

Abstract
A modified Pd-catalyzed method of forming aryl- and heteroarylboron species and a two-step, one-pot borylation/Suzuki-Miyaura cross coupling using the atom economical tetrahydroxydiboron (bis-boronic acid, BBA) is reported. By using ethylene glycol as an additive, the new method results in increased yields, lower BBA loading, faster reaction times, and a broader reaction scope, including previously problematic substrates such as heterocycles.
Keywords: bis-Boronic Acid, Tetrahydroxydiboron, Borylation, Cross-Coupling
Graphical abstract
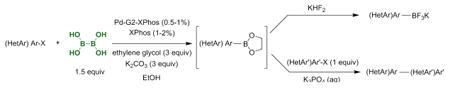
1. Introduction
The Suzuki-Miyaura reaction is one the most widely used catalytic methods for forming C(sp2)-C(sp2) bonds.1 Miyaura borylation is often used to access aryl- and heteroarylboron reagents used in this process. Although bis(pinacolato)diboron is effective as a borylating agent, its lack of atom economy and the challenges associated with removing the by-product pinacol during purification are often problematic for some applications.2 Significant improvements in borylating procedures were reported in 2010, through the development of the first direct metal-catalyzed synthesis of arylboronic acids via palladium-catalyzed cross-coupling of aryl chlorides with bis-boronic acid [BBA, tetrahydroxydiboron, B2(OH)4].3 This method offers a more efficient and atom-economical alternative to the bis(pinacolato)diboron (B2Pin2) derived approaches. By obviating the use of B2Pin2, this tactic allows facile synthesis of a variety of boronic acid derivatives and facilitates the purification of trifluoroborate salts or the desired cross-coupled products. More recently, borylations using BBA through a nickel-catalyzed reaction have also been reported.4
The palladium catalyzed method utilizing BBA to generate arylboronic acids made use of Buchwald's second generation XPhos palladium preformed catalyst (XPhos-Pd-G2).5 Bench stable XPhos-Pd-G2 allows ease of reaction set-up outside a glove box and leads to a more rapid in situ formation of the requisite Pd(0) species. The protocol initially developed was not without some limitations. Thus, although the method allowed efficient borylation of aryl chlorides, bromides as well as iodides, and triflates produced yields comparable to those published with B2Pin2, mesylates showed no conversion to the desired borylated species using BBA. For many substrates, there is an advantage in using mesylates as opposed to halides or triflates; mesylates are often less expensive to prepare and easier to handle. Additionally, phenols are sometimes more readily available than the corresponding aryl halides, and having the option to use them as starting materials greatly enhances the scope and utility of cross-coupling reactions. Another challenge in the previously reported protocol was presented by substrates embedded with reducible functional groups. Using the earlier Pd-catalyzed BBA borylation, functional groups such as aldehydes, ketones, and nitro groups generated were observed to produce reduced side products that led to lower yields of the desired trifluoroborate. The prior method was also limited in the scope of heterocycles that could be used to provide borylated products in reasonable yield.
Subsequent to the initial report of the Pd-catalyzed borylation of aryl and heteroaryl halides utilizing BBA, an efficient one-pot borylation/Suzuki cross–coupling reaction, extending even further the use of the method, was published.6 Prior to this publication, there had been few reports of one-pot, two-step couplings.7 Although these previous efforts demonstrated the ability to eliminate the need to isolate the boron species, the methods were limited by high requisite catalyst loadings and/or refluxing in ethereal solvents. The mild, one-pot conditions using the more atom economical BBA, ethanol as solvent, and low catalyst loads (0.1-1.0 mol %) was therefore an improvement to the existing methods. A useful finding evolving from the BBA study involved utilizing heteroaryl halides as the electrophillic partner in the Suzuki cross-coupling, completely eliminating the need to borylate them (Scheme 1). Although reversing the roles of the coupling partners is an option when both halides are readily accessible, this approach may not be optimal in more complex syntheses such as in the later stages of a natural product or drug candidate synthesis. Furthermore, the one-pot borylation/Suzuki cross-coupling of two heterocycles using the previous BBA protocol was unsuccessful and represented a major limitation of the method.
Scheme 1.

A comparison of previous one-pot borylation/cross-coupling methods.
Many heteroaryl systems are notoriously difficult substrates to cross-couple.8 Heteroarylboron species, especially boronic acids, are often unstable and prone to decomposition even at room temperature.9 This decomposition is exacerbated by the high temperatures and strong base sometimes needed in cross-coupling reactions, and therefore a large excess of the heterocyclic boronic acid is usually required for successful transformations.10 Boronic acids also form dimeric and cyclic trimeric anhydrides, making the amount of material actually used somewhat challenging to assess. For these reasons, the synthesis, storage, and subsequent cross-coupling of more stable, “protected” boron species in the form of boronate esters and trifluoroborates remains an active area of research. Although boronate esters, such as Suginome's 1,8-naphthalenediamineboranes [RB(dan)]11 and Burke's MIDA boronates8 eloquently address the stability associated with boronic acids, they must be hydrolyzed back to the boronic acids to be cross-coupled and therefore are not atom economical. As demonstrated frequently in the literature, trifluoroborates (RBF3K) remain the more atom economical choice for protecting boron coupling partners for storage, and numerous methods have been reported with high-yielding coupling.10
Herein is outlined a protocol for aryl- and heteroaryl borylation that resolves some of these outstanding issues in a manner that is both effective and experimentally facile.
2. Results and Discussion
To address the issues encountered with the aforementioned Pd-catalyzed BBA borylation method, a modification to the protocol that offers several advantages has been developed. It was hypothesized that a modified “hybrid” of the two commonly used borylating agents (B2Pin2 and BBA) could address some of the challenges/limitations associated with both of the reagents. In particular, the “in situ” formation of a boronate ester using BBA and ethylene glycol could potentially exhibit some of the positive qualities of both reagents, namely stability, low cost, and atom economy (Scheme 2).
Scheme 2.

Comparison of boron reagents.
To build on the existing borylation methods using this new strategy, the use of an improved borylating protocol was investigated. Upon optimization of the method, the addition of stoichiometric amounts of the inexpensive and readily available ethylene glycol during the borylation of aryl halides was determined to allow a two-fold decrease in the amount of BBA required. Furthermore, the addition of ethylene glycol resulted in a marked rate acceleration for the borylation, as well as a decrease in the amount of catalyst required for heterocyclic substrates as well as an improvement in the overall scope and yield.
As demonstrated in Table 1, substrates that were challenging under the previously reported conditions led to higher isolated yields of the RBF3K salts in substantially shorter reaction times with the addition of ethylene glycol.
Table 1.
RBF3K synthesis with and without the use of ethylene glycol additive.

| |||
|---|---|---|---|
|
| |||
| entry | product | X | % yield (time) a |
| 1 |
|
CI | 80 (20 min) |
| 77 (1.5 h) | |||
| 2 |
|
CI | 82 (2 h) |
| 68 (2 h) | |||
| 3 |
|
Br | 92 (20 min) |
| 68 (1 h) | |||
| 4 |
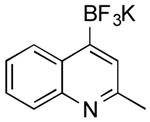
|
Cl | 52 b (30 min) |
| 47 (1.5 h) | |||
| 5 |
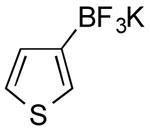
|
CI | 80 (3.5 h) |
| 0 | |||
| 6 |
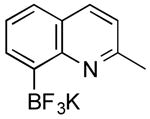
|
CI | 65 (1 h) |
| 52 (2 h) | |||
| 7 |
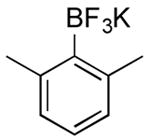
|
CI | 69 (1 h) |
| 53 (4.5 h) | |||
| 8 |
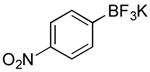
|
CI | 69 (25 min) |
| 64 (4 h) | |||
| 9 |
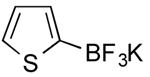
|
CI | 27 (45 min) |
| 0 | |||
| 5 c | |||
| 10 |
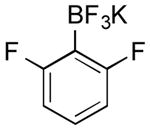
|
CI | 66 d (84 h) |
| 43 e | |||
| 37 f (22 h) | |||
| 0 g (3 h) | |||
| Br | 0 | ||
| 11 |
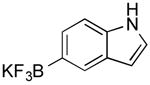
|
CI | 37 (50 min) |
| 12 |
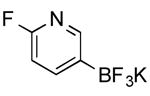
|
CI | 76 (3 h) |
| 73 h (75 min) | |||
| 13 |
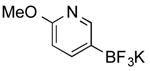
|
CI | 88 i (3 h) |
| 14 |
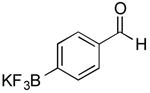
|
CI | 73 j (1 h) |
| 75k (4 h) | |||
| 15 |
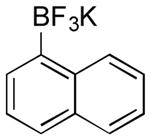
|
OMs | 60 (4 h) |
| 0 | |||
1.5 mmol scale unless otherwise noted.
As an inseparable mixture of internal salt and potassium trifluoroborate.
Using (Me2N)2B- B(NMe2)2 as the borylating agent.
0 °C.
rt.
50 °C.
80 °C.
65 °C, 7.6 mmol scale.
65 °C.
13% coupled alcohol reduction product was also generated.
30% coupled alcohol reduction product was also generated.
In general, the reaction rates were accelerated 2-3 times, with sometimes dramatically enhanced yields. For example, the yield of 3-trifluoroboratoquinoline improved dramatically from 68 to 92% in less than half the reaction time (entry 3, Table 1). Another interesting result was observed with the previously unreactive 2- and 3-substituted thiophenes (entries 5 and 9, Table 1) and 1,3-difluoro-2-chlorobenzene (entry 10, Table 1). Using the modified reaction conditions, these substrates were obtained in moderate to good yield, with the latter performing better at reduced temperatures.
Because of the reduction of reaction time, functional group tolerance also improved under the modified ethylene glycol procedure. In particular, the benzaldehyde derivative (entry 14, Table 1) was obtained in slightly higher yield, but more importantly, the observed palladium-catalyzed reduction product was reduced from 30% to 13%. The new, modified procedure also has the potential to advance the area of mesylate borylation, as the method provided product from a mesylated substrate in moderate yield (entry 15, Table 1). Additionally, this method was shown to be robust on a larger scale by obtaining comparable yields on both a 1.5 mmol scale and a 1.0 g scale (entry 12, Table 1).
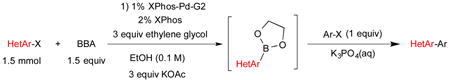
To evaluate the potential advantages of the new ethylene glycol method further, we investigated the one-pot borylation/cross-coupling of heterocyclic substrates. Previous studies indicated that when two aromatic substrates were cross-coupled, the overall yield of the process was greatly dependent on which aryl halide was borylated in situ and which aryl halide was added in the second step as the electrophile. These studies indicated that heteroaryl substrates typically react more effectively when used as the electrophilic component of the cross-coupling and are therefore added to the reaction after the borylation of the first aryl halide is complete, as attempts to use the heteroaromatic substrate as the borylated component in the cross-coupling step often resulted in low yields or no reaction at all.5 Additionally, the coupling of two heteroaromatic halides proved to be unsuccessful using the previous BBA one-pot method. To expand the utility of the current method, we applied the ethylene glycol conditions to borylate a series of heteroaromatic substrates and subsequently cross-coupled them in one pot with either an aryl or heteroaryl coupling partner (Table 2).
Table 2. One-Pot Borylation/Cross-Coupling Process.
| ||||
|---|---|---|---|---|
|
| ||||
| entry | HetAr-X | Ar-X | yield (%) | rxn temp (°C) |
| 1 |
|
|
95 | 80 |
| 2 |
|
|
84 | 80 |
| 3 |
|
|
88 a | 80 |
| 4 |
|
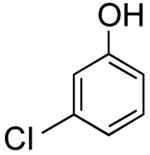
|
78; 82 a | 65 |
| 5 |
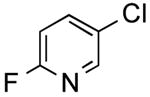
|
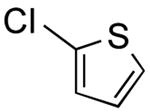
|
39 | 65 |
| 6 |
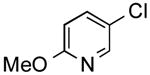
|
|
86 | 65 |
| 7 |
|
|
79 | 65 |
| 8 |
|
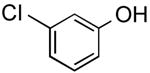
|
59 | 65 |
| 9 |
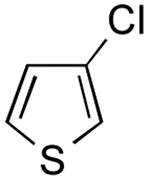
|
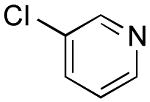
|
63 | 80 |
| 10 |
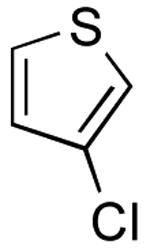
|
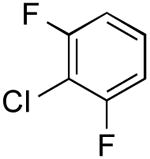
|
53 | 80 |
| 11 |
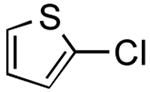
|
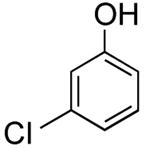
|
31 | 65 |
| 12 |
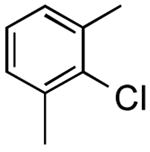
|
|
61 | 65 |
| 42 | 80 | |||
| 13 |
|
|
28 | 65 |
7.6 mmol scale, 0.75% Pd-G2-XPhos, 1.5% XPhos, 3 equiv ethylene glycol, 3 equiv KOAc, 1.5 equiv BBA
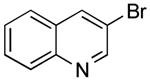
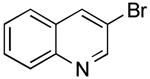
The borylation of 3-bromoquinoline (Table 1, entry 3) was improved using the ethylene glycol method and allowed a significant decrease in catalyst loading. Therefore, it was a logical starting point to test the effectiveness of the new method on the one-pot borylation/cross-coupling. The results of borylation of 3-bromoquinoline and in situ coupling with a second heteroaryl halide proved to be successful with addition of ethylene glycol. Three different heteroaryl halides were tested as electrophiles, and overall yields between 84-95% were observed (Table 2, entries 1-3).
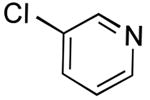
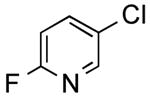
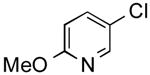
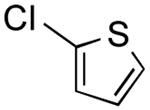
The results outlined in Table 1 (entries 12 and 13) revealed that borylation of pyridines proceeded well at 65 °C. An investigation was undertaken to determine if a two-step borylation/cross-coupling process could be achieved on pyridine derivatives at this same temperature. 5-Chloro-2-fluoropyridine and 3-chlorophenol were chosen as partners because previous methods (without ethylene glycol) were effective in borylating the phenol derivative in the first step followed by cross coupling the pyridine derivative in the second step, but attempts to reverse the order (borylation of the pyridine derivative) with the previously developed procedure proved ineffective, even when an increased Pd loading was used.5 The ethylene glycol method developed herein has now been shown to be quite effective in borylating pyridine derivatives, with subsequent cross-coupling with the phenolic halide providing the desired product in one pot in 78-82% overall yield on a 1.5 mmol or 7.6 mmol scale (Table 2, entry 4). Additionally, it was shown that the methoxy-substituted pyridine was also effective in borylation, with subsequent cross-coupling with an aryl and heteroaryl halide (Table 2, entries 6 and 7).
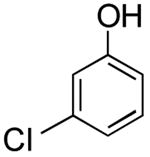
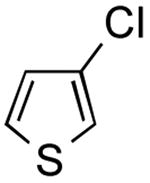
The one-pot borylation of 3-chlorothiophene and 3-chlorophenol was also limited by the role of the respective halides as the nucleophile or electrophile using the glycol-free method. Specifically, the substituted phenol could be borylated, and subsequently the 3-substituted thiophene could be utilized as the electrophile. However, the reverse order was not tolerated. Using the ethylene glycol-added method, the reverse order reaction (borylation of the thiophene and cross coupling of the resulting organoboron species with 3-chlorophenol) could be accomplished in a moderate yield (entry 8). Although the ethylene glycol significantly improved the viability for several examples, the borylation of 2-bromobenzonitrile still proved to be challenging, and a yield comparable to that previously reported was obtained (Table 2, entry 13).
Interestingly, while we were exploring the new method with ethylene glycol, a paper by Bello and Schmidt-Leithoff reported the effects of adding diols to tetrakis(dimethylamino)diboron on the borylation of halides and triflates.12 This work reported an improvement in yields in the presence of bulkier diols, which they attribute to the Thorpe-Ingold effect. The authors hypothesize that a boronate ester is formed by addition of the diol. However, in that study the reactions could not be carried out in alcoholic solvents.
3. Conclusion
The borylation and one-pot borylation/cross coupling methods previously reported utilizing BBA19 were advancements to alternative methods. However, one major limitation was the inability to use select heteroaryl halides as they often proved difficult to borylate, leading to modest yields of the trifluoroborates. Therefore, the role of heteroaryl halides in a one-pot borylation/cross-coupling sequence was limited to the electrophilic partner in reactions with aryl nucleophiles, affording the cross-coupled product in good to excellent yield. Because of this limitation, the method did not yield the cross-coupling between two heteroaryls in good yields using the one-pot method. The results of continued investigations as described in this paper support the development of an improved Pd catalyzed borylation and cross-coupling method. It is hypothesized that the success of the new method can be attributed to the formation of the bisboronate ester in situ, lending stability to the borylating agent and subsequently the newly formed arylboron species. Owing to the formation of a more stable intermediate, the amount of BBA required has been reduced by half. Further, the borylation/cross coupling using the improved method increases the flexibility in the order of the reaction (i.e., which halide is borylated and which acts as the electrophile), thus filling a previously identified gap. Also of note is the fact that ethylene glycol is inexpensive, especially when compared to its pinacol counterpart. It is also readily available and easily removed from the reaction mixture during an aqueous workup. Results indicate that this modified method involving the addition of ethylene glycol offers a significant and practical improvement to previous methods.
Experimental Section
General Considerations
New compounds were characterized by 1H NMR, 13C NMR, 11B NMR (when applicable), 19F NMR (when applicable), high-resolution mass spectrometry, and melting points for solids. Known compounds were characterized by 1H NMR, 13C NMR, 11B NMR (if relevant), and 19F NMR (if relevant). 1H and 13C NMR spectra were recorded at 500 and 125.8 MHz, respectively. Melting points are uncorrected. HRMS data were obtained using either ESI or CI and obtained by a TOF mass spectrometer.
General Procedure A: Palladium Catalyzed Borylation of Heteroaryl Halides with Ethylene Glycol Additive and Their Conversion to Trifluoroborates
To an oven dried glass vessel capable of being sealed with a Teflon cap (for microwave vials) was added XPhos-Pd-G2 (5.89 mg, 7.5 μmol), XPhos (7.14 mg, 15 μmol), B2(OH)4 (203 mg, 2.25 mmol), KOAc (441 mg, 4.5 mmol), and halide (only if a solid). The vessel was sealed, evacuated, and filled with an inert gas (4X). EtOH (15 mL, degassed) was added via syringe followed by the addition of ethylene glycol (280 mg, 250 μL, 4.5 mmol) and the halide (1.5 mmol) in a similar manner (if applicable). The reaction was heated to the specified temperature until the starting material was consumed (monitored by GC). The reaction was cooled to rt and filtered through a very thin pad of Celite and decolorizing carbon (eluting with 5 × 10 mL of EtOAc) and concentrated. The crude reaction was dissolved in EtOAc (10 mL), transferred to a separatory funnel, and then H2O (10 mL) was added. The layers were separated, and the organic layer was washed with brine (5 mL). The combined aqueous layers were extracted with EtOAc (3 × 5 mL) dried (Na2SO4), filtered, and concentrated. MeOH (∼ 15 mL) was added to concentrated crude reaction mixture, which was cooled to 0 °C. To this reaction mixture, KHF2 (1 mL of 4.5 M soln, 4.5 equiv) was added. The reaction was stirred at 0 °C for 10 min before removing the bath and allowing the reaction mixture to stir for 20 min (or until conversion to trifluoroborate is complete as observed by 11B). The resulting mixture was concentrated and then lyophilized overnight to remove any traces of H2O. The compound was purified with continuous Soxhlet extraction (8 h or by complete disappearance of RBF3K in the thimble as determined by 11B NMR). The collected solvent was concentrated until a minimum amount of acetone (∼ 3 mL) remained. The addition of Et2O (∼ 25 mL) led to the precipitation of the desired product.
General Procedure B: General Procedure for the Palladium-Catalyzed Borylation of Heteroaryl Halides with Ethylene Glycol Additive and their Suzuki Cross-Coupling with Heteroaryl Halides
To an oven dried glass vessel capable of being sealed with a Teflon cap (for microwave vials) was added XPhos-Pd-G2 (11.8 mg, 15 μmol), XPhos (14.3 mg, 30 μmol), B2(OH)4 (203 mg, 2.25 mmol), KOAc (441 mg, 4.5 mmol), and halide (only if a solid). The vessel was sealed, evacuated, and filled with an inert gas (4X). EtOH (15 mL, degassed) was added via syringe followed by the addition of ethylene glycol (280 mg, 250 μL, 4.5 mmol) and the halide (1.5 mmol) in a similar manner (if applicable). The reaction was heated to the specified temperature until the starting material was consumed (as monitored by GC). The reaction was cooled, and a needle attached to a manifold under an inert atmosphere was inserted into the septum, and 3 equivalents of degassed K3PO4 (1 M, 4.5 mL, 4.5 mmol) was added via syringe, and the reaction was allowed to sit for 5 min. Then the second halide was added in a similar manner (as a solution in 500 μL of degassed EtOH or THF if a solid). The manifold needle was removed, and the reaction was heated to the specified temperature for 24 h. The reaction was cooled to rt and filtered through a very thin pad of Celite, eluting with 5 × 10 mL of EtOAc, and then concentrated. The crude reaction was dissolved in EtOAc (10 mL), transferred to a separatory funnel, and then saturated NaHCO3 (10 mL) was added. The aqueous layer was extracted with EtOAc (3 × 5 mL). The combined organics were dried (Na2SO4), filtered, and concentrated. The desired compound was purified by silica gel column chromatography, eluting with EtOAc/hexane.
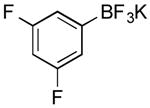
Potassium 3,5-Difluorophenyl-trifluoroborate (Table 1, Entry 1)5
General procedure A was used at 80 °C for 20 min with 1-chloro-3,5-difluorobenzene (223 mg, 168 μL, 1.5 mmol) to yield the desired compound as a white solid (265 mg, 80% yield). Spectral data were in accordance with those of published results. Mp: > 225 °C. 1H NMR (500 MHz, acetone-d6) δ 6.97 (d, J = 5.7 Hz, 2H), 6.59-6.47 (m, 1H). 13C NMR (125.8 MHz, DMSO-d6) δ 162.4 (dd, J = 243 Hz), 113.4 (d, J = 15.4 Hz), 100.6 (d, J = 25.5 Hz). 11B NMR (128.4 MHz, acetone-d6) δ 3.4 (m). 19F NMR (338.8 MHz, acetone-d6) δ -114.9, -143.2.
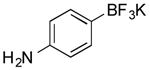
Potassium 4-Aminophenyl-trifluoroborate (Table 1, Entry 2)5
General procedure A was used at 80 °C for 2 h with 4-chloroaniline (191 mg, 1.5 mmol) to yield the desired compound as an off-white solid (244 mg, 82% yield). Spectral data were in accordance with those of published results.5 Mp: > 225 °C. 1H NMR (500 MHz, acetone-d6) δ 7.19 (d, J = 7.4 Hz, 2H), 6.45 (d, J = 7.4 Hz, 2H), 3.96 (s, 2H). 13C NMR (125.8 MHz, DMSO-d6) δ 146.0, 132.5, 113.5. 11B NMR (128.4 MHz, acetone-d6) δ 4.1 (m). 19F NMR (338.8 MHz, acetone-d6) δ -141.5.
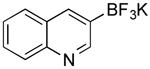
Potassium (Quinolin-3-yl)trifluoroborate (Table 1, Entry 3)13
General procedure A was used at 80 °C for 30 min with 3-bromoquinaldine (266 mg, 1.5 mmol) to yield the desired compound as a pale yellow solid (324 mg, 92% yield). Spectral data were in accordance with those of published results.13 Mp: > 225 °C. 1H NMR (500 MHz, acetone-d6) δ 9.02 (s, 1H), 8.18 (s, 1H), 7.90 (d, J = 8.1 Hz, 1H), 7.75 (d, J = 7.6 Hz, 1H), 7.52 (t, J = 6.9 Hz, 1H), 7.40 (t, J = 7.0 Hz, 1H). 13C NMR (125.8 MHz, DMSO-d6) δ 155.8, 147.3, 138.0, 129.1, 128.7, 128.2, 127.9, 125.7. 11B NMR (128.4 MHz, acetone-d6) δ 3.5 (q, J = 49.6 Hz). 19F NMR (338.8 MHz, acetone-d6) δ -141.9.
Potassium (2-Methylquinolin-4-yl)trifluoroborate(Table 1, Entry 4)13
General procedure A was used at 80 °C for 30 min with 4-chloroquinaldine (266 mg, 1.5 mmol). The desired trifluoroborate existed as an inseparable mixture of the internal salt and the potassium trifluoroborate (194 mg, 52% yield). Therefore reasonable spectra could not be obtained.
Potassium Thien-3-yltrifluoroborate (Table 1, Entry 5)14
General procedure A was used at 80 °C for 3.5 h with 3-chlorothiophene (178 mg, 1.5 mmol) to yield the desired compound as an off-white solid (227 mg, 80% yield). Spectral data were in accordance with those of published results.14 Mp: > 225 ° C. 1H NMR (500 MHz, acetone-d6) δ 7.16 (s, 1H), 6.99 (s, 1H), 6.97 (d, J = 4.6 Hz, 1H). 13C NMR (125.8 MHz, DMSO-d6) δ 132.5, 124.9, 123.2. 11B NMR (128.4 MHz, acetone-d6) δ 3.1 (q, J = 52.6 Hz). 19F NMR (338.8 MHz, acetone-d6) δ -138.6.
Potassium (2-Methylquinolin-8-yl)trifluoroborate (Table 1, Entry 6)5
General procedure A was used at 80 °C for 1 h with 8-chloro-2-methylquinoline (178 mg, 1.5 mmol) to yield the desired compound as a tan solid (243.2 mg, 65% yield). Spectral data were in accordance with those of published results.5 Mp: > 225 ° C. 1H NMR (500 MHz, DMSO) δ 8.20 (d, J = 7.6, 1H), 7.84 (s, 1H), 7.69 (d, J = 7.6, 1H), 7.39 (t, J = 6.9, 1H), 7.33 (d, J = 8.1, 1H), 2.67 (s, 3H). 13C NMR (126 MHz, DMSO) δ 172.8, 156.7, 149.3, 138.5, 134.2, 126.5, 126.5, 126.2, 125.8, 121.2, 39.5, 24.7. 11B NMR (128 MHz, DMSO-d6) δ 3.64. 19F NMR (471 MHz, DMSO-d6) δ -135.4.
Potassium (2,6-Dimethylphenyl)trifluoroborate (Table 1, Entry 7)14
General procedure A was used at 80 °C for 1 h with 2-chloro-1,3-dimethylbenzene (140.6 mg, 1.5 mmol) to yield the desired compound as an off-white solid (218.9 mg, 69% yield). Spectral data were in accordance with those of published results.14 Mp: > 225 ° C. 1H NMR (500 MHz, DMSO-d6) δ 6.80 (t, J = 7.4 Hz, 1 H), 6.71 (d, J = 7.4 Hz, 2H), 2.34 (s, 6 H). 13C NMR (126 MHz, DMSO-d6) δ 141.0, 126.6, 124.7, 23.3 (dd, J = 6.3, 3.8 Hz).
Potassium 4-Nitrophenyl-trifluoroborate (Table 1, Entry 8)5
General procedure A was used at 80 °C for 25 min with 1-chloro-4-nitrobenzene (236.33, 1.5 mmol) to yield the desired compound as a tan solid (236.5 mg, 69% yield). Spectral data were in accordance with those of published results.5 Mp: > 225 ° C. 1H NMR (500 MHz, DMSO-d6) δ 8.00 (d, J = 8.1 Hz, 2H), 7.60 (d, J = 8.0 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 146.5, 132.8, 121.8. HRMS (ESI) calcd. for C6H4NO2F3B [M]-190.0287, found 190.0296.
Potassium Thien-2-yltrifluoroborate (Table 1, Entry 9)13
General procedure A was used at 80 °C for 45 min with 2-chlorothiophene (177.9 mg, 1.5 mmol) to yield the desired compound as an off-white solid (77.3 mg, 27% yield). Spectral data were in accordance with those of published results.13 Mp: > 225 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.2 (m, 1H), 6.92-6.8 (m, 2H). 13C NMR (125.8 MHz, CDCl3) δ 127.0, 126.9, 124.4.
Potassium (2,6-Difluorophenyl)trifluoroborate (Table 1, Entry 10)14
General procedure A was used at 0 °C for 72 h with 2-chloro-1,3-difluorobenzene (222.8 mg, 1.5 mmol) to yield the desired compound as an off-white solid (217.9 mg, 66% yield). Spectral data were in accordance with those of published results.14 Mp: > 225 ° C; 1H NMR (500 MHz, DMSO-d6) δ 7.10 (m, 1H), 6.67 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 166.2 (dd, J = 241.8, 17.5 Hz), 127.9 (t, J = 10.5 Hz), 110.10 (dd, J = 22.6, 7.7 Hz). 19F NMR (471 MHz, DMSO-d6) δ -103.47, -132.20. 11B NMR (128 MHz, DMSO-d6) δ 2.02. HRMS (ESI) calcd. for C6H3F5B [M]-181.0248, found 181.0254.
Potassium 1H-Indol-5-yl-trifluoroborate (Table 1, Entry 11)13
General procedure A was used at 80 °C for 50 min with 5-chloroindole (227.4 mg, 1.5 mmol) to yield the desired compound as a tan solid (122.9 mg, 37% yield). Spectral data were in accordance with those of published results.13 Mp > 200 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.54 (brs, 1H) 7.48 (s, 1H), 7.11-7.09 (m, 3H), 6.26-6.21 (m, 1H). 13C NMR (126 MHz, DMSO-d6) 134.8, 127.0, 125.5, 123.0, 122.5, 109.0, 100.5.
Potassium 6-Fluoropyridin-3-yltrifluoroborate (Table 1, Entry 12)13
General procedure A was used at 65 °C for 3 h with 5-chloro-2-fluoropyridine (197.3 mg, 1.5 mmol) to yield the desired compound as a white solid (230.7 mg, 76% yield). Mp: > 225. 1H NMR (500 MHz, DMSO-d6) δ 8.09 (s, 1H), 7.86–7.74 (m, 1H), 6.85 (dd, J = 8.0, 1.9 Hz, 1H). 13C NMR (126 MHz, DMSO-d6) δ 162.7 (d, J = 230.7 Hz), 150.1 (d, J = 12.6 Hz), 145.18 (d, J = 6.2 Hz), 107.81 (d, J = 34.8 Hz). 11B NMR (128 MHz, DMSO-d6) δ 3.02. 19F NMR (471 MHz, DMSO-d6). δ -73.3, -138.8. HRMS (ESI) calcd. for C5H3BF4N [M]-164.0295, found 164.0301.
Potassium 6-Methoxypyridin-3-yltrifluoroborate (Table 1, Entry 13)
General procedure A was used at 65 °C for 3 h with 5-chloro-2-methoxypyridine(215.4 mg, 1.5 mmol) to yield the desired compound as a white solid (284.6 mg, 88% yield). Spectral data were in accordance with those of published results. Mp: 200-205 ° C. 1H NMR (500 MHz, DMSO-d6) δ 8.08 (s, 1H), 7.59 (d, J = 7.9 Hz, 1H), 6.58 (d, J = 8.0 Hz, 1H). 13C NMR (126 MHz, DMSO-d6) δ 162.4, 149.4, 142.6, 108.8, 52.7. 19F NMR (471 MHz, DMSO-d6) δ -138.31. 11B NMR (128 MHz, DMSO-d6) δ 3.28. HRMS (ESI) calcd. for C6H4BNOF3 [M]-176.0495, found 176.0508.
Potassium 4-Formylphenyl-trifluoroborate and Potassium 4-(Hydroxymethyl)phenyl-trifluoroborate (Table 1, Entry 14)
General procedure A was used at 80 °C for 1 hour with 4-bromobenzaldehyde (277.5 mg, 1.5 mmol) to yield a mixture of desired carbonyl and alcohol as a white solid (235.7 mg, 73% yield).
Potassium Naphthalen-1-yltrifluoroborate (Table 1, Entry 15)14
General procedure A was used at 80 °C for 4 h with naphthalen-1-yl methanesulfonate (333.5 mg, 1.5 mmol) to yield the desired compound as a white solid (210.1 mg, 60% yield). Spectral data were in accordance with those of published results.14 Mp: 205-207 ° C. 1H NMR (500 MHz, DMSO-d6) δ 8.38 (d, J = 7.2 Hz, 1H), 7.69 (dd, J = 5.8, 2.5 Hz, 1H), 7.57-7.53 (m, 2H), 7.31–7.24 (m, 3H). 13C NMR (126 MHz, DMSO-d6) δ 137.0, 133.4, 130.6, 129.0, 129.0, 127.8, 125.7, 125.3, 124.3, 123.8. 19F NMR (471 MHz, DMSO-d6) δ -135.3. 11B NMR (128 MHz, DMSO-d6) δ 3.57. HRMS (ESI) calcd. for C10H7BF3 [M]-195.0593, found 195.0593.
3-(Pyridin-3-yl)quinolone (Table 2, Entry 1)15
General Procedure B was used. Silica gel column chromatography (0-100% EtOAc/hexane) was used to obtain the desired compound as a light yellow solid (293 mg, 95% yield). Mp: 97-100 °C. 1H NMR (500 MHz, CDCl3) δ 9.54 (s, 1H), 8.77 (d, J = 5.2 Hz, 2H), 8.15 (d, J = 8.4 Hz, 1 H), 7.92 (d, J = 7.9 Hz, 1H), 7.88 (d, J = 7.9 Hz, 1H), 7.82 (t, J = 7.6 Hz, 1 H), 7.74 (t, J = 7.7 Hz, 1H), 7.57 (t, J = 7.4 Hz, 1H), 7.33-7.29 (m, 1H). 13C NMR (125.8 MHz, CDCl3) δ 154.9, 150.3, 149.4, 148.4, 137.2, 134.0, 132.0, 134.0, 132.0, 130.1, 129.4, 128.7, 128.0, 127.2, 122.9, 120.9. IR (neat) 3054, 1589, 1095 cm-1. HRMS (ES+) calcd. for C14H11N2 (M+H) 207.0922, found 207.0922.19
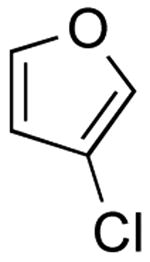
3-(Furan-3-yl)quinolone (Table 2, Entry 2)
General Procedure B was used. Silica gel column chromatography (0-100% EtOAc/hexane) was used to obtain the desired compound as a yellow solid (246 mg, 84% yield). Mp: 86-89 °C. 1H NMR (500 MHz, CDCl3) δ 9.08 (d, J = 1.9 Hz, 1H), 8.16 (s, 1H), 8.10 (d, J = 8.3 Hz, 1 H), 7.91 (s, 1H), 7.82 (d, J = 7.9 Hz, 1H), 7.68 (t, J = 7.0 Hz, 1 H), 7.60-7.52 (m, 2H), 6.84 (s, 1 H). 13C NMR (125.8 MHz, CDCl3) δ 149.1, 147.3, 144.4, 139.3, 131.5, 129.4, 129.3, 128.3, 127.8, 127.2, 125.8, 123.6, 108.8. IR (neat) 3346, 1617 cm-1. HRMS (CI+) calcd. for C13H9NO 195.0684 found 195.0682.19
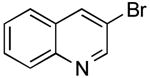
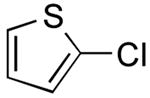
3-(Thien-2-yl)quinolone (Table 2, Entry 3)
General Procedure B was used. Silica gel column chromatography (0-100% EtOAc/hexane) was used to obtain the desired compound as a yellow solid (280 mg, 88% yield). Mp: 70-73 °C. 1H NMR (500 MHz, CDCl3) δ 9.21 (d, J = 2.0 Hz, 1H), 8.28 (s, 1H), 8.10 (d, J = 8.2 Hz, 1 H), 7.85 (d, J = 7.8 Hz, 1H), 7.70 (t, J =7.0 Hz, 1H), 7.57 (t, J =7.1 Hz, 1 H), 7.51 (d, J = 2.6 Hz, 1H), 7.41 (d, J = 4.2 Hz, 1 H), 7.22-7.14 (m, 1H). 13C NMR (125.8 MHz, CDCl3) δ 148.7, 147.4, 140.9, 131.5, 129.5, 128.6, 128.1, 127.9, 127.7, 127.4, 126.3, 124.6. IR (neat) 3066, 1492 cm-1. HRMS (CI+) calcd. for C13H9NS 211.0456, found 211.0461.19
3-(6-Fluoropyridin-3-yl)phenol (Table 2, Entry 4)15
General Procedure B was used. Silica gel column chromatography (0-100% EtOAc/hexane) was used to obtain the desired compound as a white solid (221.8 mg, 78% yield). Mp: 105 °C. 1H NMR (500 MHz, DMSO-d6) δ 9.63 (s, 1H), 8.47 (d, J = 2.6 Hz, 1H), 8.21 (td, J = 8.2, 2.6 Hz, 1H), 7.37-7.22 (m, 2H), 7.11 (dd, J = 8.2, 2.4 Hz, 1 H), 7.04 (t, J = 2.1 Hz, 1H), 6.83 (dd, J = 8.2, 2.4 Hz, 1H). 13C NMR (126 MHz, DMSO-d6) δ 163.5, 161.6, 158.0, 145.2 (d, J =15.1 Hz), 140.3 (d, J = 8.1 Hz), 134.3, 130.2, 117.6, 115.2, 113.7, 109.6 (d, J = 37.7 Hz).
2-Fluoro-5-(thien-2-yl)pyridine (Table 2, Entry 5)
General Procedure B was used. Silica gel column chromatography (0-100% EtOAc/hexane) was used to obtain the desired compound as a yellow oil (268.8 mg, 39% yield). 1H NMR (500 MHz, CDCl3) δ 8.47–8.44 (m, 1H), 7.96 (ddd, J = 8.5, 7.5, 2.6 Hz, 1H), 7.35 (dd, J = 5.1, 1.1 Hz, 1H), 7.29 (dd, J = 3.6, 1.1 Hz, 1H), 7.11 (dd, J = 5.1, 3.6 Hz, 1H), 6.96 (ddd, J = 8.5, 3.1, 0.5 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 163.0 (d, J = 240.3 Hz), 144.7 (d, J = 15.1 Hz), 139.2, 138.7 (d, J = 7.5 Hz), 128.8, 126.1, 124.4, 109.8 (d, J = 39.0 Hz). HRMS (CI+) calcd. for C9H7NSF [M+H]+ 192.0483, found 192.0484.
3-(6-Methoxypyridin-3-yl)phenol (Table 2, Entry 6)
General Procedure B was used. Silica gel column chromatography (0-100% EtOAc/hexane) was used to obtain the desired compound as an off-white solid (257.2 mg, 86% yield). Mp: 86-88 °C. 1H NMR (500 MHz, CDCl3) δ 8.41 (d, J = 2.5 Hz, 1H), 7.79 (dd, 8.6, 2.5, 1H), 7.72 (s, 1H), 7.29 (dd, 14.9, J = 7.0 Hz, 1H), 7.08-7.03 (d, J = 7.7 Hz, 1H), 6.91–6.81 (m, 3H), 3.99 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 163.8, 157.0, 144.7, 139.2, 138.2, 130.4, 130.4, 118.9, 114.9, 113.9, 110.9, 54.2. HRMS (CI+) calcd. for C10H12NO2 [M+H]+, 202.0868 found 202.0868.
2-Methoxy-5-(thien-2-yl)pyridine (Table 2, Entry 7)
General Procedure B was used. Silica gel column chromatography (0-100% EtOAc/hexane) was used to obtain the desired compound as a yellow oil (286.9 mg, 79% yield). 1H NMR (500 MHz, CDCl3) δ 8.42 (d, J = 2.3 Hz, 1H), 7.76 (dd, J = 8.6, 2.4 Hz, 1H), 7.26 (s, 1H), 7.21 (d, J = 3.5 Hz, 1H), 7.11–7.05 (m, 1H), 6.76 (d, J = 8.6 Hz, 1H), 3.96 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 163.7, 144.1, 140.9, 136.7, 128.2, 124.8, 124.2, 123.1, 111.1, 53.8. HRMS (CI+) calcd. for C10H10NOS [M+H]+ 192.0483, found 192.0484.
3-(Thien-3-yl)phenol (Table 2, Entry 8)15
General Procedure B was used. Silica gel column chromatography (0-100% EtOAc/hexane) was used to obtain the desired compound as a tan solid (156.8 mg, 59% yield). Spectral data were in accordance with those of published results.15 Mp: 94–95 ° C. 1H NMR (500 MHz, CDCl3) δ 7.43 (dd, J = 2.8, 1.3 Hz, 1H), 7.37 (m, 2H), 7.28 (m, 1H), 7.20 (m, 1H), 7.09 (m, 1H), 6.80–6.76 (m, 1H), 5.07 (s, 1H). 13C NMR (126 MHz, CDCl3) δ 155.9, 142.0, 137.7, 130.3, 126.5, 126.4, 120.8, 119.4, 114.3, 113.6.
3-(Thien-3-yl)pyridine (Table 2, Entry 9)6
General Procedure B was used. Silica gel column chromatography (0-100% EtOAc/hexane) was used to obtain the desired compound as an off-white solid (151.8 mg, 63% yield). Spectral data were in accordance with those of published results.6 Mp: 73–75 ° C. 1H NMR (500 MHz, DMSO-d6) δ 8.97 (d, J = 2.3 Hz, 1H), 8.48 (dd, J = 4.8, 1.6 Hz, 1H), 8.08 (dt, J = 8.2, 1.9 Hz, 1H), 8.01 (dd, J = 2.9, 1.5 Hz, 1H), 7.68 (dd, J = 5.0, 2.9 Hz, 1H), 7.63 (dd, J = 5.1, 1.4 Hz, 1H), 7.41 (dd, J = 8.0, 4.8 Hz, 1H). 13C NMR (126 MHz, DMSO-d6) δ 148.0, 147.1, 138.1, 133.2, 130.7, 127.6, 126.0, 123.8, 122.2.
3-(2,6-Difluorophenyl)thiophene (Table 2, Entry 10)
General Procedure B was used. Silica gel column chromatography (0-100% EtOAc/hexane) was used to obtain the desired compound as a clear oil (165 mg, 56% yield). 1H NMR (500 MHz, CDCl3) δ 7.64 (m, 1H), 7.45-7.38 (m, 2H), 7.27-7.18 (m, 1H), 7.03-6.93 (m, 2H). 13C NMR (125.8 MHz, CDCl3) δ160.1 (d, J = 242.3 Hz), 128.8 (t, J = 3.6 Hz), 128.4, 128.1 (t, J = 10.6 Hz), 126.3, 125.9 (m), 124.6, 119.7, 112.1-111.1 (m). 19F NMR (338.8 MHz, acetone-d6) δ -112.7.
3-(Thien-2-yl)phenol (Table 2, Entry 11)15
General Procedure B was used. Silica gel column chromatography (0-100% EtOAc/hexane) was used to obtain the desired compound as a tan solid (82.5 mg, 31% yield). Spectral data were in accordance with those of published results.15 1H NMR (500 MHz, CDCl3) δ 7.45 (dd, J = 2.8, 1.4 Hz, 1H), 7.39-7.36 (m, 2H), 7.25-7.32 (m, 1H), 7.21-7.19 (m, 1H), 7.09-7.08 (m, 1H), 6.80-6.77 (m, 1H), 4.78 (s, 1H). 13C NMR (125.8 MHz, CDCl3) δ 155.9, 144.0, 136.1, 130.3, 125.1, 123.5, 118.8, 119.3, 114.6, 112.9. HRMS (CI+) calcd. for C10H8SO [M]+ 176.0296, found 176.0303.
2-(2,6-Dimethylphenyl)thiophene (Table 2, Entry 12)17
General Procedure B was used at 80 °C. Silica gel column chromatography (0-100% EtOAc/hexane) was used to obtain the desired compound as a yellow oil (119.7 mg, 42% yield). 1H NMR (500 MHz, DMSO-d6) δ 7.59 (d, J = 5.1 Hz, 1H), 7.19– 7.08 (m, 4H), 6.86 (d, J = 3.3 Hz, 1H), 2.06 (s, 6H). 13C NMR (126 MHz, DMSO-d6) δ 140.3, 137.5, 133.5, 128.1, 127.4, 127.2, 126.5, 126.1, 20.4.
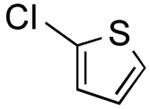
3-(2,6-Dimethylphenyl)thiophene (Table 2, Entry 13)17
General Procedure B was used at 65 °C. Silica gel column chromatography (0-100% EtOAc/hexane) was used to obtain the desired compound as a yellow oil (173.2 mg, 61% yield). 1H NMR (500 MHz, CDCl3) δ 7.42 (dd, J = 5.1, 1.2 Hz, 1H), 7.23 (dd, J = 8.3, 6.8 Hz, 1H), 7.18-7.13 (m, 3H), 6.87 (dd, J = 3.4, 1.2 Hz, 1H), 2.20 (s, 6H). 13C NMR (125.8 MHz, CDCl3) δ 141.5, 138.6, 134.3, 128.3, 127.4, 127.2, 126.5, 125.5, 21.0. HRMS (CI+) calcd. for C12H12S [M]+ 188.0660, found 188.0654.
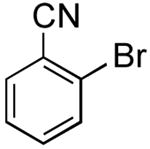
[1,1′-Biphenyl]-2-carbonitrile (Table 2, Entry 14)15
General Procedure B was used at 65 °C. Silica gel column chromatography (0-100% EtOAc/hexane) was used to obtain the desired compound as a yellow oil (75.5 mg, 28% yield). 1H NMR (500 MHz, CDCl3) δ 7.77 (dd, J = 7.8, 1.3 Hz, 1H), 7.65 (td, J = 7.7, 1.4 Hz, 1H), 7.59-7.56 (m, 2H), 7.54–7.42 (m, 5H). 13C NMR (126 MHz, CDCl3) δ 145.6, 138.3, 133.9, 133.0, 132.3, 130.3, 129.3, 128.9, 128.9, 128.9, 127.7, 118.9, 111.4. HRMS (CI+) calcd. for C13H9N [M]+ 179.0735, found 179.0733.
Supplementary Material
Acknowledgments
The authors thank the National Institutes of Health (R01-GM-081376) for their generous support of this research, and AllyChem Co., Ltd. for providing the BBA used in this study.
Footnotes
Dedicated to Professor Barry M. Trost, an inspirational mentor and chemist, on his receipt of the 2014 Tetrahedron Prize for Creativity in Organic Chemistry
Supplementary data: Supplementary data associated with this article can be found online at
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Miyaura N, Suzuki A. Chem Rev. 1995;95:2457. [Google Scholar]; (b) Miyaura N, Yamada K, Suzuki A. Tetrahedron Lett. 1979;20:3437. [Google Scholar]
- 2.(a) Ishiyama T, Murata M, Miyuara N. J Org Chem. 1995;60:7508. [Google Scholar]; (b) Ishiyama T, Itoh Y, Kitano T, Miyaura N. Tetrahedron Lett. 1997;38:3447. [Google Scholar]
- 3.Molander GA, Trice SLJ, Dreher SD. J Am Chem Soc. 2010;132:17701. doi: 10.1021/ja1089759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molander GA, Cavalcanti LN, García-García C. J Org Chem. 2013;78:6427. doi: 10.1021/jo401104y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Molander GA, Trice SLJ, Kennedy SM, Dreher SD, Tudge MT. J Am Chem Soc. 2012;134:11667. doi: 10.1021/ja303181m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Molander GA, Trice SLJ, Kennedy SM, Dreher SD, Tudge MT. J Org Chem. 2012;77:8678. doi: 10.1021/jo301642v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Ishiyama T, Itoh Y, Kitano T, Miyaura N. Tetrahedron Lett. 1997;38:3447. [Google Scholar]; (b) Baudoin O, Cesario M, Guénard D, Guéritte F. J Org Chem. 2002;67:1199. doi: 10.1021/jo0160726. [DOI] [PubMed] [Google Scholar]; (c) Baudoin O, Guénard D, Guéritte F. J Org Chem. 2000;65:9268. doi: 10.1021/jo005663d. [DOI] [PubMed] [Google Scholar]; (d) Billingsley KL, Barder TE, Buchwald SL. Angew Chem Int Ed. 2007;46:5359. doi: 10.1002/anie.200701551. [DOI] [PubMed] [Google Scholar]; (e) Wang L, Cui X, Zhu Z, Wu Y, Li J, Wu Y. Eur J Org Chem. 2012;595 [Google Scholar]
- 8.Kinzel T, Zhang Y, Buchwald SL. J Am Chem Soc. 2010;132:14073. doi: 10.1021/ja1073799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knapp DM, Gillis EP, Burke MD. J Am Chem Soc. 2009;131:6961. doi: 10.1021/ja901416p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Molander GA, Canturk B. Angew Chem Int Ed. 2009;48:9240. doi: 10.1002/anie.200904306. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Molander GA, Jean-Gérard L. Org React. 2013;79:1. [Google Scholar]
- 11.Noguchi N, Hojo K, Suginome M. J Am Chem Soc. 2007;129:758. doi: 10.1021/ja067975p. [DOI] [PubMed] [Google Scholar]
- 12.Bello CS, Schmidt-Leithoff J. Tetrahedron Lett. 2012;53:6230. [Google Scholar]
- 13.Molander GA, Canturk B, Kennedy L. J Org Chem. 2009;74:973. doi: 10.1021/jo802590b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santos-Filho EF, Jokderléa SC, Bezzerra NMM, Menezes PH, Oliveira RA. Tetrahedron Lett. 2011;52:5288. [Google Scholar]
- 15.Fu XL, Wu LL, Fu HY, Chen H, Li RX. Eur J Org Chem. 2009;2051 [Google Scholar]
- 16.Billingsley K, Buchwald SL. J Am Chem Soc. 2007;129:3358. doi: 10.1021/ja068577p. [DOI] [PubMed] [Google Scholar]
- 17.Bey E, Marchais-Oberwinkler S, Werth R, Negri M, Al-Soud Y, Krutchten P, Oster A, Frotscher M, Birk B, Hartmann RW. J Med Chem. 2008;51:6725. doi: 10.1021/jm8006917. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt A, Rahimi A. Chem Commun. 2010;46:2995. doi: 10.1039/c001362e. [DOI] [PubMed] [Google Scholar]
- 19.bis-Boronic acid (BBA, tetrahydroxydiboron) is commercially available from Sigma-Aldrich (catalog number 754242) and other vendors.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.