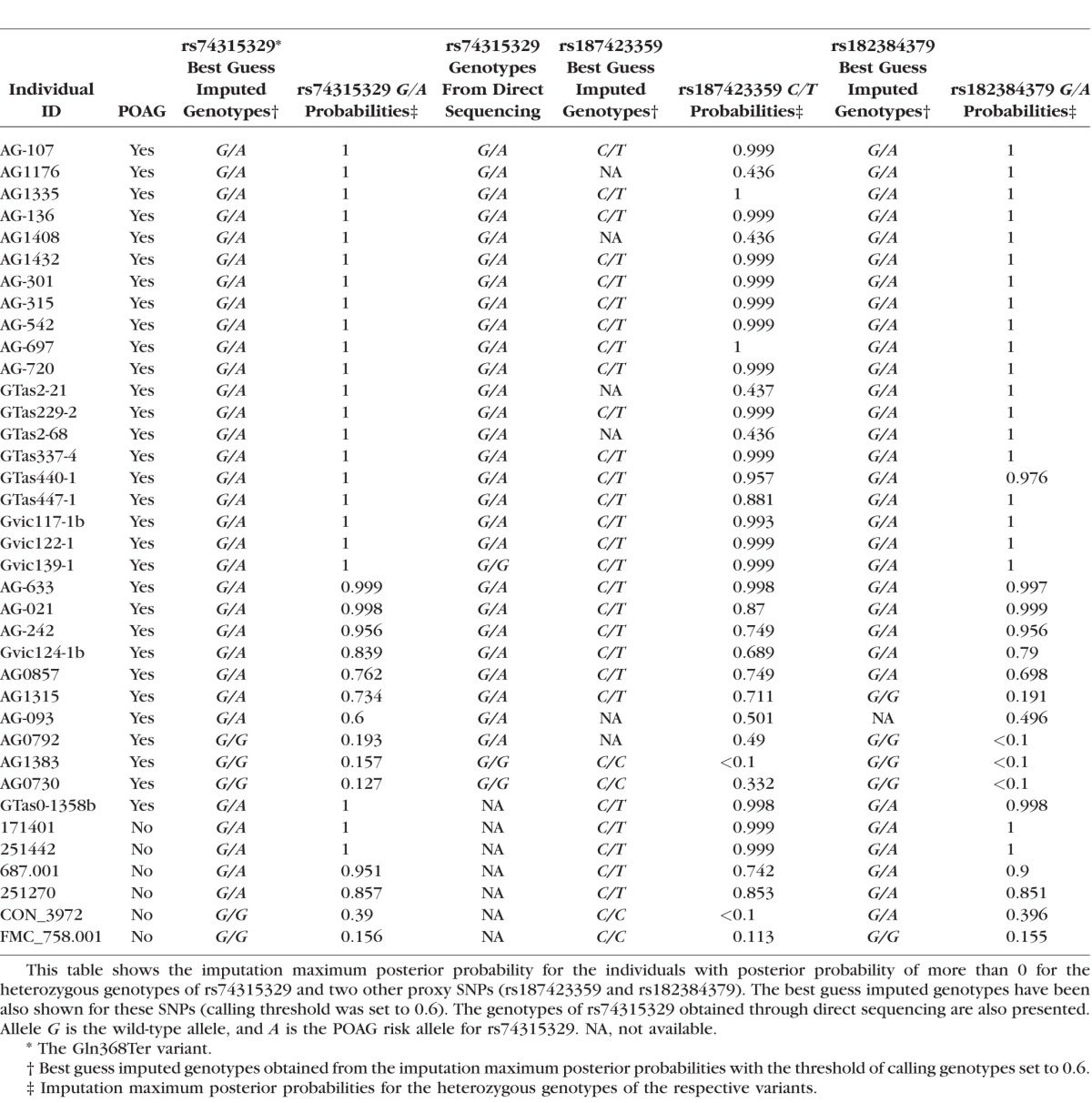
Table 2.
Accuracy of Imputation Compared With Direct Sequencing for the Individuals With Imputation Posterior Probabilities > 0.9 for Gln368Ter Variant

Table 1 also shows the best guess imputed genotypes and imputation posterior probabilities for the heterozygous genotypes of rs187423359 and rs182384379 (the proxy rare variants in r2 = 0.5 with rs74315329). Although the imputation results for rs187423359 and rs182384379 were consistent with the imputation results for rs74315329, the results were not identical because those SNPs are not in complete LD with rs74315329 (r2 = 0.5).
We also investigated whether rs74315329 can be imputed accurately using the other commonly used genotyping arrays. rs74315329 was well imputed with INFO = 0.83 in 1801 individuals of European descent (all unscreened for POAG) genotyped on the Illumina HumanHap610 array, with 1000 Genomes phase 1 as the reference panel. Three individuals in this dataset were carriers of the risk allele with high confidence (imputation posterior probability of 100%). The lower imputation accuracy for rs74315329 in that study may be due in part to a lower frequency of the risk allele in individuals without POAG (frequency of 0.1% estimated from the imputed data in this dataset compared with 0.5% in the ANZRAG dataset), or may be due to lower SNP coverage on the HumanHap610 array. However, while lower, INFO 0.83 still represents high-quality imputation, and suggests that the HumanHap610 arrays, in addition to Omni1M, OmniExpress HumanOmni1- Quad, and HumanOmniExpressExome arrays, can be used for imputation of rs74315329 Gln368Ter.
The other POAG-associated MYOC variants (http://www.omim.org/entry/601652 [in the public domain]) either were monomorphic in 1000 Genomes phase 1 European population and were filtered out during imputation or were not present in the reference panel. Thus, this study could not investigate the imputation of other POAG-associated MYOC variants.
Association analysis for the imputed variants was performed using an additive model adjusted for the sex and the first six principal components. The genomic inflation factor λ was 1.06 after including sex and the first six principal components as covariates. The P values obtained from the association analysis were corrected for the genomic inflation factor λ. In addition to the common variants previously reported,7 the only genome-wide significant rare variant (MAF < 1%) associated with POAG in this study was rs74315329, the Gln368Ter mutation (odds ratio = 15.53 and P = 1.07 × 10−9).
Discussion
In this study we report the accurate imputation of a rare variant (Gln368Ter mutation in the myocilin gene [MYOC] with MAF = 0.5% in our study population) imputed to the 1000 Genomes phase 1 reference panel. The imputed variant was successfully used in a GWAS to detect an association with POAG using standard allelic association analysis. This study suggests that rare variants can be accurately imputed using dense reference panels such as the 1000 Genomes project data and high-coverage microarrays such as HumanHap610, OmniExpress, Omni1M, HumanOmniExpressExome, and HumanOmni1-Quad. Imputation of rare variants is currently far more cost-effective than sequencing methods for genotyping a large numbers of variants. This in its own right is important given the prohibitive costs of whole-genome sequencing and the resultant small sample sizes, which are poorly powered to detect an association with complex traits.23 Although detecting associations of imputed rare variants by single variant tests in standard GWAS may not be powerful due to the low allele frequency and low imputation accuracy,25 the results of this study suggest that some clinically important rare variants can be imputed with high accuracy to detect an association with complex traits in standard GWAS.
This study used a mix of arrays with the sets genotyped separately; they were combined and thinned to a common set of SNPs with appropriate QC. We were able to accurately impute Gln368Ter with SNPs from the intersection of a number of arrays, suggesting that the method to impute Gln368Ter might be robust to array choice.
Could this approach be used for screening other pathogenic variants with lower MAF? Imputation effectiveness is dependent on the existence of a haplotype that tags the target SNP, that haplotype being properly captured/characterized in the reference panel, and the genotyping array containing SNPs in that haplotype. Thus, while imputation difficulty is inversely proportional to MAF (as linkage disequilibrium is limited by the relative allele frequency difference, and rarer SNPs have a smaller range of allele frequencies that can tag them), there isn't a simple cut off. Also the rarer the target SNP, the larger the reference panel required to capture the correct haplotypes to impute it, should such a haplotype exist. Accordingly, the lower limit of this approach is proportional to the MAF, the size of the reference panel, and the SNP array coverage.
It was demonstrated previously that using dense genotyping arrays (such as Illumina Omin1M and HumanHap 610 arrays) and dense reference panels (such as 1000 Genomes) will increase the accuracy of imputation for common and rare variants.24 While none of the rare variants with MAF < 0.3% were well imputed (INFO > 0.4) in that study, we could accurately impute 53% of rare variants with MAF < 0.1% (INFO > 0.4). The likely reason for this poor imputation of rare variants with MAF < 0.3% could be the smaller sample size used for imputation (153 individuals versus 3147 individuals used in the ANZRAG dataset) as well as the greater coverage in the 1000 Genomes phase 1 release. These data suggest that using dense reference panels and genotyping arrays along with a large number of people for imputation can improve the imputation accuracy of rare variants.
HapMap-based imputation has a higher proportion of well-imputed rare SNPs than 1000 Genomes pilot (not phase 1) imputation.24 This may be due to the larger number of rare variants (including very rare variants with MAF < 0.3%) in the 1000 Genomes panel compared to the HapMap panel, which in turn may result in an overall reduction in the proportion of well-imputed rare variants. On the other hand, the 1000 Genomes pilot reference panel contains a relatively small population (60 CEU [Utah Residents with Northern and Western European Ancestry] individuals, 62 Han Chinese in Bejing + Japanese in Tokyo (CHB+JPT) individuals, and 59 Yoruba in Ibadan, Nigeria (YRI) individuals) compared to the following release (phase 1) of the 1000 Genomes data. Moreover, since genotypes in 1000 Genomes have been derived using low pass sequencing, the genotyping quality of the reference panel may be low for very rare SNPs. However, 1000 Genomes may be a better source for imputation of rare variants compared to HapMap due to the increased density and inclusion of a larger number of rare variants.24
Screening rare variants associated with complex traits can be clinically important for prediction of risk and diagnosis and treatment. Here, we could accurately screen the Gln368Ter mutation in the MYOC gene, which is associated with POAG, by imputing this mutation using genotypes available on common genotyping arrays. The penetrance of the Gln368Ter mutation is high and increases with aging.39–41 The frequency of this mutation has been estimated to be 0.1% to 0.3% in the European population, which means that at least 2 people in every 1000 are expected to be carriers. As such this represents a relatively high number of people at risk who can be accurately screened for the mutation using a relatively cheap array typing. We have found that detecting the Gln368Ter MYOC mutation using imputation can accurately identify people at high risk of developing POAG. This can in turn result in early diagnosis and timely treatment, thereby preventing the development of irreversible blindness. Of the total of 3793 individuals without POAG in this study, 6 people were found with high confidence (imputation posterior probability > 0.95) to carry the Gln368Ter mutation. These people are at high risk of developing POAG later in their life. Similarly, a large number of individuals have had their genome scanned using arrays (e.g., almost a million 23andMe customers); being able to predict which of those individuals carry a high-risk MYOC mutation would be of considerable significance as it would lead directly to many individuals seeking appropriate clinical advice.
One limitation of this study is that we did not use other reference panels or imputation tools to compare the results and investigate whether high accuracy of imputation will also be obtained using those panels and tools. In addition, although we validated the imputation results for the Gln368Ter variant using Sanger sequencing in the 1155 POAG cases, we did not have DNA available for the controls and hence did not Sanger sequence controls to verify any controls that were carriers.
In summary, we showed that imputation using common SNPs directly genotyped on genotyping arrays could be an accurate and less expensive (compared to direct sequencing) approach for detecting some clinically important rare variants such as Gln368Ter. These results are clinically important in terms of early detection and treatment of patients at high risk.
Acknowledgments
The authors acknowledge Stuart L. Graham, Robert J. Casson, Ivan Goldberg, and Andrew J. White for clinical sample collection. The authors also acknowledge the support of Bronwyn Usher-Ridge in patient recruitment and data collection; Patrick Danoy and Johanna Hadler for genotyping; Rhys Fogarty for sample and data management and cleaning; Nicholas G. Martin for providing genetic data for the twin 610K study; and David C. Whiteman for providing Australian Cancer Study (ACS) genetic data for use with the ANZRAG controls. The authors also thank the following organizations for their financial support: Genotyping for part of the Australian sample was funded by an Australian National Health and Medical Research Council (NHMRC) Medical Genomics Grant; genotyping for the remainder was performed by the National Institutes of Health (NIH) Center for Inherited Research (CIDR) as part of an NIH/National Eye Institute (NEI) Grant 1RO1EY018246. The authors are grateful to Camilla Day and staff. The authors also thank Scott D. Gordon, Sarah E. Medland, Anjali K. Henders, Brian McEvoy, Dale R. Nyholt, Margaret J. Wright, Megan J. Campbell, and Anthony Caracella for their assistance in processing the Australian genotyping data.
Supported by the Royal Australian and New Zealand College of Ophthalmology (RANZCO) Eye Foundation for recruitment of Australian and New Zealand Registry of Advanced Glaucoma (ANZRAG). Genotyping was funded by the National Health and Medical Research Council (NHMRC) of Australia (Nos. 535074 and 1023911). This work was also supported by funding from NHMRC Nos. 1031362 (JEC), 1037838 (AWH), 1048037, 1009844, and 1031920; an Alcon Research Institute grant (DAM); an Allergan unrestricted grant (AJW); the BrightFocus Foundation; and a Ramaciotti establishment grant. Controls for the ANZRAG cohort were drawn from the Australian Cancer Study (ACS), the Study of Digestive Health and a study of inflammatory bowel diseases. The Australian Cancer Study was supported by the Queensland Cancer Fund and the NHMRC of Australia program Nos. 199600 and 552429. The Study of Digestive Health was supported by Grant 5 R01 CA001833 from the US National Cancer Institute. The Barrett's and Esophageal Adenocarcinoma Genetic Susceptibility Study (BEAGESS) sponsored the genotyping of cases with esophageal cancer and Barrett's esophagus, which were used as unscreened controls in the ANZRAG cohort. BEAGESS was funded by Grant R01 CA136725 from the US National Cancer Institute. SM is supported by an Australian Research Council Future Fellowship. GWM, KPB, and JEC are supported by Australian NHMRC Fellowships. GR-S was funded by NHMRC during the period of this study. The Australian Twin Registry was supported by an NHMRC enabling grant (2004–2009).
Disclosure: P. Gharahkhani, None; K.P. Burdon, None; A.W. Hewitt, None; M.H. Law, None; E. Souzeau, None; G.W. Montgomery, None; G. Radford-Smith, None; D.A. Mackey, None; J.E. Craig, None; S. MacGregor, None
References
- 1. Quigley HA,, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006; 90: 262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Casson RJ,, Chidlow G,, Wood JP,, Crowston JG,, Goldberg I. Definition of glaucoma: clinical and experimental concepts. Clin Experiment Ophthalmol. 2012; 40: 341–349. [DOI] [PubMed] [Google Scholar]
- 3. Stone EM,, Fingert JH,, Alward WL,, et al. Identification of a gene that causes primary open angle glaucoma. Science. 1997; 275: 668–670. [DOI] [PubMed] [Google Scholar]
- 4. Thorleifsson G,, Walters GB,, Hewitt AW,, et al. Common variants near CAV1 and CAV2 are associated with primary open-angle glaucoma. Nat Genet. 2010; 42: 906–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burdon KP,, Macgregor S,, Hewitt AW,, et al. Genome-wide association study identifies susceptibility loci for open angle glaucoma at TMCO1 and CDKN2B-AS1. Nat Genet. 2011; 43: 574–578. [DOI] [PubMed] [Google Scholar]
- 6. Wiggs JL,, Yaspan BL,, Hauser MA,, et al. Common variants at 9p21 and 8q22 are associated with increased susceptibility to optic nerve degeneration in glaucoma. PLoS Genet. 2012; 8: e1002654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gharahkhani P,, Burdon KP,, Fogarty R,, et al. Common variants near ABCA1, AFAP1 and GMDS confer risk of primary open-angle glaucoma. Nat Genet. 2014; 46: 1120–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rezaie T,, Child A,, Hitchings R,, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science. 2002; 295: 1077–1079. [DOI] [PubMed] [Google Scholar]
- 9. Fingert JH,, Robin AL,, Stone JL,, et al. Copy number variations on chromosome 12q14 in patients with normal tension glaucoma. Hum Mol Genet. 2011; 20: 2482–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gong G,, Kosoko-Lasaki O,, Haynatzki GR,, Wilson MR. Genetic dissection of myocilin glaucoma. Hum Mol Genet. 2004; 13 Spec No 1: R91–R102. [DOI] [PubMed] [Google Scholar]
- 11. Fingert JH,, Heon E,, Liebmann JM,, et al. Analysis of myocilin mutations in 1703 glaucoma patients from five different populations. Hum Mol Genet. 1999; 8: 899–905. [DOI] [PubMed] [Google Scholar]
- 12. Souzeau E,, Burdon KP,, Dubowsky A,, et al. Higher prevalence of myocilin mutations in advanced glaucoma in comparison with less advanced disease in an Australasian disease registry. Ophthalmology. 2013; 120: 1135–1143. [DOI] [PubMed] [Google Scholar]
- 13. Resch ZT,, Fautsch MP. Glaucoma-associated myocilin: a better understanding but much more to learn. Exp Eye Res. 2009; 88: 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baird PN,, Craig JE,, Richardson AJ,, et al. Analysis of 15 primary open-angle glaucoma families from Australia identifies a founder effect for the Q368STOP mutation of myocilin. Hum Genet. 2003; 112: 110–116. [DOI] [PubMed] [Google Scholar]
- 15. Baird PN,, Richardson AJ,, Mackey DA,, Craig JE,, Faucher M,, Raymond V. A common disease haplotype for the Q368STOP mutation of the myocilin gene in Australian and Canadian glaucoma families. Am J Ophthalmol. 2005; 140: 760–762. [DOI] [PubMed] [Google Scholar]
- 16. Baird PN,, Richardson AJ,, Craig JE,, Rochtchina E,, Mackey DA,, Mitchell P. The Q368STOP myocilin mutation in a population-based cohort: the Blue Mountains Eye Study. Am J Ophthalmol. 2005; 139: 1125–1126. [DOI] [PubMed] [Google Scholar]
- 17. Welter D,, MacArthur J,, Morales J,, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014; 42: D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eichler EE,, Flint J,, Gibson G,, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010; 11: 446–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maher B. Personal genomes: the case of the missing heritability. Nature. 2008; 456: 18–21. [DOI] [PubMed] [Google Scholar]
- 20. Manolio TA,, Collins FS,, Cox NJ,, et al. Finding the missing heritability of complex diseases. Nature. 2009; 461: 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Charlesworth J,, Kramer PL,, Dyer T,, et al. The path to open-angle glaucoma gene discovery: endophenotypic status of intraocular pressure, cup-to-disc ratio, and central corneal thickness. Invest Ophthalmol Vis Sci. 2010; 51: 3509–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Metzker ML. Sequencing technologies—the next generation. Nat Rev Genet. 2010; 11: 31–46. [DOI] [PubMed] [Google Scholar]
- 23. Ladouceur M,, Dastani Z,, Aulchenko YS,, Greenwood CM,, Richards JB. The empirical power of rare variant association methods: results from sanger sequencing in 1998 individuals. PLoS Genet. 2012; 8: e1002496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zheng HF,, Ladouceur M,, Greenwood CM,, Richards JB. Effect of genome-wide genotyping and reference panels on rare variants imputation. J Genet Genomics. 2012; 39: 545–550. [DOI] [PubMed] [Google Scholar]
- 25. Asimit JL,, Zeggini E. Imputation of rare variants in next-generation association studies. Hum Hered. 2012; 74: 196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Magi R,, Asimit JL,, Day-Williams AG,, Zeggini E,, Morris AP. Genome-wide association analysis of imputed rare variants: application to seven common complex diseases. Genet Epidemiol. 2012; 36: 785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Souzeau E,, Goldberg I,, Healey PR,, et al. Australian and New Zealand Registry of Advanced Glaucoma: methodology and recruitment. Clin Experiment Ophthalmol. 2012; 40: 569–575. [DOI] [PubMed] [Google Scholar]
- 28. Purcell S,, Neale B,, Todd-Brown K,, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007; 81: 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Patterson N,, Price AL,, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006; 2: e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Price AL,, Patterson NJ,, Plenge RM,, Weinblatt ME,, Shadick NA,, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006; 38: 904–909. [DOI] [PubMed] [Google Scholar]
- 31. Howie BN,, Donnelly P,, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009; 5: e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. 1000 Genomes Project Consortium. Abecasis GR,, Altshuler D, et al. A map of human genome variation from population-scale sequencing. Nature. 2010; 467: 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007; 447: 661–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marchini J,, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet. 2010; 11: 499–511. [DOI] [PubMed] [Google Scholar]
- 35. Souzeau E,, Glading J,, Keane M,, et al. Predictive genetic testing experience for myocilin primary open-angle glaucoma using the Australian and New Zealand Registry of Advanced Glaucoma. Genet Med. 2014; 16: 558–563. [DOI] [PubMed] [Google Scholar]
- 36. Young TK,, Souzeau E,, Liu L,, et al. Compound heterozygote myocilin mutations in a pedigree with high prevalence of primary open-angle glaucoma. Mol Vis. 2012; 18: 3064–3069. [PMC free article] [PubMed] [Google Scholar]
- 37. Zhu G,, Duffy DL,, Eldridge A,, et al. A major quantitative-trait locus for mole density is linked to the familial melanoma gene CDKN2A: a maximum-likelihood combined linkage and association analysis in twins and their sibs. Am J Hum Genet. 1999; 65: 483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McGregor B,, Pfitzner J,, Zhu G,, et al. Genetic and environmental contributions to size, color, shape, and other characteristics of melanocytic naevi in a sample of adolescent twins. Genet Epidemiol. 1999; 16: 40–53. [DOI] [PubMed] [Google Scholar]
- 39. Craig JE,, Baird PN,, Healey DL,, et al. Evidence for genetic heterogeneity within eight glaucoma families, with the GLC1A Gln368STOP mutation being an important phenotypic modifier. Ophthalmology. 2001; 108: 1607–1620. [DOI] [PubMed] [Google Scholar]
- 40. Allingham RR,, Wiggs JL,, De La Paz MA,, et al. Gln368STOP myocilin mutation in families with late-onset primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 1998; 39: 2288–2295. [PubMed] [Google Scholar]
- 41. Angius A,, Spinelli P,, Ghilotti G,, et al. Myocilin Gln368stop mutation and advanced age as risk factors for late-onset primary open-angle glaucoma. Arch Ophthalmol. 2000; 118: 674–679. [DOI] [PubMed] [Google Scholar]