

Abstract
Chemoresistance is a major obstacle to successful chemotherapy for glioma. Formononetin is a novel herbal isoflavonoid isolated from Astragalus membranaceus and possesses antitumorigenic properties. In the present study, we investigated the anti-proliferative effects of formononetin on human glioma cells, and further elucidated the molecular mechanism underlying the anti-tumor property. We found that formononetin enhanced doxorubicin cytotoxicity in glioma cells. Combined treatment with formononetin reversed the doxorubicin-induced epithelial-mesenchymal transition (EMT) in tumor cells. Moreover, we found that formononetin treatment significantly decreased the expression of HDAC5. Overexpression of HDAC5 diminished the suppressive effects of formononetin on glioma cell viability. Furthermore, knockdown of HDAC5 by siRNA inhibited the doxorubicin-induced EMT in glioma cells. Taken together, these results demonstrated that formononetin-combined therapy may enhance the therapeutic efficacy of doxorubicin in glioma cells by preventing EMT through inhibition of HDAC5.
Keywords: Glioma, epithelial-mesenchymal transition, HDAC5, combination treatment
Introduction
Glioblastoma multiforme (GBM) is the most common primary brain tumor, which is characterized by high aggressiveness and poor prognosis [1,2]. The lethality of this malignancy is mainly due to the abnormal proliferation and invasiveness of glioma cells. The current strategy for the treatment of GBM is general palliative treatment, including surgical palliative resection, standard chemotherapy, and focal radiotherapy [3-5]. However, the development of acquired drug resistance to conventional chemotherapeutics has become a major obstacle in GBM treatment [6]. Such limitation highlights the imperative need for identifying novel treatment strategies which may help overcome drug resistance and enhance tumor cell response to anti-cancer drugs.
It has been acknowledged that the pathogenesis of glioma is a multistep process regulated by aberrantly protein expression and alterations of morphological and molecular features during malignant progression [7,8]. Epithelial-mesenchymal transition (EMT) is a complex, reversible process which induces epithelial cells to transform to mesenchymal phenotype [9,10]. Accumulating evidences suggest that EMT plays an important role in regulating the chemoresistance properties of glioma [11].
Traditional Chinese herbs are significant sources of drugs that serve as potential therapeutic compounds for cancer treatment. Astragalus membranaceus (Radix Astragali) has a long history of medicinal use in traditional Chinese medicine as an immunomodulating agent to treat diarrhea, anorexia and fatigue [12]. Formononetin, an active component isolated from Astragalus membranaceus, possesses diverse pharmacological benefits such as anti-inflammatory and immuno-modulatory activity [13]. Accumulating evidences also demonstrated the anticancer activity of formononetin on several cancer types such as breast cancer, prostate cancer and cervical cancer [14-16]. However, the effect of formononetin on human glioma has not been elucidated. Thus, the present study aimed to explore the anti-proliferative effects of formononetin on glioma cells, and further elucidate the molecular mechanism underlying the anti-tumor property on human glioma.
Materials and methods
Cell culture and reagents
Human glioma cell lines U87MG, U251MG, and T98G were purchased from the ATCC (Manassas, VA, USA) and cultured in DMEM (Gibco, Carlsbad, CA, USA) supplemented with 10% FBS and 1% penicillin/streptomycin. All cells were maintained at 37°C in 5% CO2 incubator. Formononetin (purity > 99%) and doxorubicin (Dox) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The HDAC5 plasmid, HDAC5 siRNA and negative control siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
CCK-8 assay
Tumor cells were seeded onto 96-well plates at 3000 cells/well. The medium was replaced with the corresponding serum-free medium for 24 h to synchronize the cell cycle, and then serum-free medium was replaced with complete medium containing the drugs at the indicated concentrations for 48 h. Then 10 μL/well CCK8 solution (Dojindo, Kumamoto, Japan) was added, the plates incubated for 3 h, and absorbance was measured at 450 nm using an MRX II microplate reader (Dynex, Chantilly, VA, USA).
Transfection
Cells were transfected with HDAC5 plasmid, HDAC5 siRNA or negative control siRNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. The transfection medium (Opti-MEM; Gibco) was replaced with complete medium 12 h after transfection, and the cells were incubated for the indicated times.
Western blot analysis
Cells were lysed in 50 μl cell lysis buffer (Cell Signaling, Danvers, MA, USA) containing protease inhibitors (Sigma). The protein concentration was quantified using the BCA Protein Kit (Thermo, Rockford, IL, USA). Cell lysates were separated by 10% SDS-PAGE and proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The membranes were then incubated with primary antibodies (E-cadherin, Vimentin or HDAC5, diluted 1:1000; Abcam, Cambridge, USA) at 4°C overnight. The membranes were washed three times with TBS/T and then incubated with the appropriate HRP-conjugated secondary antibodies for 1 h at room temperature. Protein expression was detected by chemiluminescence (GE Healthcare, Piscataway, NJ, USA).
Statistical analysis
Each experiment was performed in triplicate, and repeated at least three times. All the data were presented as means ± SD and treated for statistics analysis by SPSS program. Comparison between groups was made using ANOVA and statistically significant difference was defined as P < 0.05.
Results
Formononetin enhanced doxorubicin sensitivity in glioma cells
Firstly, CCK-8 assay was performed to determine the appropriate concentration of formononetin for combined treatment with doxorubicin. A series of formononetin concentrations ranging from 0~ 200 μM were incubated with three glioma cells lines (U87MG, U251MG and T98G) and data from CCK-8 assay showed that formononetin exerted little cytotoxicity in cancer cells between 0 and 100 µM. However, higher concentrations of formononetin (150, 200 µM) significantly inhibited the viability of the three cell lines (Figure 1A-C). Therefore, 100 µM formononetin was used for further co-administration with doxorubicin. To evaluate the synergistic cytotoxic effects of doxorubicin combined with formononetin, we used CCK-8 assay to measure cell viability treated with doxorubicin alone or in combination with formononetin for 48 h. At a result, the doxorubicin sensitivity was increased in U87MG, U251MG and T98G cells after co-administration with formononetin (Figure 2A-C). These results suggested that formononetin could enhance the sensitivity of doxorubicin in glioma cells.
Figure 1.
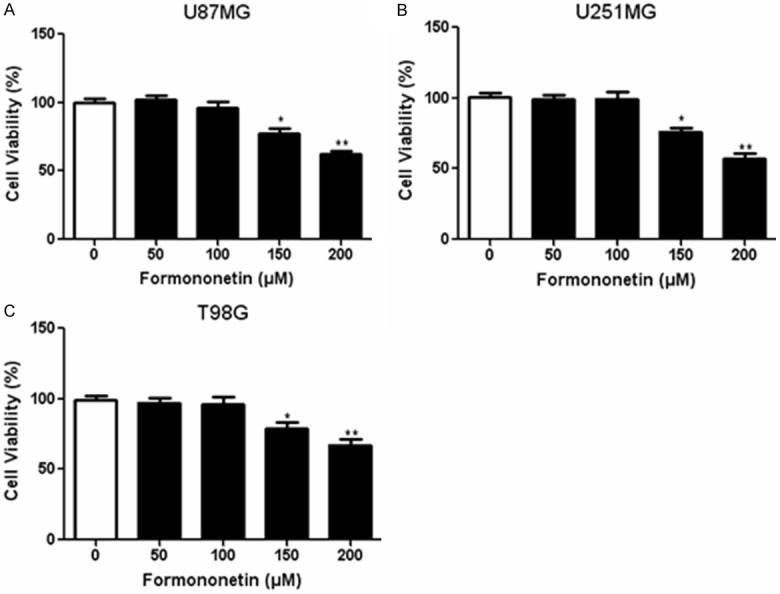
Cytotoxic effects of formononetin on glioma cells. Three glioma cell lines including U87MG (A), U251MG (B) and T98G (C) were incubated with different concentrations (0~ 200 μM) of formononetin for 48 h. The CCK-8 values of the treated glioma cells were normalized to the control group with the absence of formononetin. *P < 0.05; **P < 0.01.
Figure 2.
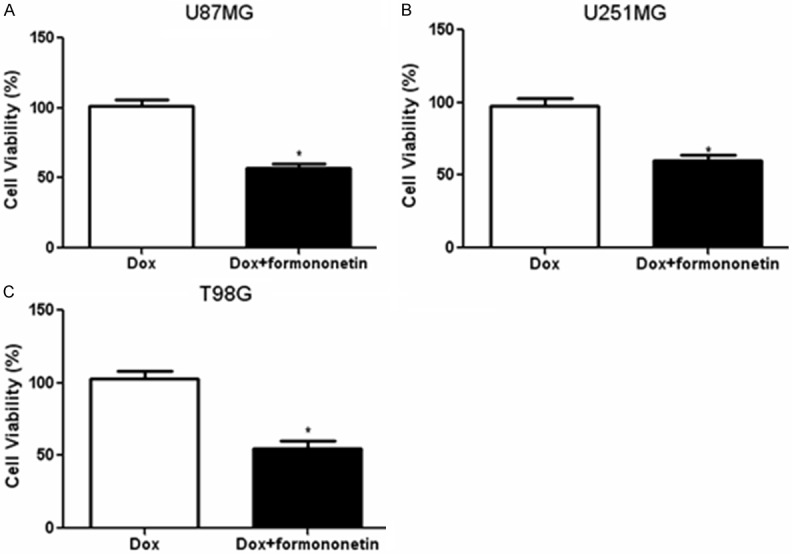
Formononetin enhanced doxorubicin sensitivity in glioma cells. Three glioma cell lines including U87MG (A), U251MG (B) and T98G (C) were incubated with doxorubicin alone or in combination with formononetin (100 μM) for 48 h. CCK-8 assay was performed to measure the cell viability of different glioma cell lines. *P < 0.05; **P < 0.01.
Formononetin inhibited doxorubicin-induced EMT
In order to investigate whether doxorubicin can induce EMT in tumor cells, we evaluated the expression of epithelial/mesenchymal markers in glioma cells treated with doxorubicin for 48 h. Results showed that administration of doxorubicin significantly enhanced the expression of vimentin and decreased the expression of E-cadherin in U87MG cells (Figure 3A and 3B). However, combine treatment with formononetin decreased the expression of vimentin and increased the E-cadherin levels, indicating that formononetin reversed the doxorubicin-induced EMT in glioma cells (Figure 3C, 3D).
Figure 3.
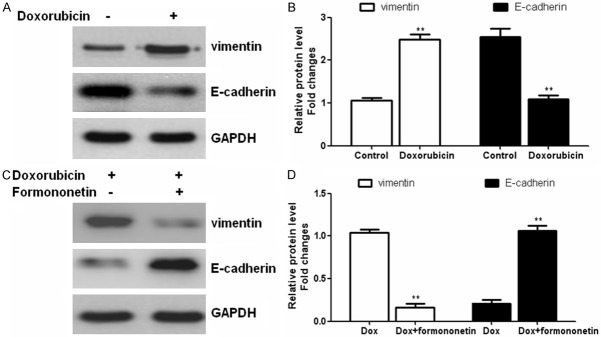
Formononetin treatment altered the expression of doxorubicin-induced EMT-markers. U87MG cells were incubated with doxorubicin alone (A and B) or in combination with Formononetin (100 μM) (C and D) for 48 h. Western blot was performed to determine the expression of EMT markers E-cadherin and vimentin. *P < 0.05; **P < 0.01.
Formononetin treatment suppressed the expression of HDAC5
Our previous study showed that HDAC5 promoted glioma cell proliferation [17]; we further examined the relevance of HDAC5 with chemoresistance in tumor cells. We found that HDAC5 was significantly increased in doxorubicin-treated glioma cells (Figure 4A and 4B). However, formononetin co-treatment reduced the expression of HDAC5 in glioma cells (Figure 4C and 4D). We than transfected U87MG cells with the plasmid encoding HDAC5 (Figure 4E). As a result, overexpression of HDAC5 diminished the suppressive effects of formononetin on glioma cell viability (Figure 4F). These data implied that formononetin sensitized glioma cells through inhibition of HDAC5.
Figure 4.

Formononetin treatment decreased the expression of HDAC5. U87MG cells was treated with doxorubicin or in combination with formononetin (100 μM) for 48 h. Western blot was performed to determine the expression of HDAC5 in glioma cells incubated with doxorubicin (A and B) or in combination with formononetin (100 μM) (C and D). U87MG cells were transfected with the plasmid encoding HDAC5 (E) and cell viability was determined by CCK-8 methods after incubation with doxorubicin alone or combined with formononetin (F). *P < 0.05; **P < 0.01.
Knockdown of HDAC5 diminished the doxorubicin-induced EMT
Nest we investigated whether HDAC5 regulated doxorubicin-induced EMT, RNAi was applied to knockdown the expression of HDAC5 in glioma cells. The HDAC5 siRNA-transfected glioma cells were incubated with doxorubicin alone or in combination with formononetin for 48 h. CCK-8 assay revealed that the cell viability of formononetin plus doxorubicin-treated cells was not significantly different compared to the doxorubicin-treated cells transfected with HDAC5 siRNA (Figure 5A-C), suggesting that HDAC5 was involved in the sensitivity to doxorubicin in glioma. Western blotting showed the upregulation of E-cadherin and downregulation of vimentin in HDAC5 siRNA-transfected U87MG cells (Figure 5D and 5E). Taken together, these data demonstrated that knockdown of HDAC5 by siRNA could alter the doxorubicin-induced EMT in glioma cells.
Figure 5.
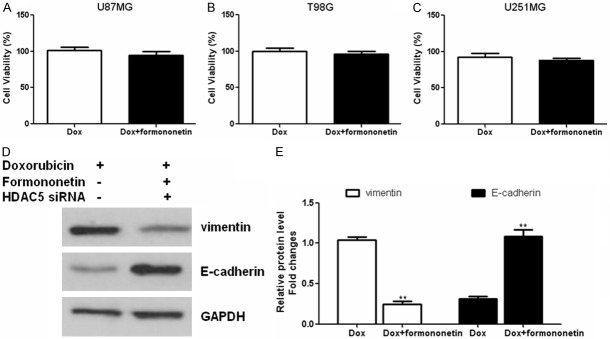
Knockdown of HDAC5 diminished the doxorubicin-induced EMT in glioma cells. The HDAC5 siRNA-transfected glioma cells were incubated with doxorubicin alone or in combination with formononetin for 48 h. CCK-8 assay was used to determine the cell viability in different glioma cell lines including U87MG (A), U251MG (B) and T98G (C). Western blot analysis of E-cadherin and vimentin expression in HDAC5-siRNA or negative-siRNA transfected glioma cells (D and E). Relative protein expression of HDAC5 was quantified by band density with GAPDH served as control. *P < 0.05; **P < 0.01.
Discussion
Accumulating evidences suggest that the acquired drug resistance to traditional chemotherapeutics has become a major obstacle to the triumph of chemotherapy [18]. In this study, we examined whether formononetin could enhance the cytotoxicity of doxorubicin in human glioma cells. Our data showed that combination treatment with formononetin increased the doxorubicin sensitivity in glioma cells.
Many studies have indicated that EMT plays a key role in carcinogenicity, metastasis, progression and acquired chemoresistance in various types of cancers [19,20]. During EMT, epithelial markers such as E-cadherin decrease, while mesenchymal markers such as vimentin increase [21]. In our study, we found that doxorubicin treatment induced EMT in glioma cells. However, formononetin co-administration reversed the EMT induced by doxorubicin in glioma cells. These results demonstrated that formononetin regulated doxorubicin-induced EMT in glioma cells.
The histone deacetylase (HDACs) family contains a family of 18 proteins and these proteins are classified into classes I-IV based on their homology and structure [22]. Accumulating evidences suggested that HDAC family functions as an important regulator in tumor progression and metastasis [23]. Recently, several HDAC inhibitors have been shown to exhibit anti-tumor activity in cancer cells and animal models [24]. In addition, clinical studies have also shown the potential application of HDAC inhibitors as anti-cancer agents [25]. HDAC5, a member of the class II histone deacetylase family, has been shown play critical roles in cell proliferation, cell cycle progression and apoptosis [26-28]. A recent study reported that HDAC5 promoted the twist 1 expression and highlighted a potential link between HDAC5 and osteosarcoma progression [29]. Our previous study demonstrated that HDAC5 promoted glioma cells proliferation via up-regulation of Notch 1 expression and might provide novel therapeutic targets in the treatment of gliomas [17]. In the current research, we found that the expression of HDAC5 was significantly up-regulated in doxorubicin-treated glioma cells, which was reduced after formononetin co-treatment. In addition, overexpression of HDAC5 diminished the inhibitory effects of formononetin on glioma cells, suggesting that formononetin sensitized tumor cells through inhibition of HDAC5.
Furthermore, we investigated the molecular mechanism underlying the reversal of EMT by formononetin. The etiology of glioma involves a complex interplay of various factors, of which the accumulation of oncogenes and loss of tumor repressors are crucial events in the initiation and progression of cancer [30,31]. Recently studies found that HDAC5 was aberrantly expressed in several types of tumor cells, such as glioma [17], osteosarcoma [29] and colon cancer [32]. In our previous study, we measured HDAC5 expression in glioma tumor tissues and showed that HDAC5 plays an important role in cell proliferation [17]. In the current study, our data showed that knockdown of HDAC5 by siRNA could alter cell phenotypes, suggesting that HDAC5 was a key factor in doxorubicin-induced EMT in glioma cells.
In conclusion, the present study showed that combined treatment with formononetin enhances the cytotoxicity of doxorubicin in glioma cells through suppressing HDAC5 and preventing doxorubicin-induced EMT. Therefore, combination therapy with formononetin may contribute to a better therapeutic effect in doxorubicin-based chemotherapy for patients with gliomas.
Disclosure of conflict of interest
None.
References
- 1.Stupp R, Hegi ME, van den Bent MJ, Mason WP, Weller M, Mirimanoff RO, Cairncross JG. Changing paradigms--an update on the multidisciplinary management of malignant glioma. Oncologist. 2006;11:165–180. doi: 10.1634/theoncologist.11-2-165. [DOI] [PubMed] [Google Scholar]
- 2.Giordano FA, Brehmer S, Abo-Madyan Y, Welzel G, Sperk E, Keller A, Schneider F, Clausen S, Herskind C, Schmiedek P, Wenz F. INTRAGO: intraoperative radiotherapy in glioblastoma multiforme - a Phase I/II dose escalation study. BMC Cancer. 2014;14:992. doi: 10.1186/1471-2407-14-992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin L, Gaut D, Hu K, Yan H, Yin D, Koeffler HP. Dual targeting of glioblastoma multiforme with a proteasome inhibitor (Velcade) and a phosphatidylinositol 3-kinase inhibitor (ZSTK474) Int J Oncol. 2014;44:557–562. doi: 10.3892/ijo.2013.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson DR, Chang SM. Recent medical management of glioblastoma. Adv Exp Med Biol. 2012;746:26–40. doi: 10.1007/978-1-4614-3146-6_3. [DOI] [PubMed] [Google Scholar]
- 5.Preusser M, de Ribaupierre S, Wohrer A, Erridge SC, Hegi M, Weller M, Stupp R. Current concepts and management of glioblastoma. Ann Neurol. 2011;70:9–21. doi: 10.1002/ana.22425. [DOI] [PubMed] [Google Scholar]
- 6.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 7.Knizhnik AV, Roos WP, Nikolova T, Quiros S, Tomaszowski KH, Christmann M, Kaina B. Survival and death strategies in glioma cells: autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS One. 2013;8:e55665. doi: 10.1371/journal.pone.0055665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 9.Grant CM, Kyprianou N. Epithelial mesenchymal transition (EMT) in prostate growth and tumor progression. Transl Androl Urol. 2013;2:202–211. doi: 10.3978/j.issn.2223-4683.2013.09.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim EM, Jeong MH, Kim DW, Jeong HJ, Lim ST, Sohn MH. Iodine 125-labeled mesenchymal-epithelial transition factor binding peptide-click-cRGDyk heterodimer for glioma imaging. Cancer Sci. 2011;102:1516–1521. doi: 10.1111/j.1349-7006.2011.01983.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan YR, Xie Q, Li F, Zhang Y, Ma JW, Xie SM, Li HY, Zhong XY. Epithelial-to-mesenchymal transition is involved in BCNU resistance in human glioma cells. Neuropathology. 2014;34:128–134. doi: 10.1111/neup.12062. [DOI] [PubMed] [Google Scholar]
- 12.Cho WC, Leung KN. In vitro and in vivo immunomodulating and immunorestorative effects of Astragalus membranaceus . J Ethnopharmacol. 2007;113:132–141. doi: 10.1016/j.jep.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 13.Park J, Kim SH, Cho D, Kim TS. Formononetin, a phyto-oestrogen, and its metabolites up-regulate interleukin-4 production in activated T cells via increased AP-1 DNA binding activity. Immunology. 2005;116:71–81. doi: 10.1111/j.1365-2567.2005.02199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou R, Xu L, Ye M, Liao M, Du H, Chen H. Formononetin inhibits migration and invasion of MDA-MB-231 and 4T1 breast cancercells by suppressing MMP-2 and MMP-9 through PI3K/AKT signaling pathways. Horm Metab Res. 2014;46:753–760. doi: 10.1055/s-0034-1376977. [DOI] [PubMed] [Google Scholar]
- 15.Li T, Zhao X, Mo Z, Huang W, Yan H, Ling Z, Ye Y. Formononetin promotes cell cycle arrest via downregulation of Akt/Cyclin D1/CDK4 in human prostate cancer cells. Cell Physiol Biochem. 2014;34:1351–1358. doi: 10.1159/000366342. [DOI] [PubMed] [Google Scholar]
- 16.Jin YM, Xu TM, Zhao YH, Wang YC, Cui MH. In vitro and in vivo anti-cancer activity of formononetin on human cervical cancer cell line HeLa. Tumour Biol. 2014;35:2279–2284. doi: 10.1007/s13277-013-1302-1. [DOI] [PubMed] [Google Scholar]
- 17.Liu Q, Zheng JM, Chen JK, Yan XL, Chen HM, Nong WX, Huang HQ. Histone deacetylase 5 promotes the proliferation of glioma cells by upregulation of Notch 1. Mol Med Rep. 2014;10:2045–2050. doi: 10.3892/mmr.2014.2395. [DOI] [PubMed] [Google Scholar]
- 18.Loscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci. 2005;6:591–602. doi: 10.1038/nrn1728. [DOI] [PubMed] [Google Scholar]
- 19.Liu X, Fan D. The Epithelial-Mesenchymal Transition and Cancer Stem Cells: Functional and Mechanistic Links. Curr Pharm Des. 2015;21:1279–91. doi: 10.2174/1381612821666141211115611. [DOI] [PubMed] [Google Scholar]
- 20.Su Y, Wang J, Zhang X, Shen J, Deng L, Liu Q, Li G. Targeting SIM2-s decreases glioma cell invasion through mesenchymal--epithelial transition. J Cell Biochem. 2014;115:1900–1907. doi: 10.1002/jcb.24859. [DOI] [PubMed] [Google Scholar]
- 21.Bogachek MV, De Andrade JP, Weigel RJ. Regulation of epithelial-mesenchymal transition through SUMOylation of transcription factors. Cancer Res. 2015;75:11–5. doi: 10.1158/0008-5472.CAN-14-2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang JC, Kafeel MI, Avezbakiyev B, Chen C, Sun Y, Rathnasabapathy C, Kalavar M, He Z, Burton J, Lichter S. Histone deacetylase in chronic lymphocytic leukemia. Oncology. 2011;81:325–329. doi: 10.1159/000334577. [DOI] [PubMed] [Google Scholar]
- 23.Francisco R, Perez-Perarnau A, Cortes C, Gil J, Tauler A, Ambrosio S. Histone deacetylase inhibition induces apoptosis and autophagy in human neuroblastoma cells. Cancer Lett. 2012;318:42–52. doi: 10.1016/j.canlet.2011.11.036. [DOI] [PubMed] [Google Scholar]
- 24.Greco TM, Yu F, Guise AJ, Cristea IM. Nuclear import of histone deacetylase 5 by requisite nuclear localization signal phosphorylation. Mol Cell Proteomics. 2011;10:M110–M4317. doi: 10.1074/mcp.M110.004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wagner JM, Hackanson B, Lubbert M, Jung M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenetics. 2010;1:117–136. doi: 10.1007/s13148-010-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsieh T, Hsu C, Tsai C, Long C, Wu C, Wu D, Lee J, Tsai E. HDAC inhibitors target HDAC5, upregulate MicroRNA-125a-5p, and induce Apoptosis in Breast Cancer Cells. Mol Ther. 2015;23:656–66. doi: 10.1038/mt.2014.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peixoto P, Castronovo V, Matheus N, Polese C, Peulen O, Gonzalez A, Boxus M, Verdin E, Thiry M, Dequiedt F, Mottet D. HDAC5 is required for maintenance of pericentric heterochromatin, and controls cell-cycle progression and survival of human cancer cells. Cell Death Differ. 2012;19:1239–1252. doi: 10.1038/cdd.2012.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polese C, Mottet D. [HDAC5 inhibition: a tool to stop cancer cell immortality] . Med Sci (Paris) 2014;30:730–732. doi: 10.1051/medsci/20143008005. [DOI] [PubMed] [Google Scholar]
- 29.Chen J, Xia J, Yu YL, Wang SQ, Wei YB, Chen FY, Huang GY, Shi JS. HDAC5 promotes osteosarcoma progression by upregulation of Twist 1 expression. Tumour Biol. 2014;35:1383–1387. doi: 10.1007/s13277-013-1189-x. [DOI] [PubMed] [Google Scholar]
- 30.Adams S, Braidy N, Bessede A, Brew BJ, Grant R, Teo C, Guillemin GJ. The kynurenine pathway in brain tumor pathogenesis. Cancer Res. 2012;72:5649–5657. doi: 10.1158/0008-5472.CAN-12-0549. [DOI] [PubMed] [Google Scholar]
- 31.Jung S, Moon KS, Kim ST, Ryu HH, Lee YH, Jeong YI, Jung TY, Kim IY, Kim KK, Kang SS. Increased expression of intracystic matrix metalloproteinases in brain tumors: relationship to the pathogenesis of brain tumor-associated cysts and peritumoral edema. J Clin Neurosci. 2007;14:1192–1198. doi: 10.1016/j.jocn.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 32.Stypula-Cyrus Y, Damania D, Kunte DP, Cruz MD, Subramanian H, Roy HK, Backman V. HDAC up-regulation in early colon field carcinogenesis is involved in cell tumorigenicity through regulation of chromatin structure. PLoS One. 2013;8:e64600. doi: 10.1371/journal.pone.0064600. [DOI] [PMC free article] [PubMed] [Google Scholar]