

Abstract
Purpose
Retinal mitochondria are dysfunctional in diabetes, and mitochondrial DNA (mtDNA) is damaged and its transcription is compromised. Our aim was to investigate the role of mtDNA methylation in the development of diabetic retinopathy.
Methods
Effect of high glucose (20 mM) on mtDNA methylation was analyzed in retinal endothelial cells by methylation-specific PCR and by quantifying 5-methylcytosine (5mC). Dnmt1 binding at the D-loop and Cytb regions of mtDNA was analyzed by chromatin immunoprecipitation. The role of mtDNA methylation in transcription and cell death was confirmed by quantifying transcripts of mtDNA-encoded genes (Cytb, ND6, and CoxII) and apoptosis, using cells transfected with Dnmt1-small interfering RNA (siRNA), or incubated with a Dnmt inhibitor. The key parameters were validated in the retinal microvasculature from human donors with diabetic retinopathy.
Results
High glucose increased mtDNA methylation, and methylation was significantly higher at the D-loop than at the Cytb and CoxII regions. Mitochondrial accumulation of Dnmt1 and its binding at the D-loop were also significantly increased. Inhibition of Dnmt by its siRNA or pharmacologic inhibitor ameliorated glucose-induced increase in 5mC levels and cell apoptosis. Retinal microvasculature from human donors with diabetic retinopathy presented similar increase in D-loop methylation and decrease in mtDNA transcription.
Conclusions
Hypermethylation of mtDNA in diabetes impairs its transcription, resulting in dysfunctional mitochondria and accelerated capillary cell apoptosis. Regulation of mtDNA methylation has potential to restore mitochondrial homeostasis and inhibit/retard the development of diabetic retinopathy.
Keywords: diabetic retinopathy, DNA methylation, epigenetic modifications, mitochondria, mitochondrial DNA
Hypermethylation of mitochondrial DNA in diabetes impairs the transcription of genes important in functioning of the electron transport system. Compromised electron transport system fuels into the vicious cycle of superoxide generated by the hyperglycemic milieu.
In the pathogenesis of diabetic retinopathy, retinal mitochondria become dysfunctional, initiating the apoptotic machinery, and capillary cell apoptosis precedes the development of retinal histopathology associated with diabetic retinopathy.1–3 Mammalian mitochondria are equipped with their own double-stranded circular DNA (mtDNA), and its two strands are separated by a noncoding regulatory region, the displacement loop (D-loop), which contains essential transcription and replication elements.4 Due to lack of supporting histones, and close proximity to the superoxide-generating electron transport chain (ETC), mtDNA is prone to oxidative damage.5,6 Our previous studies7–10 have shown that diabetes damages retinal mtDNA and reduces its transcription and biogenesis, and the damage is more extensive at the D-loop than at the other regions of the mtDNA.
Mitochondrial DNA encodes 37 genes, and 13 of these genes make proteins essential in the ETC system for oxidative phosphorylation.11 In diabetic retinopathy, the transcription of mtDNA-encoded NADH dehydrogenase 1 and 6 (ND1 and ND6) of complex I and cytochrome b (Cytb) of complex III of the ETC system becomes subnormal,8,9 and the activity of the complex III is compromised, fueling into a self-perpetuating cycle of superoxide accumulation.8,12
Recent studies13,14 have shown that diabetic environment favors methylation of CpG dinucleotides forming 5-methylcytosine (5mC). DNA methylation is catalyzed by DNA methyltransferases (Dnmts) and is commonly associated with gene silencing.15 The Dnmts themselves are redox-sensitive enzymes, and superoxide can enhance DNA methylation via deprotonating the cytosine molecule and accelerating the reaction of DNA with the S-adenosyl-L-methionine.16 In diabetes, the activity of retinal Dnmt is increased, and the mtDNA replication enzyme, polymerase γ-1 (POLG1), is hypermethylated and its binding at the D-loop is impaired, resulting in decreased mtDNA biogenesis.10,17
Although mtDNA represents <1% of the total cellular DNA, it has ∼440 CpG sites, and methylation of mtDNA has been shown to regulate mitochondria copy numbers18 and is associated with chronic diseases, including amyotrophic lateral sclerosis and cancer.19 Dnmt1, the major enzyme responsible for maintenance of DNA methylation patterns, has a mitochondrial targeting sequence, and D-loop methylation in colorectal cancer is associated with altered expression of mtDNA-encoded ND2.20 How diabetes affects methylation of retinal mtDNA remains to be elucidated.
The aim of this study was to investigate the role of mtDNA hypermethylation in the development of diabetic retinopathy. Using retinal endothelial cells, we investigated the effect of hyperglycemia on methylation of mtDNA, especially its D-loop region. The specific role of Dnmt1 in the regulation of mtDNA methylation and its transcription was evaluated by using Dnmt1-small interfering RNA (siRNA) transfected endothelial cells. The results were confirmed in the retinal microvasculature from human donors with documented diabetic retinopathy.
Materials and Methods
Retinal Endothelial Cells
Bovine retinal endothelial cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 15% fetal calf serum (heat inactivated), 5% replacement serum (Nu-serum; BD Bioscience, San Jose, CA, USA), heparin (50 μg/mL), and endothelial growth supplement (50 μg/mL; BD Bioscience). Cells from fourth to sixth passage were transfected with Dnmt1-siRNA by using transfection reagent (sc-29528; Santa Cruz Biotechnology, Santa Cruz, CA, USA) as routinely performed in our laboratory.21–24 Parallel incubations were performed by using nontargeting scrambled RNA. After transfection, the cells were rinsed with PBS, and incubated in 5 or 20 mM glucose media for 4 days. Cells incubated in 20 mM mannitol, instead of 20 mM glucose, served as osmotic control.
To rule out the possibility that mtDNA methylation in bovine and human retina are different, retinal endothelial cells isolated from human donors were incubated in 5 or 20 mM glucose in DME-F12 medium (Sigma-Aldrich Corp., St. Louis, MO, USA) containing 1% fetal calf serum (heat inactivated), 9% replacement serum (Nu-serum), 1% glutamax (Invitrogen, Carlsbad, CA, USA), and 0.5 μg/mL endothelial growth supplement (BD Bioscience), as described previously.25 To investigate the effect of regulation of Dnmt on mtDNA transcription, a group of cells were also incubated in the presence of a Dnmt inhibitor, 5-aza-2′-deoxycytidine (1 μM).26
Human Retinal Microvasculature
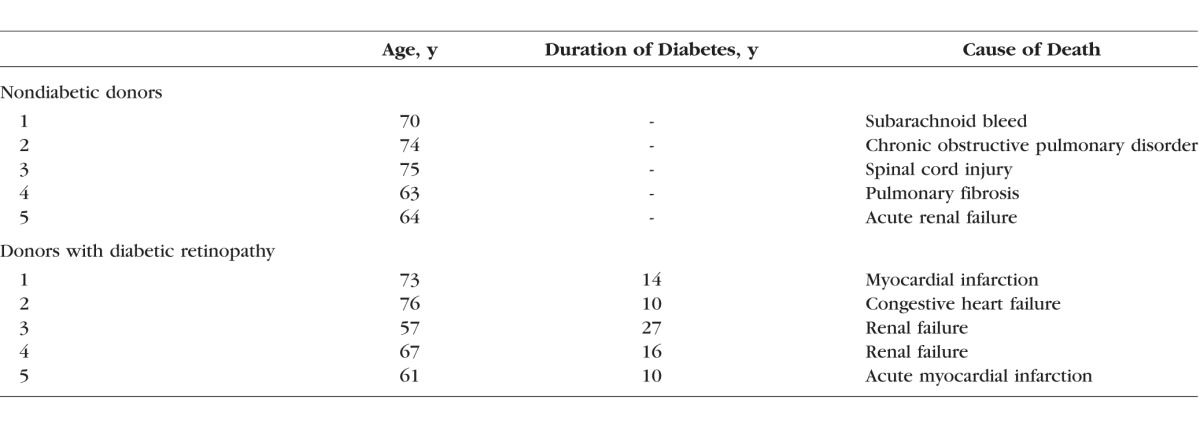
Retina was isolated from eye globes (enucleated within 8 hours of death; obtained from Midwest Eye Banks, Ann Arbor, MI, USA) of human donors (55–76 years of age) with diabetes of more than 10 years' duration and clinically documented retinopathy. Age matched (45–75 years of age) nondiabetic donors served as controls (Table 1).21,27 Retinal microvasculature was prepared by osmotic shock method by incubating a small portion of the retina in distilled water for 1 hour at 37°C, as described previously.10,28,29 The debris was cleaned by repetitive aspiration and ejection through Pasteur pipette, and the microvasculature, generally devoid of nonvascular contamination, was prepared.
Table 1.
Patient Characteristics
Bisulfite Conversion of DNA and Methylation-Specific PCR (MS-PCR)
DNA methylation was quantified by MS-PCR. After performing bisulfite reaction using EpiTect Plus Lyse All Bisulfite Kit (Qiagen, Valencia, CA, USA), the converted DNA was eluted with nuclease-free water. The PCR reaction was performed for methylated (M) and unmethylated (U) regions by using 4 μL GoTaq reaction buffer, 0.8 mM dNTPs (Promega, Madison, WI, USA), 0.5 μM each of the forward and reverse primers, GoTaq DNA polymerase, and 1 μL converted DNA. Thermal cycling included 94°C for 5 minutes, 35 cycles of 94°C for 1 minute, 55°C for 30 seconds, 72°C for 1 minute, and final extension at 72°C for 10 minutes. The PCR products were analyzed on 2% agarose gel, and the intensity of M to U bands was quantified, as reported previously.17
Gene Expression
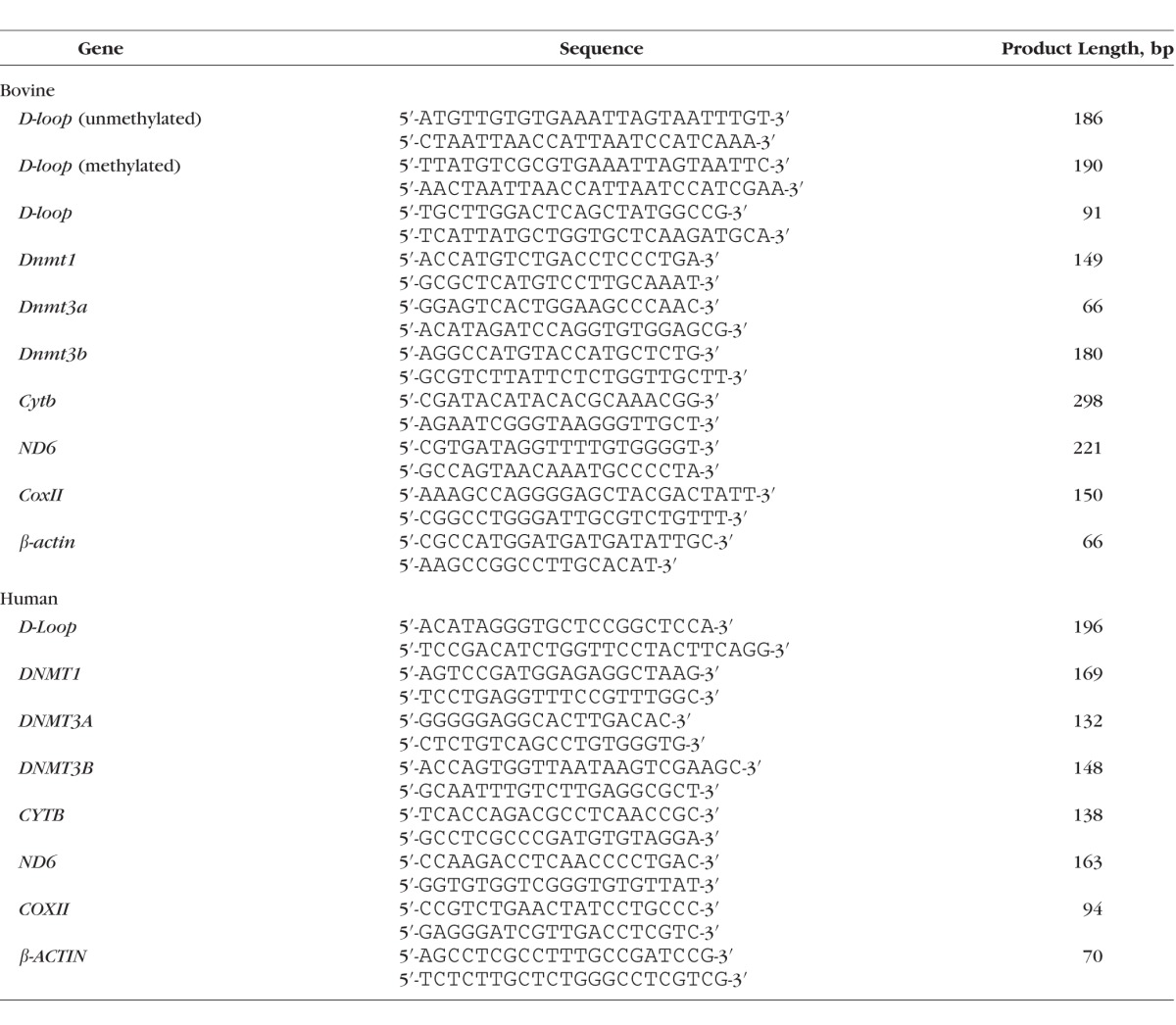
Gene expression was quantified by SYBR green–based quantitative PCR (q-PCR). Amplification was performed in 1 μL chromatin immunoprecipitation (ChIP) purified DNA or cDNA with target-specific primers (Table 2). The PCR conditions included denaturation at 95°C for 10 minutes, 40 cycles of denaturation at 95°C for 15 seconds, and annealing and extension at 60°C for 60 seconds. This was followed by 95°C for 15 seconds, 60°C for 60 seconds, 95°C for 15 seconds, and 60°C for 15 seconds. The specific products were confirmed by SYBR green single melting curve. Values in the immunoprecipitates were normalized to the Ct value from the input sample, and those in cDNA were normalized to the cycle threshold (Ct) value from β-actin in the same sample by using the ddCt method. Relative fold changes were calculated by setting the mean fraction of 5 mM glucose cells as 1.17,21,23
Table 2.
List of Primers
Isolation of Mitochondria and Protein Expression
Mitochondria were isolated by using mitochondria isolation kit (Invitrogen) as reported previously.17,30,31 Mitochondrial protein (30 μg) was separated on a 10% SDS-PAGE, and Dnmt1 expression was normalized to that of the loading protein cytochrome oxidase IV (Cox IV, sc-58348; Santa Cruz Biotechnology). The values for 5 mM glucose were considered as 100%.
Mitochondrial Localization of Dnmt1
Fluorescence microscopy was performed to confirm the effect of high glucose on mitochondrial translocation of Dnmt1 by using MitoTracker green for mitochondrial staining (Invitrogen). The cells were rinsed with PBS and incubated with 500 nM MitoTracker green for 45 minutes at 37°C, and then fixed in 1% formaldehyde for 15 minutes. After blocking with BSA, they were incubated with anti–rabbit-Dnmt1 antibody (ab13537; Abcam, Cambridge, MA, USA), rinsed with PBS, and incubated with Texas Red–conjugated anti-rabbit IgG. The cells were rinsed with PBS and mounted with 4′,6-diamidino-2-phenylindole (DAPI)–containing Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA) and imaged with ZEISS ApoTome fluorescence microscope at ×40 magnification (Carl Zeiss, Chicago, IL, USA).9,30
Dnmt1 Binding at the mtDNA
Dnmt1 binding at D-loop, and for comparison, at Cytb region, was assessed by ChIP assay. Cells or retinal microvessels were cross-linked with 1% paraformaldehyde, and after sonication, the protein extract was precleared with protein-A agarose/salmon sperm DNA slurry. Protein–DNA complex (120 μg) was immunoprecipitated overnight at 4°C with Dnmt1 antibody. The antibody–protein-DNA complex was pulled down by protein-A agarose and washed with low salt and high salt buffers, followed by lithium chloride buffer. This was followed by washing twice with Tris-EDTA buffer, and reverse cross-linking. The complex was digested with proteinase K, and DNA was purified by phenol–chloroform–isoamyl alcohol. After ethanol precipitation, it was resuspended in 14 μL water for real-time q-PCR system. Normal rabbit IgG (2729S; Cell Signaling, Cambridge, MA, USA) was used as negative antibody control. The specificity of ChIP assay was confirmed by analyzing the products on a 2% agarose gel. The input DNA (40 μg protein–DNA complex) was used as an internal control to calculate the fold change; these conditions are routinely used in our laboratory.9,17,32
Quantification of 5mC
Sonicated DNA (100 ng) was immunoprecipitated for 5mC by using methylated DNA immunoprecipitation kit from Epigentek (P-1015-48; Epigentek, Farmingdale, NY, USA). The enriched 5mC fraction was analyzed by using primers for the D-loop, Cytb, and CoxII regions by following the manufacturer's instructions.
Cell Apoptosis
Capillary cell apoptosis was determined by Cell Death Detection ELISAPLUS kit from Roche Diagnostics (Indianapolis, IN, USA). Formation of oligonucleosome in the cytoplasmic fractions was quantified by using monoclonal antibodies directed against DNA and histones, followed by peroxidase-conjugated anti-DNA and biotin-labeled anti-histone. The samples were then incubated with 2,20-azino-di-[3-ethylbenzthiazoline sulfonate] diammonium salt, and absorbance was measured at 405 and 490 nm.22
Statistical Analysis
Statistical analysis was performed with SigmaStat statistical software (Systat Software, Chicago, IL, USA) and the data are expressed as mean ± SD. For multiple comparisons, one-way ANOVA followed by Student-Newman-Keuls test was performed for the data with normal distribution, and for the data that did not qualify, the normal distribution pattern was analyzed by Kruskal-Wallis one-way analysis followed by Dunn's test. A P value < 0.05 was considered as statistically significant.
Results
Bovine Retinal Endothelial Cells
Effect of high glucose on methylation of mtDNA was investigated in the bisulfite-converted DNA. The ratio of methylated to unmethylated DNA was increased by ∼2-fold in the cells incubated in high glucose compared to the cells in normal glucose (Fig. 1).
Figure 1.
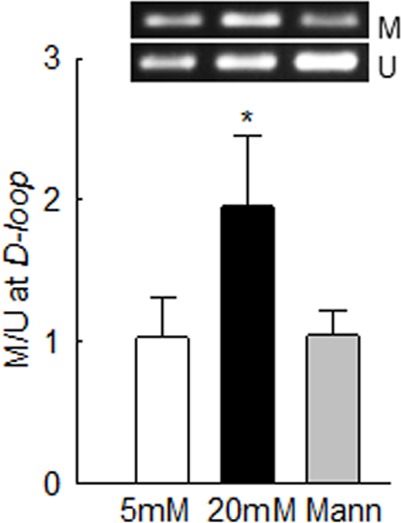
In retinal endothelial cells, glucose hypermethylates mtDNA. Methylation of mtDNA was determined in bovine retinal endothelial cells, incubated in 5 or 20 mM glucose for 4 days, by methylation-specific PCR using bisulfite-converted DNA, and M and U primers for the D-loop region. The ratio of the M and U bands from 5 mM glucose was considered as one. 5 mM and 20 mM, cells in 5 mM glucose or 20 mM glucose. *P < 0.05 vs. 5 mM glucose. M, methylated; Mann, 20 mM mannitol; U, unmethylated.
To investigate the effect of high glucose on DNA methylating machinery, the gene transcripts of Dnmts were quantified. Although high glucose increased Dnmt1 mRNA by 4-fold, it had no significant effect on Dnmt3a and Dnmt3b gene transcripts. As an osmotic control, incubation of cells with 20 mM mannitol, instead of 20 mM glucose, also had no effect on gene transcripts of Dnmts (Fig. 2a). Dnmt1, with a mitochondrial targeting sequence, is considered as the major enzyme responsible for maintenance of DNA methylation pattern,33 and to facilitate DNA methylation in the mitochondria, nuclear-encoded Dnmts translocate to the mitochondria; mitochondrial levels of Dnmt1 were quantified by Western blot technique. Figure 2b shows a significant increase in Dnmt1 levels in the mitochondria of cells in high glucose compared to the cells in normal glucose. Consistent with the Western blot results, high glucose also increased Dnmt1 staining (red) in the mitochondria (Fig. 2c).
Figure 2.

Gene expression and mitochondrial accumulation of Dnmt1 are increased by high glucose. (a) Dnmt1, Dnmt3a, and Dnmt3b gene transcripts were quantified in bovine retinal endothelial cells by SYBR green–based q-PCR using β-actin as the housekeeping gene. Mitochondrial expression of Dnmt1 was determined by (b) Western blot using CoxIV as the loading control, and (c) immunofluorescence using MitoTracker Green (for mitochondria) and Texas Red–conjugated secondary antibody. Values are mean ± SD from three to six samples per group. *P < 0.05 vs. 5 mM glucose.
To understand the role of Dnmt1 in mtDNA methylation, Dnmt1 binding with mtDNA was quantified; Figure 3a clearly demonstrates that glucose increased Dnmt1 binding at the D-loop region by more than 3.5-fold compared to the cells in normal glucose. Although in the same preparations Dnmt1 binding at the Cytb region was also increased, it was ∼2-fold less than that at the D-loop region. As shown in Figure 3b, the products on agarose gel also showed higher Dnmt1 binding at the D-loop region than at the Cytb region. The input DNA (DNA before ChIP) had almost equal abundance of D-loop or Cytb in all of the experimental groups, and the products produced by IgG were almost negligible.
Figure 3.

High glucose increases Dnmt1 binding at the mtDNA. (a) Dnmt1 binding was quantified in bovine retinal endothelial cells by immunoprecipitating the cross-linked cells with Dnmt1 antibody, followed by amplification of the D-loop and Cytb regions by q-PCR using IgG as antibody control (indicated as ^). (b) The specificity of ChIP products was verified on a 2% agarose gel. Input represents the abundance of D-loop or Cytb regions in the DNA before subjecting to ChIP. Each measurement was made in duplicate in three to four samples per group. *P < 0.05 vs. 5 mM glucose.
The levels of 5mC were increased by more than 3-fold at the D-loop region compared to the values obtained from the cells in normal glucose (Fig. 4a), further confirming mtDNA hypermethylation. Consistent with the Dnmt1 binding, 5mC levels at the Cytb region were also increased, but this increase was ∼2-fold less than the one observed at the D-loop region. In contrast, 5mC levels remained insignificant at CoxII region, suggesting that the effect of high glucose on mtDNA methylation is region-specific. To understand the specific role of Dnmt1 in mtDNA methylation, Dnmt1-siRNA transfected cells were used. Dnmt1-siRNA significantly decreased glucose-induced increase in 5mC levels at the D-loop and Cytb regions, compared to the scrambled RNA. The values obtained from cells transfected with Dnmt1-siRNA or scrambled RNA, incubated in normal glucose, were similar to those obtained from untransfected cells incubated in normal glucose. Figures 4b and 4c are included to show the transfection efficiency by quantifying Dnmt1 mRNA and protein expressions, respectively.
Figure 4.

High glucose elevates 5mC levels in the mtDNA in bovine retinal endothelial cells. (a) The 5mC levels were quantified by using methylated DNA immunoprecipitation technique. Fold change was calculated relative to the values obtained from cells in 5 mM glucose. Transfection efficiency was determined by quantifying Dnmt1 (b) gene transcripts and (c) protein expression. Data are represented as mean ± SD from each measurement made in duplicate in three to five cell preparations. 5 mM and 20 mM, 5 mM and 20 mM glucose; si-D and SC, Dnmt1-siRNA– or scrambled RNA–transfected cells; 5+si-D, scrambled RNA–transfected cells incubated in 5 mM glucose. *P < 0.05 vs. 5 mM glucose; #P < 0.05 vs. 20 mM glucose.
The effect of mtDNA methylation on its transcription was evaluated by quantifying mtDNA-encoded genes; the cells transfected with Dnmt1-siRNA, but not with scrambled RNA, were prevented from glucose-induced decrease in mtDNA-encoded Cytb, ND6, and CoxII transcripts (Fig. 5a). Incubation of Dnmt1-siRNA– or scrambled RNA–transfected cells in normal glucose had no effect on the expression of Cytb, ND6, and CoxII. In the same transfected cells, apoptosis was also ameliorated (Fig. 5b); the values obtained from Dnmt1-siRNA cells were significantly different from those obtained from scrambled RNA or untransfected cells incubated in high glucose.
Figure 5.

Regulation of Dnmt1 in bovine retinal endothelial cells by its siRNA ameliorates glucose-induced decrease in mtDNA transcription and cell apoptosis. (a) Gene transcripts of Cytb, ND6, and CoxII were quantified by q-PCR using β-actin as the housekeeping gene, and (b) apoptosis was detected by an ELISA kit for histone-associated DNA fragments. Each measurement was made in duplicate in three to four preparations, and results are represented as mean ± SD. 5 mM and 20 mM, 5 mM and 20 mM glucose; si-D and SC, cells transfected with Dnmt1-siRNA and scrambled RNA, respectively; Mann, cells in 20 mM mannitol. *P < 0.05 vs. 5 mM glucose; #P < 0.05 vs. 20 mM glucose.
Human Retinal Endothelial Cells
Consistent with the results from bovine retinal endothelial cells, although incubation of human retinal endothelial cells with high glucose increased DNMT1 expression by 2-fold, no significant increase in DNMT3A and DNMT3B was observed (Fig. 6a). In the same cell preparations, the levels of 5mC in the D-Loop region were elevated by more than 2.5-fold compared to ∼70% in the CYTB region (Fig. 6b). To investigate the effect of D-Loop methylation on mtDNA transcription, CYTB, ND6, and COXII transcripts were quantified. As expected, glucose significantly decreased mRNA levels of these mtDNA-encoded genes (Fig. 6c), and increased cell apoptosis (Fig. 6d). To further confirm the effect of mtDNA methylation on its transcription, cells incubated with Dnmt inhibitor 5-Azacytidine were analyzed. Addition of 5-Azacytidine ameliorated decrease in the transcription of CYTB, ND6, and COXII, and significantly reduced glucose-induced cell apoptosis (Figs. 6c, 6d). As a control, addition of 5-Azacytidine in normal glucose medium had no effect on mtDNA transcription or apoptosis.
Figure 6.

High glucose exposure of human retinal endothelial cells alters mtDNA methylation and its transcription, and increases cell apoptosis. Human retinal endothelial cells, incubated with high glucose, in the presence or absence of 1 μM 5-Azacytidine were analyzed for (a) gene transcripts of DNMTs by q-PCR using β-ACTIN as the housekeeping gene, and (b) 5mC levels at the D-Loop and CYTB regions of the mtDNA by using methylated DNA immunoprecipitation technique. (c) Expression of CYTB, ND6, and COXII was determined by q-PCR, and β-ACTIN was used as the housekeeping gene. (d) Capillary cell apoptosis was quantified by an ELISA kit. Fold change was calculated relative to the values obtained from cells in 5 mM glucose. Results are represented as mean ± SD from three to four cell preparations per group. 5-Aza, cells incubated with 5-aza-2′-deoxycytidine. *P < 0.05 vs. 5 mM glucose; #P < 0.05 vs. 20 mM glucose.
Retinal Microvessels From Human Donors
To support our in vitro results, methylation of mtDNA was investigated in the retinal microvessels prepared from human donors with established diabetic retinopathy. Transcripts of DNMT1 were significantly increased in the microvasculature of diabetic retinopathy donors compared with age-matched nondiabetic donors (Fig. 7). As with the retinal endothelial cells, DNMT3A and DNMT3B gene transcripts were not increased in diabetic retinopathy donors. Owing to limitations with the osmotic shock method to yield intact mitochondria, Dnmt1 localization in the mitochondria could not be performed. However, mitochondria from the whole retina from donors with diabetic retinopathy had increased Dnmt1 accumulation, compared with their age-matched nondiabetic controls (data not shown).
Figure 7.
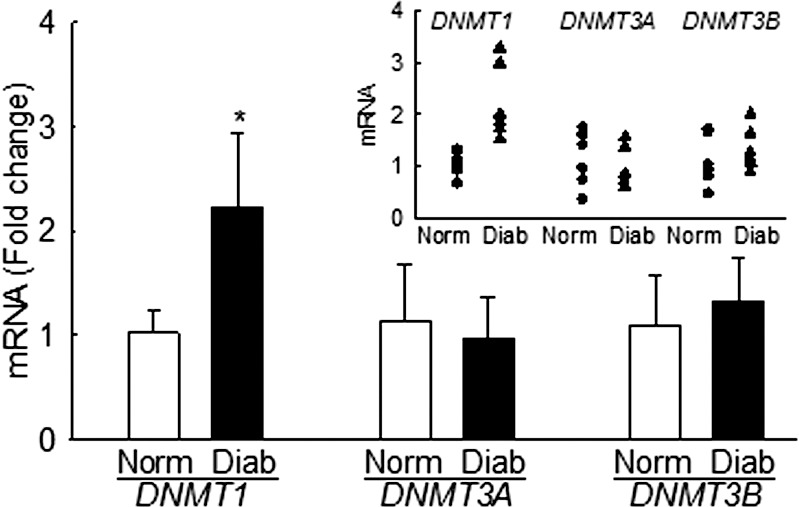
Retinal microvessels from human donors with diabetic retinopathy have increased Dnmt1. Gene transcripts of DNMTs were quantified by q-PCR using β-actin as the housekeeping gene. Values are mean ± SD from four to five donors in each group. Individual values are shown in the scatterplot inset. Norm and Diab, microvasculature from nondiabetic and diabetic donors with retinopathy, respectively. *P < 0.05 versus age-matched nondiabetic donors.
Dnmt1 binding at the D-Loop region was increased by ∼2-fold; however, contrary to the results from retinal endothelial cells, its binding at the CYTB region was not altered (Fig. 8a). Dnmt1 binding was also confirmed by analyzing the products on a 2% agarose gel, and Figure 8b shows that diabetic donors had increased Dnmt1 binding at the D-loop region, compared to their nondiabetic counterparts, but their abundance was equal in the input DNA. However, the products produced by IgG were almost undetectable. Consistent with the Dnmt1 binding, 5mC levels were also elevated by ∼3-fold at the D-Loop region, compared to ∼70% in the CYTB region, and normal rabbit IgG values were <1% when compared to those obtained from Dnmt1 antibody (Fig. 8c). In the same retinal microvasculature, the transcripts of CYTB, ND6, and COXII were also decreased by more than 2-fold compared to the values obtained from nondiabetic donors (Fig. 8d), further confirming the importance of D-Loop methylation in mtDNA transcription.
Figure 8.

Dnmt1 binding and 5mC levels are increased at the D-loop region and transcription of mtDNA-encoded genes is decreased in donors with diabetic retinopathy. (a) Dnmt1 binding was performed by using ChIP technique using IgG as a negative antibody control (indicated as ^). (b) The products were analyzed on an agarose gel. Input represents the abundance of D-loop or Cytb regions in the DNA before ChIP. (c) The levels of 5mC were quantified by methylated DNA immunoprecipitation kit. (d) CYTB, ND6, and COXII transcripts were quantified in the retinal microvasculature by q-PCR using β-ACTIN as the housekeeping gene. Results are represented as mean ± SD from four to five preparations per group, and inset scatterplots are included to show interindividual variations. *P < 0.05 versus nondiabetic donors.
Discussion
Convincing evidence exists that mitochondrial superoxide levels are elevated in the retina in diabetes, that the ETC system is compromised, and that dysfunctional mitochondria accelerate apoptosis of retinal cells,34,35 which precedes the histopathology associated with diabetic retinopathy.36 Mitochondrial DNA is damaged, and its replication and transcription are impaired, further fueling into the damage of ETC system.7,9,17,37 Diabetic environment also brings about epigenetic modifications in the retina, and enzymes responsible for modifications of histones and DNA are altered.17,21,23,38 Using both in vitro model and retinal microvasculature from human donors with diabetic retinopathy, here we showed that the retinal mtDNA is hypermethylated in diabetes, and compared to other regions of mtDNA, its regulatory region showed higher degree of methylation. Dnmt1 appears to be the member of the Dnmt family playing an active role in mtDNA methylation, as its expression is increased in the mitochondria, and inhibition of Dnmt1 by its siRNA ameliorated hyperglycemia-induced decrease in mtDNA transcription and increase in apoptosis, thus suggesting a critical role of D-loop methylation in the development of diabetic retinopathy.
DNA is a “highly dynamic” entity, and it can respond to the external stimuli by adapting to changes and modifying its properties. The biochemical process of addition of a methyl group to the cytosine at CpG DNA dinucleotide is influenced by external factors, and aberrant gene regulation by DNA methylation can also develop as a result of pathologic processes including diabetes and cancer.39,40 Methylation of CpG islands changes protein–DNA interactions, leading to alterations in chromatin structure, and this interferes with the binding of the transcriptional machinery, resulting in gene suppression.41,42 Here we showed that methylation of D-loop region was significantly increased in the retinal capillary cells as evident from increased ratio of methylated to unmethylated mtDNA. Since methylation of cytosine forms 5mC, we showed that the levels of 5mC and binding of the methylating enzyme Dnmt1 also increased significantly in diabetes. Although the Cytb region also showed increased methylation in high glucose, the extent of methylation was significantly lower than that of the D-loop, and methylation of CoxII region was not affected by diabetes. In support, we have shown higher oxidative damage and increased sequence variants at the D-loop region in diabetes, compared to the other regions of mtDNA,10,17,35 and others43 have shown different degree of methylation on different loci of mtDNA in individuals exposed to airborne pollutants.
DNA methylation is catalyzed by a family of methyltransferases, and among those, Dnmt1, Dnmt3a, and Dnmt3b are catalytically active enzymes. While Dnmt3a and Dnmt3b are de novo enzymes, Dnmt1 is the major enzyme responsible for maintenance of the DNA methylation.44 We have shown that the activity of retinal Dnmt is increased in diabetes, and the regulatory region of POLG1 is hypermethylated, decreasing its expression and compromising mtDNA biogenesis.17 To identify the specific member responsible for mtDNA methylation, the expressions of Dnmts were quantified. Our data clearly demonstrated that, while diabetes had no effect on the expression of Dnmt3a and Dnmt3b, Dnmt1 was significantly increased in the retinal microvasculature. Although the exact mechanism by which Dnmt1 is increased is not clear, activation of this redox-sensitive enzyme by oxidative stress45 cannot be ruled out, and oxidative stress is closely associated with the development of diabetic retinopathy.12,34,46 To methylate mtDNA, nuclear-encoded Dnmt translocates inside the mitochondria, and Dnmt1 has a mitochondrial targeting sequence, which is considered important for its transport.18,33 Our results clearly showed increased mitochondrial accumulation of Dnmt1 in diabetes, further supporting its role in mtDNA methylation. To strengthen the role of Dnmt1, we showed that its binding at the D-loop was also increased significantly. Although the presence of Dnmt3a and Dnmt3b inside the mitochondria is also reported in other pathologic conditions,19,47 our results showed that in the pathogenesis of diabetic retinopathy, Dnmt1 is the major enzyme responsible for hypermethylation of mtDNA. Consistent with this, others33 have shown involvement of Dnmt1 in the methylation-induced suppression of ND6 expression.
DNA methylation is generally associated with the suppression of gene transcription, as it inhibits the binding of transcription factor(s) to the DNA, or recruits proteins involved in gene repression.41,42 In diabetic retinopathy, the transcription of mtDNA is compromised, and the transcripts of mtDNA-encoded genes become subnormal, damaging the ETC system and disturbing mitochondrial homeostasis, and this accelerates capillary cell apoptosis.8,9,17 Dnmt1-siRNA or pharmacologic inhibitor 5-Azacytidine restored hyperglycemia-induced decrease in Cytb, CoxII, and ND6, suggesting that the transcription of mtDNA-encoded genes could be under the control of mtDNA methylation. Interestingly, our results also showed that, despite no change in 5mC levels at CoxII region, the transcripts of CoxII were significantly decreased, and Dnmt1-siRNA also prevented decrease in CoxII. This strongly suggests that hypermethylation of D-loop, the region with essential transcription and replication elements, could be playing a central role in the transcription of mtDNA. In support, in nonalcoholic fatty liver disease, increase in Dnmt1 expression is correlated with decreased ND6 expression.48 Others have also shown both CpG and non-CpG methylated sites in the D-loop region,49 and the role of non-CpG methylation in the impaired retinal mtDNA transcription in diabetes cannot be ruled out.
Recent case-control study has shown higher global DNA methylation in diabetic patients with retinopathy compared to those with no retinopathy,50 and our results showed that mtDNA methylation, especially that of the D-loop, was also higher in the human donors with diabetic retinopathy with increased Dnmt1 in the mitochondria. Although isolated retinal endothelial cells (human or bovine) had significant, but substantially reduced, hypermethylation in the CYTB region compared to the D-Loop region of the mtDNA, CYTB region in the retinal microvasculature from donors with diabetic retinopathy did not show significant increase in methylation. The reason for such discrepancy is not clear, since the vasculature also contains basement membrane and pericytes; the possibility that methylation of their CYTB regions is not affected in diabetes, however, cannot be ruled out. Thus, our results using isolated retinal endothelial cells in high glucose and retinal microvasculature from human donors with diabetic retinopathy strongly support the role of mtDNA methylation, especially that of the D-loop region, in the pathogenesis of diabetic retinopathy. In addition, we do recognize that the retina has a complex cellular structure, and diabetes affects many of its cells, including vascular and neuronal cells, and our study focused on the endothelial cells, one of the cell types in the retinal microvasculature that is the target of histopathology associated with diabetic retinopathy. However, we cannot rule out similar or additional epigenetic changes, brought up by the diabetic environment, in other retinal cell types.
In conclusion, we provided a novel mechanism of imbalance in retinal mitochondrial homeostasis in the development of diabetic retinopathy, and showed the significant role of mtDNA methylation. Diabetes facilitates the translocation of Dnmt1 inside the retinal mitochondria, and Dnmt1 in the mitochondria hypermethylates mtDNA. Methylation is much higher at the D-loop region, the region with transcription and replication elements, than at other regions of mtDNA. Due to hypermethylation of the D-loop, the transcription of the mtDNA-encoded genes that are important in the maintenance of ETC system is compromised, and the leakage of electrons in the ETC complexes is increased. These side reactions of the mitochondrial ETC directly generate superoxide radicals, further fueling into the vicious cycle of superoxide radicals generated by the hyperglycemic milieu. Thus, modulation of Dnmt1 by pharmaceutical or molecular means could help maintain mitochondrial integrity and serve as a potential strategy to inhibit/halt the development of diabetic retinopathy.
Acknowledgments
The authors sincerely appreciate the technical assistance of Mangayarkarasi ThandampallayamAjjeya and Doug Putt of the Wayne State University for their help with cell culture and the maintenance of diabetic animal colony.
Supported in part by grants from the National Institutes of Health, Juvenile Diabetes Research Foundation, the Thomas Foundation, and an unrestricted grant to the Department of Ophthalmology from the Research to Prevent Blindness.
Disclosure: M. Mishra, None; R.A. Kowluru, None
References
- 1. Kern TS,, Tang J,, Mizutani M,, Kowluru R,, Nagraj R,, Lorenzi M. Response of capillary cell death to aminoguanidine predicts the development of retinopathy: comparison of diabetes and galactosemia. Invest Ophthalmol Vis Sci. 2000; 41: 3972–3978. [PubMed] [Google Scholar]
- 2. Kowluru RA. Mitochondria damage in the pathogenesis of diabetic retinopathy and in the metabolic memory associated with its continued progression. Curr Med Chem. 2013. ; 20: 3226–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kowluru RA,, Chan PS. Metabolic memory in diabetes—from in vitro oddity to in vivo problem: role of apoptosis. Brain Res Bull. 2010; 87: 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Falkenberg M,, Larsson NG,, Gustafsson CM. DNA replication and transcription in mammalian mitochondria. Annu Rev Biochem. 2007. ; 76: 679–699. [DOI] [PubMed] [Google Scholar]
- 5. Alexeyev M,, Shokolenko I,, Wilson G,, LeDoux S. The maintenance of mitochondrial DNA integrity—critical analysis and update. Cold Spring Harb Perspect Biol. 2013; 5: a012641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shokolenko I,, Venediktova N,, Bochkareva A,, Wilson GL,, Alexeyev MF. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009. ; 37: 2539–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Madsen-Bouterse S,, Zhong Q,, Mohammad G,, Ho YS,, Kowluru RA. Oxidative damage of mitochondrial DNA in diabetes and its protection by manganese superoxide dismutase. Free Radic Res. 2010; 44: 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Madsen-Bouterse SA,, Mohammad G,, Kanwar M,, Kowluru RA. Role of mitochondrial DNA damage in the development of diabetic retinopathy and the metabolic memory phenomenon associated with its progression. Antioxid Redox Signal. 2010. ; 13: 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Santos JM,, Tewari S,, Goldberg AFX,, Kowluru RA. Mitochondria biogenesis and the development of diabetic retinopathy. Free Radic Biol Med. 2011; 51: 1849–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tewari S,, Santos JM,, Kowluru RA. Damaged mitochondrial DNA replication system and the development of diabetic retinopathy. Antioxid Redox Signal. 2012. ; 17: 492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rebelo AP,, Dillon LM,, Moraes CT. Mitochondrial DNA transcription regulation and nucleoid organization. J Inherit Metab Dis. 2011. ; 34: 941–951. [DOI] [PubMed] [Google Scholar]
- 12. Kanwar M,, Chan PS,, Kern TS,, Kowluru RA. Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Invest Ophthalmol Vis Sci. 2007. ; 48: 3805–3811. [DOI] [PubMed] [Google Scholar]
- 13. Maghbooli Z,, Larijani B,, Emamgholipour S,, Amini M,, Keshtkar A,, Pasalar P. Aberrant DNA methylation patterns in diabetic nephropathy. J Diabetes Metab Disord. 2014. ; 13: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zou L,, Yan S,, Guan X,, Pan Y,, Qu X. Hypermethylation of the prkcz gene in type 2 diabetes mellitus. J Diabetes Res. 2013. ; 2013: 721493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000. ; 9: 2395–2402. [DOI] [PubMed] [Google Scholar]
- 16. Afanas'ev I. New nucleophilic mechanisms of ROS-dependent epigenetic modifications: comparison of aging and cancer. Aging Dis. 2014; 5: 52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tewari S,, Zhong Q,, Santos JM,, Kowluru RA. Mitochondria DNA replication and DNA methylation in the metabolic memory associated with continued progression of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2012; 53: 4881–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Iacobazzi V,, Castegna A,, Infantino V,, Andria G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol Genet Metab. 2013. ; 110: 25–34. [DOI] [PubMed] [Google Scholar]
- 19. Wong M,, Gertz B,, Chestnut BA,, Martin LJ. Mitochondrial dnmt3a and DNA methylation in skeletal muscle and cns of transgenic mouse models of als. Front Cell Neurosci. 2013. ; 7: 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Feng S,, Xiong L,, Ji Z,, Cheng W,, Yang H. Correlation between increased nd2 expression and demethylated displacement loop of mtDNA in colorectal cancer. Mol Med Rep. 2012. ; 6: 125–130. [DOI] [PubMed] [Google Scholar]
- 21. Mishra M,, Zhong Q,, Kowluru RA. Epigenetic modifications of Nrf2-mediated glutamate-cysteine ligase: implications for the development of diabetic retinopathy and the metabolic memory phenomenon associated with its continued progression. Free Radic Biol Med. 2014; 75C: 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mohammad G,, Kowluru RA. Diabetic retinopathy and signaling mechanism for activation of matrix metalloproteinase-9. J Cell Physiol. 2012. ; 227: 1052–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhong Q,, Kowluru RA. Epigenetic modification of Sod2 in the development of diabetic retinopathy and in the metabolic memory: role of histone methylation. Invest Ophthalmol Vis Sci. 2013; 54: 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Santos JM,, Mishra M,, Kowluru RA. Posttranslational modification of mitochondrial transcription factor a in impaired mitochondria biogenesis: implications in diabetic retinopathy and metabolic memory phenomenon. Exp Eye Res. 2014; 121: 168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kowluru RA,, Kanwar M. Oxidative stress and the development of diabetic retinopathy: contributory role of matrix metalloproteinase-2. Free Radic Biol Med. 2009; 46: 1677–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Manev H,, Uz T. DNA hypomethylating agents 5-aza-2′-deoxycytidine and valproate increase neuronal 5-lipoxygenase mrna. Eur J Pharmacol. 2002. ; 445: 149–150. [DOI] [PubMed] [Google Scholar]
- 27. Zhong Q,, Mishra M,, Kowluru RA. Transcription factor Nrf2-mediated antioxidant defense system in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2013. ; 54: 3941–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kowluru RA,, Kowluru A,, Veluthakal R,, et al. Tiam1-Rac1 signalling axis-mediated activation of NADPH oxidase-2 initiates mitochondrial damage in the development of diabetic retinopathy. Diabetologia. 2014. ; 57: 1047–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kowluru RA,, Santos JM,, Zhong Q. Sirt1, a negative regulator of matrix metalloproteinase-9 in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2014. ; 55: 5653–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kowluru RA. Role of matrix metalloproteinase-9 in the development of diabetic retinopathy and its regulation by h-ras. Invest Ophthalmol Vis Sci. 2010. ; 51: 4320–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Santos JM,, Kowluru RA. Impaired transport of mitochondrial transcription factor A (TFAM) and the metabolic memory phenomenon associated with the progression of diabetic retinopathy. Diabetes Metab Res Rev. 2013. ; 29: 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kowluru RA,, Mohammad G,, dos Santos JM,, Zhong Q. Abrogation of MMP-9 gene protects against the development of retinopathy in diabetic mice by preventing mitochondrial damage. Diabetes. 2011. ; 60: 3023–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shock LS,, Thakkar PV,, Peterson EJ,, Moran RG,, Taylor SM. DNA methyltransferase 1 cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci U S A. 2011. ; 108: 3630–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kowluru RA,, Abbas SN. Diabetes-induced mitochondrial dysfunction in the retina. Invest Ophthalmol Vis Sci. 2003; 44: 5327–5334. [DOI] [PubMed] [Google Scholar]
- 35. Mishra M,, Kowluru RA. Retinal mitochondrial DNA mismatch repair in the development of diabetic retinopathy, and its continued progression after termination of hyperglycemia. Invest Ophthalmol Vis Sci. 2014. ; 55: 6960–6967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kern TS,, Engerman RL. Pharmacological inhibition of diabetic retinopathy: aminoguanidine and aspirin. Diabetes. 2001; 50: 1636–1642. [DOI] [PubMed] [Google Scholar]
- 37. Santos JM,, Tewari S,, Kowluru RA. A compensatory mechanism protects retinal mitochondria from initial insult in diabetic retinopathy. Free Radic Biol Med. 2012; 53: 1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mishra M,, Zhong Q,, Kowluru RA. Epigenetic modifications of keap1 regulate its interaction with the protective factor Nrf2 in the development of diabetic retinopa. Invest Ophthalmol Vis Sci. 2014. ; 55: 7256–7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hinoue T,, Weisenberger DJ,, Lange CP,, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012. ; 22: 271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kowluru RA,, Santos JM,, Mishra M. Epigenetic modifications and diabetic retinopathy. Biomed Res Int. 2013. ; 2013: 635284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Deaton AM,, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011. ; 25: 1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jones PA. Functions of DNA methylation: islands start sites, gene bodies and beyond. Nat Rev Genet. 2012; 13: 484–492. [DOI] [PubMed] [Google Scholar]
- 43. Byun HM,, Panni T,, Motta V,, et al. Effects of airborne pollutants on mitochondrial DNA methylation. Part Fibre Toxicol. 2013. ; 10: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kar S,, Deb M,, Sengupta D,, et al. An insight into the various regulatory mechanisms modulating human DNA methyltransferase 1 stability and function. Epigenetics. 2012. ; 7: 994–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guo W,, Yang D,, Xu H,, et al. Mutations in the D-loop region and increased copy number of mitochondrial DNA in human laryngeal squamous cell carcinoma. Mol Biol Rep. 2013. ; 40: 13–20. [DOI] [PubMed] [Google Scholar]
- 46. Kowluru RA,, Tang J,, Kern TS. Abnormalities of retinal metabolism in diabetes and experimental galactosemia VII: effect of long-term administration of antioxidants on the development of retinopathy. Diabetes. 2001; 50: 1938–1942. [DOI] [PubMed] [Google Scholar]
- 47. Chestnut BA,, Chang Q,, Price A,, Lesuisse C,, Wong M,, Martin LJ. Epigenetic regulation of motor neuron cell death through DNA methylation. J Neurosci. 2011. ; 31: 16619–16636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pirola CJ,, Gianotti TF,, Burgueno AL,, et al. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut. 2013. ; 62: 1356–1363. [DOI] [PubMed] [Google Scholar]
- 49. Bellizzi D,, D'Aquila P,, Scafone T,, et al. The control region of mitochondrial DNA shows an unusual cpg and non-CpG methylation pattern. DNA Res. 2013. ; 20: 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Maghbooli Z,, Hossein-Nezhad A,, Larijani B,, Amini M,, Keshtkar A. Global DNA methylation as a possible biomarker for diabetic retinopathy. Diabetes Metab Res Rev. 2015. ; 31: 183–189. [DOI] [PubMed] [Google Scholar]