

Abstract
Researches have revealed several stressors, which could activate unfolded protein response (UPR) in cells. However, the survival or death pathway was determined by the duration of UPR exposure. Based on the UPR mediated death pathway, our study was aimed to investigate role of UPR on c-Jun N-terminal kinase (JNK)/activator protein-1 (Ap-1) signal transduction in diindolylmethane (DIM) treated ovarian cancer cell lines. Activation of UPR proteins, UPR mediated apoptotic signaling proteins and expression level of EpCAM, JNK, Ap-1, Caspase-3 and Bcl-2 were measured. Protein and gene expression, transcription factor activity, and protein phosphorylation were measured using standard molecular biology techniques. Our results demonstrated DIM treatment had significantly increased the expression of Endoplasmic reticulum (ER) stress regulators such as Bip, IRE1, CHOP and activation of UPR related apoptotic proteins in ovarian cancer cells. Decreased expression of EpCAM and activity of AP-1 transcription factor were observed in DIM treated cells. The pharmacologic inhibitors of the JNK signal transduction pathway, suggest that the impact of EpCAM expression on AP-1 transcription factor activity is mediated through the JNK pathway. Taken together, these results suggest that UPR mediated JNK/Ap-1 signal transduction has a significant role in the regulation of apoptosis in human ovarian cancer cells, and is a potential molecular target to enhance sensitivity of ovarian cancer to chemotherapy.
Keywords: SKOV3 cells, diindolylmethane, EpCAM, endoplasmic reticulum stress
Introduction
The endoplasmic reticulum (ER) has an important role in the cellular response to stressors such as nutrient deprivation, hypoxia, DNA damage and disruption of ER Ca2+ homeostasis [1]. These stressors lead to accumulation and aggregation of unfolded proteins in the ER lumen, inducing the phenomenon of ER stress, and ultimately lead to apoptosis [2,3]. The unfolded protein response (UPR) is a pro-survival response to reduce the accumulation of unfolded proteins and restore normal ER functioning [4] through transcriptional induction of genes that enhance the protein folding capacity of the ER such as Glucose-Regulated Protein 78 (GRP78) or Bip, and promotion of ER-associated protein degradation to remove misfolded proteins. However, when the stress is beyond the capacity of the compensatory machinery, stressed cells will eventually activate an evolutionary conserved death program, ER stress-mediated apoptosis [5].
Activation of JNK occurs following recruitment of TRAF2 by the cytosolic kinase of IRE1 in response to ER stress [6]. Moreover, an apoptosis signal-regulating kinase (ASK1) is required for TRAF2-dependent JNK activation during TNF-induced apoptosis [7]. ASK1-deficient cells are resistant to ER stress-induced apoptosis, as well as JNK activation, indicating that ASK1 acts upstream of the site of JNK activation, and that the IRE1/TRAF2/ASK1-signaling pathway is important for ER stress-induced apoptosis [8]. Importantly, it is likely JNK is responsible for PS-341-induced apoptosis through activation of JNK/AP-1/Gadd153-signaling via inhibition of NF-κB and Bcl-2, which are regulated by Akt/CREB-signaling as part of the UPR to DNA damage [9].
AP-1 is a dimer of Fos and Jun proteins implicated in the regulation of cell proliferation, transformation and apoptosis [10]. AP-1 target genes involved in both cell cycle progression (e.g. cyclin D1), and tumour cell invasion (e.g. collagenase/MMP-1) have been identified. However, we are far from understanding the entire AP-1-regulated transcriptional programme that is important in cell transformation and cancer progression. It has been shown that epithelial cell adhesion molecule (EpCAM) expression can influence the JNK/AP-1 signal transduction pathway, and suggest that modulation of AP-1 transcription factor activity contributes to EpCAM dependent breast cancer invasion [11]. EpCAM is a type I transmembran protein that is localized to the basolateral membrane in the majority of normal epithelial tissues [12]. EpCAM is perhaps best known for the fact that it is over expressed in the majority of human epithelial cancers including colorectal, breast, gastric, prostate, ovarian, and lung cancers [13,14].
Ovarian cancer is one of the leading gynecological cancers in well-developed countries with high mortality rates, which is usually detected in malignant stages with poor prediction. In the present study, we have demonstrated the importance of UPR mediated JNK/Ap-1 signal transduction in various ovarian cancer cell lines.
Materials and methods
Cell lines, culture conditions and reagents
Human ovarian cancer cell lines SKOV3 (American Type Culture Collection, Manassas, VA), A2780 (Sigma-Aldrich Corp.) and COLO-316 (Georgia Chenevix-Trench, QIMR, Australia) were maintained in RPMI-1640 media (10% fetal calf serum, 2 mM L-glutamine, 10 mM HEPES) purchased from Life Technologies (Mulgrave, VIC, Australia). Cell lines were routinely shown to be negative for mycoplasma. During experiments, cells were plated with a density of 0.3 × 106 cells per well in a six-well plate and allowed to attach overnight. Cells were then treated with or without 75 μM diindolylmethane (DIM; Sigma-Aldrich Co.) for 24 h, where DIM act as the activator of ER stress or UPR.
Western blot analysis
Rabbit polyclonal antibodies to Bip, IRE1, CHOP and goat polyclonal AP-1 were from Santa Cruz (Santa Cruz, CA, USA). Polyclonal antibodies against JNK and ATF4, cleaved caspase-3 and monoclonal antibodies against P-c-Jun, c-Jun, EpCAM and Bcl2 were from Cell Signalling Technology Inc. (Danvers, MA, USA). A monoclonal antibody to β-actin was used as a loading control (Sigma-Aldrich Co.). After incubation with primary antibodies and washes, blots were incubated with horseradish peroxidase-conjugated donkey anti-mouse or anti-rabbit or anti-goat secondary antibodies (1:5000). Blots were visualized with a chemiluminescent detection system (ECL; GE Healthcare Australia, Rydalmere, NSW, Australia) according to the manufacturer’s protocol.
Flow cytometry
EpCAM expression levels were measured by flow cytometry using phycoerythrin labeled EpCAM antibody with a FACScan flow cytometer (BD Biosciences, San Jose, CA, USA; #347211). EpCAM expression was quantified as mean fluorescence intensity (MFI).
Acridine orange assay
Ovarian cancer cell lines were plated at a density of 0.3 × 106 cells per well in a six-well plate and allowed to attach overnight. Cells were then treated with or without DIM. After 24 h, cells were washed, suspended in binding buffer and incubated for 15 minutes with 0.1 μg/ml acridine orange. Fluorescence was measured using FACS can flow cytometer to determine the level of autophagy.
Transmission electron microscopy
SKOV-3 cells treated with or without DIM were harvested by trypsinization and fixed in ice cold 3% glutaraldehyde overnight. After washing in PBS, cells were post fixed in Oso4 and embedded in EPON resin. Embedded samples were further processed for ultra-structural analysis as described previously [15].
Statistical analyses
Statistical analyses were conducted using SigmaPlot software, and values are presented as mean ± SD. Significant differences between groups were determined using an unpaired Student’s t-test. A value of *P < 0.05 was accepted as an indication of statistical significance.
Results
DIM induced UPR dependent autophagy or cell death
Treatment of ovarian cancer cells (SKOV-3, A2780 and COLO-316) with various concentrations (25, 50 and 75 μM) of DIM for 24 h resulted in a concentration dependent increase in percentage of autophagy detected by measuring acridine orange fluorescence (Figure 1A). Our results showed that DIM-induced autophagy was nearly 4 to 6 fold in SKOV-3 (30%), 2 to 4 fold in A2780 (23%) and 2 to 4 fold in COLO-316 cells (18%), when compared with their respective controls (Figure 1A). DIM induced protein aggregation was further confirmed by electron microscopy. The intercellular protein aggregation, an indicator of UPR was clearly represented in the transmission electron microscopic images taken from the DIM exposed SKOV3 cells (Figure 1B).
Figure 1.
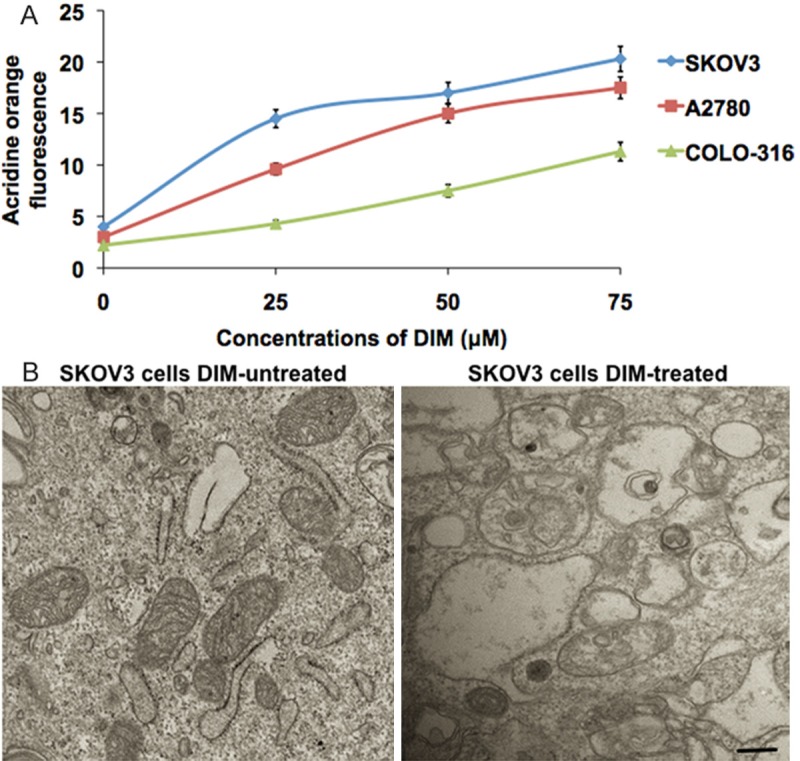
DIM induces changes in ovarian cancer cells. A. The ovarian cancer cells (SKOV-3, A2780 and COLO-316) were exposed to various concentrations of DIM for 24 hours. The fluorescence intensity of acridine orange from the cells were measured and graphically represented. B. Electron microscopy images of control and DIM treated SKOV-3 cells. The scale bar denotes 50 mm.
Activation of UPR in ovarian cancer cells
Initially, we demonstrated the DIM-induced activation of UPR in various ovarian cancer cells (SKOV3, A2780 and COLO-316). The cells were harvested at 6, 12 and 24 h time points were subjected to the analysis. The UPR related proteins (Bip, IRE1 and ATF4) and apoptotic UPR proteins (CHOP, JNK, Ap-1, caspase-3 and Bcl-2) were detected and quantified in western blot analysis. The early increase in the level of UPR proteins was observed in SKOV3 cells at 6 h and significantly (P < 0.001) increased thereafter. In A2780 and COLO-316 cells, the UPR proteins were elevated from 12 h exposure to DIM (Figure 2). Similarly, the UPR mediated apoptotic proteins were elevated depending on the cell type and the duration of DIM exposure (at least minimum 12 h). Our results showed, a significant (P < 0.001) increase in the apoptotic proteins CHOP, JNK, Ap-1, caspase-3 and decreased level of Bcl-2 at 12 h DIM exposure in SKOV3 cells. However, these effects were observed at 24 h exposure of DIM in A2780 and COLO-316 cells (Figure 3). These data suggest that prolonged UPR activation triggers the JNK/Ap-1 mediated apoptotic pathway.
Figure 2.
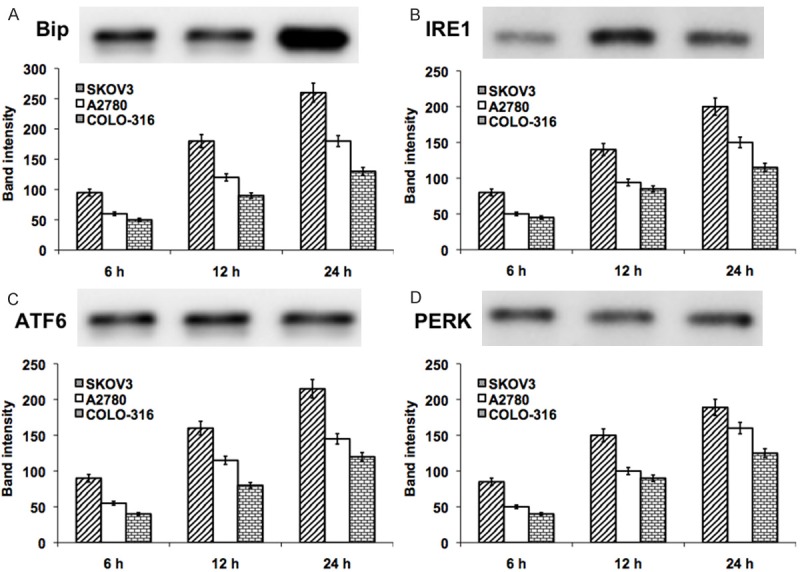
Represent the western blot analysis of UPR related proteins Bip (A), IRE1 (B), ATF6 (C) and PERK (D) in DIM exposed ovarian cancer cells (SKOV-3, A2780 and COLO-316) for 24 hours. The represented blot pictures are correspondent to SKOV-3 cells. The graphical representation shows the quantifications of band intensity from the blots correspondent to all three ovarian cancer cell types.
Figure 3.
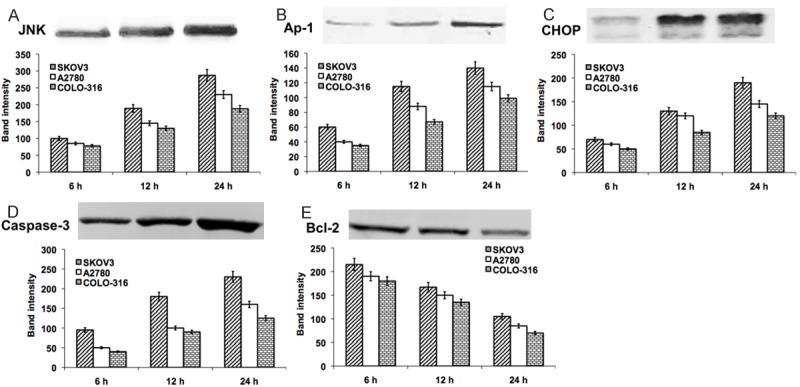
Represent the western blot analysis of UPR mediated apoptotic related proteins JNK (A), Ap-1 (B), CHOP (C), Caspase-3 (D) and Bcl-2 (E) in DIM exposed ovarian cancer cells (SKOV-3, A2780 and COLO-316) for 24 hours. The represented blot pictures are correspondent to SKOV-3 cells. The graphical representation shows the quantifications of band intensity from the blots correspondent to all three ovarian cancer cell types.
EpCAM expression in ovarian cancer cells
To better understand the involvement of EpCAM in JNK/Ap-1 signal transduction, we have measured the EpCAM expression in DIM exposed ovarian cancer cells for 24 h in qualitatively (western blot), and the metabolic activity of EpCAM for 6, 12 & 24 h in quantitatively (FACS). Increased level of EpCAM was observed in SKOV3 than that of A2780 and COLO-316 cells (Figure 4A). These results were mimicked in quantitative analysis (Figure 4B). Taken together this data provide evidence that EpCAM expression can modulate the JNK signal transduction pathway, and Ap-1 transcription factor activity.
Figure 4.
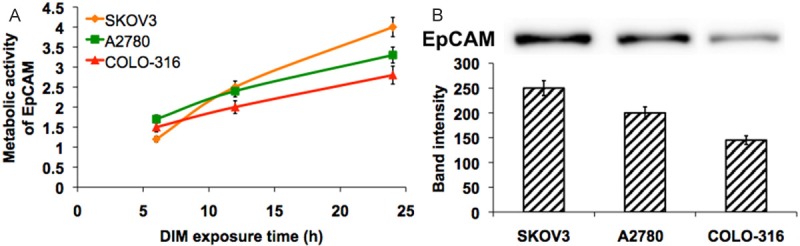
Represent the quantification of the metabolic activity of EpCAM (A) and the western blot analysis of EpCAM protein (B) with corresponding band quantification from DIM exposed ovarian cancer cells for 24 hours.
Pharmacological inhibition of JNK
To confirm the JNK signal transduction-mediated apoptosis, we performed the inhibitor study using the SP600125, a pharmacological inhibitor of JNK. To confirm that SP600125 inhibited JNK activity, western blot analysis was conducted using p-c-Jun antibodies in SKOV3 cells. Treatment with 20 µM SP600125 almost completely abolished c-Jun phosphorylation after 12 h, but total c-Jun protein levels had no influence on the expression status (Figure 5). These results indicate that SP600125 causes anti-proliferative effects with enlarged cell morphology in SKOV3 cells through suppression of JNK mediated pro-survival signaling.
Figure 5.
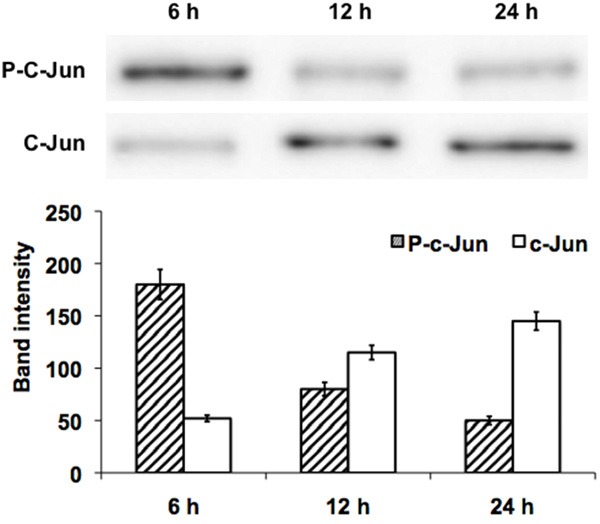
Represent the western blot analysis of JNK inhibition study. The pharmacological JNK inhibitor SP600125 added to SKOV-3 cells exposed to DIM for 6, 12 and 24 hours. The corresponding band intensity was quantified and plotted as bar graph.
Discussion
A pilot study previously demonstrated that oncogenic ER stress induces activation PERK, ATF4 and CHOP protects human tumour cells during hypoxia through autophagy [16]. However, further studies are needed to elucidate the complex crosstalk between these processes and reveal their requirement in all stages of cancer development and progression. In the present, we had demonstrated the UPR mediated JNK/AP-1 signal transduction as a target for the treatment of ovarian cancer, based on the previous studies on DIM induces cellular stress in ovarian cancer cells [17]. Previous report states that DIM induces cellular stress leading to DNA damage in ovarian cancer cells [17]. Hence, initially, we evaluated the level of DIM induced UPR mediated autophagy or apoptosis in ovarian cancer cells (SKOV-3, A2780 or COLO-316). Similar to the previous report [17], our present results showed DIM induces autophagy in human ovarian cancer cells in a concentration dependent manner as analyzed by flowcytometry and electron microscopy.
Next we demonstrated the DIM induced UPR activation. These processes are carried out by three transmembrane ER stress sensors, RNA activated protein kinase like ER kinase (PERK), the basic leucine-zipper activating transcription factor ATF6, and the kinase endoribonuclease IRE1 [18]. Activation of ER stress in our model was associated with an increase in the expression of Bip, IRE1, PERK and ATF6. Both Bip and IRE1 were significantly up regulated by DIM treatment in all the three ovarian cancer cells. Bip is a chaperone that folds protein into its proper confirmation. Bip acts as a sensor in ER and it binds to IRE1 under normal conditions [18]. However, during ER stress, it releases IRE1, which further leads to expression of a transcriptional factor GADD 153 that continues the response of unfolded proteins [18,19]. In evidence to the previous report, our results showed increased the level of UPR-mediated apoptotic proteins such as CHOP, JNK, Ap-1 and caspase-3. Moreover, DIM exposure also decreased the level of Bcl-2, suggesting the importance of JNK/Ap-1 signal transduction.
Recent study had demonstrated that EpCAM expression modulates the JNK signal transduction pathway, and AP-1 transcription factor activity in breast cancer cells. Functional studies confirm that AP-1 is a key downstream mediator of EpCAM biology, contributing to EpCAM-dependent breast cancer invasion [11]. In support of this report, our results showed the elevated level of EpCAM activity as well as protein level. AP-1 induction appears to pass exclusively via the JNK kinase pathway and inhibition of JNK leads to apoptosis of infected B cells, where as interfering with AP-1 transcriptional activity reduces tumour invasiveness [20,21]. AP-1 is a dimer of Fos and Jun proteins implicated in the regulation of cell proliferation, transformation and apoptosis [10]. AP-1 is a major target of the JNK signal transduction pathway, and activated JNK can bind and phosphorylate c-Jun [22]. Further we demonstrated the JNK-inhibitor study to examine the involvement of the JNK pathway. Our present results showed time dependent increase in P-c-Jun protein in SP600125 treated SKOV-3 cells exposed to DIM for 24 h. Our results are consistent with the previous report increase in AP-1 transcription factor binding activity as well as the increase in the phosphorylation of c-Jun. Addition of SP600125, an anthrapyrazolone inhibitor of JNK [22], abrogates the increase in AP-1 transcription factor activity induced by rEpCAM, suggesting a role for the JNK signal transduction pathway [11].
In conclusion, chronic UPR activation is associated with increased risk of UPR mediated death pathway in which JNK/Ap-1 signal transduction plays a major role. In our present study, we demonstrate that DIM exposure in human ovarian cancer cell lines enhanced UPR related proteins, pro-apoptotic or autophagy, increased EpCAM expression thereby modulating JNK/Ap-1 signal transduction pathway. JNK inhibitor study further supports the involvement of JNK/Ap-1 signal transduction. Further study on this signaling pathway in in-vivo model is necessary to facilitate the rational design and application of molecular therapies for ovarian cancer targeting JNK/Ap-1 signal transduction.
Disclosure of conflict of interest
None.
References
- 1.Gorlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid Redox Signal. 2006;8:1391–1418. doi: 10.1089/ars.2006.8.1391. [DOI] [PubMed] [Google Scholar]
- 2.Ermak G, Davies KJ. Calcium and oxidative stress: from cell signaling to cell death. Mol Immunol. 2002;38:713–721. doi: 10.1016/s0161-5890(01)00108-0. [DOI] [PubMed] [Google Scholar]
- 3.Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe? Nat Rev Cancer. 2004;4:966–977. doi: 10.1038/nrc1505. [DOI] [PubMed] [Google Scholar]
- 4.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 5.Perez-Sala D, Mollinedo F. Inhibition of N-linked glycosylation induces early apoptosis in human promyelocytic HL-60 cells. J Cell Physiol. 1995;163:523–531. doi: 10.1002/jcp.1041630312. [DOI] [PubMed] [Google Scholar]
- 6.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 7.Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kadowaki H, Nishitoh H, Urano F, Sadamitsu C, Matsuzawa A, Takeda K, Masutani H, Yodoi J, Urano Y, Nagano T, Ichijo H. Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2005;12:19–24. doi: 10.1038/sj.cdd.4401528. [DOI] [PubMed] [Google Scholar]
- 9.Nozaki S, Sledge GW Jr, Nakshatri H. Repression of GADD153/CHOP by NF-kappaB: A possible cellular defense against endoplasmic reticulum stress-induced cell death. Oncogene. 2001;20:2178–2185. doi: 10.1038/sj.onc.1204292. [DOI] [PubMed] [Google Scholar]
- 10.Jochum W, Passegue E, Wagner EF. AP-1 in mouse development and tumorigenesis. Oncogene. 2001;20:2401–2412. doi: 10.1038/sj.onc.1204389. [DOI] [PubMed] [Google Scholar]
- 11.Sankpal NV, Mayfield JD, Willman MW, Fleming TP, Gillanders WE. Activator protein 1 (AP-1) contributes to EpCAM-dependent breast cancer invasion. Breast Cancer Res. 2011;13:R124. doi: 10.1186/bcr3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baeuerle PA, Gires O. EpCAM (CD326) finding its role in cancer. Br J Cancer. 2007;96:417–423. doi: 10.1038/sj.bjc.6603494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spizzo G, Went P, Dirnhofer S, Obrist P, Simon R, Spichtin H, Maurer R, Metzger U, von Castelberg B, Bart R, Stopatschinskaya S, Köchli OR, Haas P, Mross F, Zuber M, Dietrich H, Bischoff S, Mirlacher M, Sauter G, Gastl G. High Ep-CAM expression is associated with poor prognosis in nodepositive breast cancer. Breast Cancer Res Treat. 2004;86:207–213. doi: 10.1023/B:BREA.0000036787.59816.01. [DOI] [PubMed] [Google Scholar]
- 14.Went PT, Lugli A, Meier S, Bundi M, Mirlacher M, Sauter G, Dirnhofer S. Frequent EpCam protein expression in human carcinomas. Hum Pathol. 2004;35:122–128. doi: 10.1016/j.humpath.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 15.Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11:448–457. doi: 10.1038/sj.cdd.4401359. [DOI] [PubMed] [Google Scholar]
- 16.Rouschop KM, Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW, Lambin P, Kogel AJ, Koritzinsky M, Wouters BG. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. 2010;120:127–141. doi: 10.1172/JCI40027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kandala PK, Srivastava SK. Activation of checkpoint kinase 2 by 3,3’-diindolylmethane is required for causing G2/M cell cycle arrest in human ovarian cancer cells. Mol Pharmacol. 2010;78:297–309. doi: 10.1124/mol.110.063750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Denmeade SR, Isaacs JT. The SERCA pump as a therapeutic target: making a “smart bomb” for prostate cancer. Cancer Biol Ther. 2005;4:14–22. doi: 10.4161/cbt.4.1.1505. [DOI] [PubMed] [Google Scholar]
- 19.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lizundia R, Sengmanivong L, Guergnon J, Muller T, Schnelle T, Langsley G, Shorte SL. Use of micro-rotation imaging to study JNK-mediated cell survival in Theileria parva-infected B-lymphocytes. Parasitology. 2005;130:629–635. doi: 10.1017/s0031182004007097. [DOI] [PubMed] [Google Scholar]
- 21.Lizundia R, Chaussepied M, Huerre M, Werling D, Di Santo JP, Langsley G. c-Jun NH2-terminal kinase/c-Jun signaling promotes survival and metastasis of B lymphocytes transformed by Theileria. Cancer Res. 2006;66:6105–6110. doi: 10.1158/0008-5472.CAN-05-3861. [DOI] [PubMed] [Google Scholar]
- 22.Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]