

Abstract
The direct installation of the C4 and C10 methyl groups present in the 6,12-guaianolide framework using a Rh(I)-catalyzed cyclocarbonylation reaction of methyl subsituted allenes and alkynes is described. High yields of bicyclo[5.3.0]decanes are afforded when low reaction concentrations involving syringe pump addition of the allene-yne to the catalyst are used.
Keywords: Pauson-Khand, Allene-yne, Bicyclo[5.3.0]decadienone, Guaianolide, High Dilution, Solid-supported Scavenger
Graphical Abstract
Guaianolides are the largest class of sesquiterpene lactones (SLs) and possess a wide range of biological activities, of which the anti-inflammatory and antitumor properties are especially interesting.1,2 Despite their potential value as medicinal agents only one guaianolide, arglabin (1, Figure 1), has been approved as a drug, marketed for lung, liver, and ovarian cancer treatment,3 and another, thapsigargin (2), is being developed as a prodrug for prostate cancer.4 This dearth of medicinally relevant guaianolides is largely due to the presence of one or more α, β-unsaturated carbonyl groups in the substructures of these natural products. These electrophilic motifs commonly include α-methylene-γ-butyrolactones, cyclopentenones, and acrylic esters which can react with biological thiols via a 1,4-conjugate addition reaction; a reactivity profile that may be responsible for both the biological activity and the toxicity of these compounds.2,5
Figure 1.

Examples of naturally occurring 6,12-guaianolides.
The molecularly complex chemical structures of the guaianolides further hamper their inclusion in the drug discovery process, in part due to the synthetic difficulties associated with analog and natural product preparation.1 For example, guaianolides commonly have reactive functional groups that require incorporation at a late-stage in the synthetic sequence or alternatively incorporation of the groups at an early-stage, necessitating a series of protecting group manipulations.6 The former approach limits the number of synthetic strategies available for compound preparation, while the latter approach is not step economical. Guaianolide structures also feature a bicyclo[5.3.0]decane system for which there are limited synthetic methods for accessing fused seven-membered rings in comparison to those available to prepare fused five- and sixmembered rings.7 Finally, guaianolides possess one or more stereocenters, a stereochemical complication that is further amplified by the conformational mobility of five- and seven-membered rings. New synthetic methods that address these challenges will facilitate our understanding of the value of guaianolides in modern medicine.
Previous results from our group have established the synthetic utility of the allenic Pauson-Khand reaction (APKR) for accessing the key portion of the 6,12-guaianolide framework. Hallmarks of this APKR approach include obtaining the 5-7-5 tricyclic framework stereoselectively, in high yield by incorporating the α-methylene-γ-butyrolactone into the alleneyne precursor.8 Moreover, the dienone, a motif resulting from the APKR is well placed for the eventual conversion to functionality present in a number of naturally occurring 6,12-guaianolides. This APKR approach has been used for the preparation of novel guaianolide analogs that inhibit NF-κB at activity levels on par with the most potent naturally occurring SLs.9 Early stage inclusion of the α-methylene-γ-butyrolactone allowed preparation of analogs in nearly half the number of synthetic steps typically required for the de novo synthesis of other compounds of this class. Curiously, low yielding APKRs were observed when using precursors with an allene substituted with a methyl group on the proximal double bond or for methyl substituted alkynes.8,10 These methylated sites of the allene-yne precursor ultimately translate into the C4 and C10 positions of the 6,12-guaianolides, which are nearly always occupied by methyl or methylene substituents in the natural products. Thus, a high yielding cycloaddition reaction is needed to provide the requisite C4 and C10 substitution pattern. Herein, we report our studies achieving this goal.
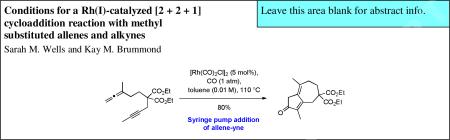
Investigations were initiated with the preparation of allene-yne 8a, an APKR precursor that was selected for its synthetic accessibility. The synthesis of 8a was accomplished in 5 steps from allene 3 which was available using a Johnson-Claisen rearrangement from 2-butyn-1-ol and triethylorthoacetate in 71% yield.11 Reduction of allenyl ester 3 with lithium aluminum hydride, followed by conversion of the resulting hydroxyl group to a mesylate afforded compound 4 in 89% yield over the two steps. In turn, mesylate 4 was reacted with sodium triethyl methanetricarboxylate (5) to give the corresponding triester in 69% yield, which was decarboxylated by reacting it with sodium ethoxide to afford malonate 6 in 97% yield. Mesylate 4 was not reacted with sodium diethyl malonate directly because of previously reported problems with dialkylation.12 Deprotonation of malonate 6 with sodium hydride followed by the addition of 1-bromo-2-butyne (7) gave allene-yne 8a in 86% yield (Scheme 1, Method A). Interestingly, when the allene and alkyne building blocks were introduced in reverse order, overall yields of alleneyne 8a were significantly lower. For example, reaction of sodium triethyl methanetricarboxylate (5) with 1-bromo-2-butyne (7) followed by decarboxylation gave diester 9 in an overall yield of 89%.10a However, deprotonation and alkylation of 9 with mesylate 4 produced 8a in only 39% yield (Scheme 1, Method B). We attribute this lower yield to competing substitution and elimination reactions resulting from higher basicity of the sodium salt of diester 9 compared to sodium triethyl methanetricarboxylate (5).
Scheme 1.

Synthesis of allene-yne 8a.
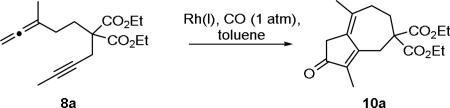
With allene-yne 8a in hand, investigations were initiated to increase the yield of the APKR of methyl substituted allenes and alkynes (Table 1). First, for the conversion of allene-yne 8a to the cyclocarbonylation adduct 10a, we used our standard APKR conditions of 0.1 M toluene, balloon pressure of carbon monoxide, and 90 °C, with 15 mol% rhodium biscarbonyl chloride dimer ([Rh(CO)2Cl]2) rather than the previously reported 10 mol%.8,9 After reacting for 1.5 h, these conditions gave dienone 10a in 27% yield along with substantial quantities of a byproduct (entry 1, Table 1). Analysis of the byproduct by 1H NMR spectroscopy (see Supplementary Information for spectra) revealed signals with chemical shifts similar to those present in the 1H NMR spectra for dienone 10a because signals corresponding to the allene hydrogens were missing. However, additional resonances were observed in the aromatic region (8.22-7.07 ppm) of the spectrum. Information gained from the relatively large integration values of the signals for the OCH2CH3 of the ethyl ester in the 1H NMR, along with the downfield resonances led to a hypothesis that the byproduct may be the result of a competing intermolecular cycloaddition or dimerization reaction. This hypothesis was further supported by ESI mass spectroscopy analysis of the byproduct in the positive ion mode which revealed a base peak with an exact mass of 581.3099; the same analysis technique applied to 8a and 10a show [M+H] molecular ion peaks with masses 321.1687, of 293.1751 and respectively. While the mass of the base peak does not provide conclusive evidence for a byproduct structure, the near doubling of the mass supports this hypothesis.13
Table 1.
Optimization of APKR for the synthesis of dienone 10a.
| ||||||
|---|---|---|---|---|---|---|
| Entry | Rh(I) | Catalyst Loading (mol%) | Temperature (°C) | Concentration (M) | Time | Yield (%) |
| 1 | [Rh(CO)2Cl]2 | 15 | 90 | 0.1 | 1.5 h | 27 |
| 2 | [Rh(CO)2Cl]2 | 15a | 90 | 0.1 | 1.5 h | 32 |
| 3 | [Rh(CO)2Cl]2 | 15a | 90 | 0.01 | 1.5 h | 53 |
| 4 | [Rh(CO)2Cl]2 | 15a | 110 | 0.01 | 25 min | 57 |
| 5 | [Rh(CO)2Cl]2 | 10a | 110 | 0.01b | c | 81 |
| 6 | [Rh(CO)2Cl]2 | 5a | 110 | 0.01b | c | 80 |
| 7 | [Rh(CO)2Cl]2 | 2a | 110 | 0.01b | c | 62 |
| 8 | [Rh(CO)2Cl]2 | 1a | 110 | 0.01b | c | 63 |
| 9 | [Rh(CO)(dppp)2]Cl | 10a | 110 | 0.1 | 21 hd | 27 |
| 10 | [Rh(CO)(dppp)2]Cl | 10a | 110 | 0.01 | 21 hd | 46 |
| 11 | [Rh(CO)Cl(dppp)]2 | 10a | 110 | 0.01 | 6 h | 29 |
Triphenylphosphine polymer bound was used in the reaction work up.
Syringe pump addition of allene-yne to Rh(I) solution was utilized.
Dropwise addition over 1.5 h and an additional 15 min of reaction time.
Reaction was not monitored between 8-21 h.
In addition to the byproduct (Rf = 0.38, 20% EtOAc/hexanes) observed in the APKR reaction of allene-yne 8a, baseline impurities were visible as brown spots by TLC even after filtering the reaction through a celite plug. Because of the spot discoloration, rhodium-containing contaminants were suspected. Thus, the metal scavenger triphenylphosphine polymer bound was added to the reaction prior to concentration and after stirring for approximately 14 h at rt, the impurities were no longer visible by TLC.14 Despite a marginal increase in the yield of dienone 10a (compare entries 1 and 2, Table 1) these conditions were used for subsequent reactions to remove baseline impurities.
Next, the APKR reaction of 8a was performed at a concentration of 0.01 M in toluene, a ten-fold dilution, affording a 53% yield of 10a with less byproduct (entry 3, Table 1). This result supports our hypothesis of a bimolecular reaction as the cause for the byproduct. The temperature of this reaction was increased to 110 °C to limit exposure time of the product to the rhodium catalyst, resulting in only a slight increase in the yield of dienone 10a (entry 4). Decreasing the concentration of the reaction further was accomplished by adding a solution of alleneyne 8a in toluene dropwise over 1.5 h to a preheated solution of [Rh(CO)2Cl]2 in toluene with a final concentration of 0.01 M.15 Upon completion of the addition, the reaction is stirred for an additional 15 min until allene-yne 8a was no longer visible by TLC. Dienone 10a was afforded in 81% yield with no byproduct observed (entry 5). Lowering the catalyst loading to 5 mol% gave a nearly identical yield of 10a (entry 6) while attempts to reduce the catalyst loading further gave significantly lower yields for these reactions (entries 7-8).
A comparison was made between these newly developed high dilution reaction conditions and Mukai's previously reported conditions where the catalyst [Rh(CO)(dppp)2]Cl proved advantageous for methyl substituted allenes and alkynes.16 Reaction of 8a with Mukai's conditions of 10 mol% rhodium (1,5-cyclooctadiene) chloride dimer ([Rh(cod)Cl]2) and 50 mol% 1,3-bis(diphenylphosphino)propane (dppp), to produce the Rh (I) monomer [Rh(CO)(dppp)2]Cl, afforded dienone 10a in 27% yield (entry 9, Table 1). Lowering the concentration of this reaction to 0.01 M gave a 46% yield of dienone 10a (entry 10). Finally, [Rh(CO)Cl(dppp)]2, a dimeric Rh(I) catalyst produced from 10 mol% [Rh(cod)Cl]2 and 20 mol% dppp, also used by Mukai for methyl substituted allenes and alkynes,16 was used for the cyclocarbonylation of allene-yne 8a at 0.01 M, but this reaction only produced 10a in 29% yield (entry 11). The operational simplicity of the cyclocarbonylation reaction with rhodium biscarbonyl chloride dimer versus either of Mukai's catalyst systems led us to focus our efforts on the former.
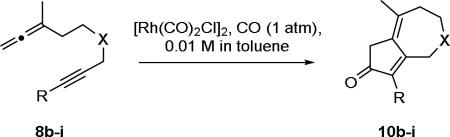
Next, the generality of these high dilution reaction conditions17 was examined on a number of allene-ynes (Table 2). Exposure of allene-yne 8b, containing a diester in the tether and a terminal alkyne, to the high dilution conditions produced dienone 10b in 92% yield (entry 1). This yield represents a 42% increase when compared to the yield of the cyclocarbonylation reaction where 8b is added all at once (entry 2). Similarly, allene-yne 8c, with an acetonide in the tether and a methyl substituted alkyne, afforded 10c in 87% yield (entry 3) a 60% yield increase when compared to entry 4. Acetonide 8d with a terminal alkyne afforded dienone 10d in 78% yield (entry 5). Dienone 10e was generated from diol containing allene-yne tether 8e in 59% yield (entry 6). We attribute this lower yield to the diol functioning as a catalyst poison.
Table 2.
Result of high dilution conditions on a variety allene-ynes.
| |||||
|---|---|---|---|---|---|
| Entry | Allene-yne | X | R | Conditions | Yield 10 (%) |
| 1 | 8b | C(CO2Et)2 | H | a | 92 |
| 2 | 8b | b | 50 | ||
| 3 | 8c |
|
Me | a | 87 |
| 4 | 8c | b | 27 | ||
| 5 | 8d | H | a | 78 | |
| 6 | 8e | C(CH2OH)2 | Me | a | 59 |
| 7 | 8f | NTs | Me | a | 92 |
| 8 | 8g | H | a | 73 | |
| 9 | 8h | O | Me | a | 49 |
| 10 | 8i | H | a | 44 | |
Syringe pump addition of allene-yne to Rh(I) solution was utilized.
Allene-yne was added to the solution all at once.
Next, a tosylamide was incorporated into the allene-yne tether. Both the methyl substituted alkyne 8f and terminal alkyne 8g afforded high yielding cyclocarbonylation reactions; producing 10f and 10g in 92% and 78% yield respectively (entries 7 and 8, Table 2). Finally, ether containing allene-ynes 8h and 8i afforded moderate yields of their respective dienones; 10h was obtained in 49% yield and 10i was obtained in 44% yield (entries 9 and 10). Previous examples have been reported where allene-ynes containing ether tethers afforded diminished yields of APKR dienone product when compared to analogous all carbon- and tosylamide-containing tethers.10c, 18 Moreover, all examples discussed within reacted quickly upon exposure to the reaction conditions, with full consumption of the allene-yne observed within 15 min of the completed dropwise addition.
In summary, this study provides optimized reaction conditions for the Rh(I)-catalyzed APKR with methyl substituted allenes and alkynes, as well as terminal alkynes. High yields of bicyclo[5.3.0]decadienones were obtained by implementing a dropwise addition of the allene-yne precursors to a dilute solution of [Rh(CO)2Cl]2 to minimize competing intermolecular processes. A variety of APKR precursors were subjected to these optimized conditions. Allene-yne tethers with ester, acetonide and tosylamide functionalities afforded high yields of bicyclo[5.3.0]decadienones while allene-yne tethers with ether and diol functionalities afforded moderate yields of cyclocarbonylation products. We expect these newly developed APKR conditions to facilitate the preparation of naturally occurring 6,12-guaianolides and their structurally related analogs. In addition, the high dilution conditions may provide general improvement for other transition metal catalyzed reactions.
Supplementary Material
Acknowledgments
We thank the National Institute of Health (GM54161) for financially supporting this work. We also acknowledge Bhaskar Godugu (University of Pittsburgh) for assisting with mass spectrometry analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Data
Supplementary data associated with this article can be found in the online version, at
References and notes
- 1.Drew DP, Krichau N, Reichwald K, Simonsen HT. Phytochem. Rev. 2009;8:581–599. [Google Scholar]
- 2.Merfort I. Curr. Drug Targets. 2011;12:1560–1573. doi: 10.2174/138945011798109437. [DOI] [PubMed] [Google Scholar]
- 3.a Csuk R, Heinold A, Siewart B, Schwarz S, Barthel A, Kluge R, Ströhl D. Arch. Pharm. Chem. Life. Sci. 2012;345:215–222. doi: 10.1002/ardp.201100065. [DOI] [PubMed] [Google Scholar]; b Musulmanbekow KJ. Chem. Abs. 1999;132:30823. WO9964003A1. [Google Scholar]; c Musulmanbekow KJ. Chem. Abs. 1999;131:332093. WO9958148A1. [Google Scholar]; d Adekenov SM. Chem. Abs. 1998;130:480. WO9848789A1. [Google Scholar]
- 4.Søhoel H, Jensen AL, Møller JV, Nissen P, Denmeade SR, Isaacs JT, Olsen CE, Christensen SB. Bioorg. Med. Chem. 2006;14:2810–2815. doi: 10.1016/j.bmc.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Siedle B, García-Piñeres AJ, Murillo R, Schulte-Mönting J, Castro V, Rüngeler P, Klaas CA, Da Costa FB, Kisiel W, Merfort I. J. Med. Chem. 2004;47:6042–6054. doi: 10.1021/jm049937r. [DOI] [PubMed] [Google Scholar]
- 6.Schall A, Reiser O. Eur. J. Org. Chem. 2008:2353–2364. [Google Scholar]
- 7.Battiste MA, Pelphrey PM, Wright DL. Chem. Eur. J. 2006;12:3438–3447. doi: 10.1002/chem.200501083. [DOI] [PubMed] [Google Scholar]
- 8.Grillet F, Huang C, Brummond KM. Org. Lett. 2011;13:6304–6307. doi: 10.1021/ol2028515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wen B, Hexum JK, Widen JC, Harki DA, Brummond KM. Org. Lett. 2013;15:2644–2647. doi: 10.1021/ol400904y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a Brummond KM, Chen H, Fisher KD, Kerekes AD, Rickards B, Sill PC, Geib S. J. Org. Lett. 2002;4:1931–1934. doi: 10.1021/ol025955w. [DOI] [PubMed] [Google Scholar]; b Brummond KM, Chen D, Davis MM. J. Org. Chem. 2008;73:5064–5068. doi: 10.1021/jo8007258. [DOI] [PubMed] [Google Scholar]; c Grillet F, Brummond KM. J. Org. Chem. 2013;78:3737–3754. doi: 10.1021/jo4002432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z, Liu C, Kinder RE, Han X, Qian H, Widenhoefer RA. J. Am. Chem. Soc. 2006:9066–9073. doi: 10.1021/ja062045r. [DOI] [PubMed] [Google Scholar]
- 12.Padgett HC, Csendes IG, Rapoport HJ. Org. Chem. 1979;44:3492–3496. [Google Scholar]
- 13.a Murakami M, Ubukata M, Itami K, Ito Y. Angew. Chem. Int. Ed. 1998;37:2248–2250. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2248::AID-ANIE2248>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]; b Wegner HA, de Meijere A, Wender PA. J. Am. Chem. Soc. 2005;127:6530–6531. doi: 10.1021/ja043671w. [DOI] [PubMed] [Google Scholar]; c Hong X, Stevens MC, Liu P, Wender PA, Houk KN. J. Am. Chem. Soc. 2014;136:17273–17283. doi: 10.1021/ja5098308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.For examples of solid-supported phosphine scavengers of ruthenium complexes see: Skowerski K, Gułajski Ł. In: Olefin Metathesis: Theory and Practice. Grela K, editor. Wiley; Somerset, NJ: 2014. pp. 559–570.
- 15.Denmark SE, Muhuhi JM. J. Am. Chem. Soc. 2010;132:11768–11778. doi: 10.1021/ja1047363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a Hirose T, Miyakoshi N, Mukai C. J. Org. Chem. 2008;73:1061–1066. doi: 10.1021/jo702330y. [DOI] [PubMed] [Google Scholar]; b Hayashi Y, Ogawa K, Inagaki F, Mukai C. Org. Biomol. Chem. 2012;10:4747–4751. doi: 10.1039/c2ob25500f. [DOI] [PubMed] [Google Scholar]
- 17.General Procedure: A High Dilution Allenic Pauson-Khand Reaction (APKR): A flame-dried, 2-necked, round-bottomed flask equipped with a stir bar, a condenser topped with a septum pierced with a nitrogen inlet needle, and a septum in the side arm was charged with rhodium biscarbonyl chloride dimer (0.05 equiv) and toluene (0.0013 M with respect to rhodium catalyst). The apparatus was evacuated through a needle connected to the vacuum gas manifold and then filled with carbon monoxide gas with a balloon (3 x). The flask was placed in a pre-heated oil bath (110 °C). In a separate flask, allene-yne (1 equiv) was dissolved in toluene (0.04 M with respect to allene-yne). The allene-yne solution was drawn into a syringe and added to the Rh(I) solution dropwise over 1.5 h using a syringe pump. After the addition was complete, heating and stirring were maintained for an additional 15 min at which time the reaction was judged complete as evidenced by TLC. The oil bath was removed and the reaction was allowed to cool to rt. Triphenylphosphine polymer bound (~3.0 mmol/g, 1 equiv) was added and the reaction was stirred for 14 h. The polymer was removed by vacuum filtration and rinsed with diethyl ether (10 mL). The filtrate was concentrated under reduced pressure using rotary evaporation, and the residue was purified by silica gel flash column chromatography to yield the bicyclo[5.3.0]decadienone.
- 18.Mukai C, Inagaki F, Yoshida T, Yoshitani K, Hara Y, Kitagaki S. J. Org. Chem. 2005;70:7159–7171. doi: 10.1021/jo050770z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.