

Abstract
Chemokine (C-X-C motif) ligand 12 (CXCL12) and its receptor chemokine receptor 4 (CXCR4) have been recognized to play a crucial role in the pathogenesis of bronchial asthma, but the underlying molecular mechanisms are yet to be fully addressed. In the present report we demonstrated that CXCL12/CXCR4 signaling mediates allergic airway inflammation through induction of matrix metalloproteinase 9 (MMP-9) in a murine asthmatic model. We noted that administration of AMD3100, a specific CXCR4 antagonist, significantly attenuated OVA-induced asthmatic responses along with reduced epithelial MMP-9 expression. Our studies in a bronchial epithelial cell line, 16HBE cells, further revealed that CXCL12/CXCR4 signaling synergizes with IL-13 to enhance epithelial MMP-9 expression. Our mechanistic studies demonstrated that CXCL12/CXCR4 enhances epithelial MMP-9 expression by inducing ERK1/2 expression and activation. Together, these studies would bring novel insight into the understanding for the role of CXCL12/CXCR4 signaling in asthmatic responses during the course of bronchial asthma development.
Keywords: Asthma, CXCR4, CXCL12, MMP-9
Introduction
Chemokine (C-X-C motif) ligand 12 (CXCL12) or stromal cell-derived factor (SDF)-1 is a chemokine with a board range of functions. CXCL12 binds to its receptor, chemokine receptor 4 (CXCR4), and by which it attracts a variety of immune cells such as T lymphocytes and eosinophils implicated in allergic inflammation [1,2]. Previous studies revealed that blockade of CXCR4 attenuates pathological features relevant to asthmatic inflammation in a murine asthma model [3,4]. Significantly higher levels of CXCL12 have also been noted in bronchoalveolar lavage fluids (BALF) of asthmatic patients, and the concentration of CXCL12 is correlated with the number of inflammatory cells in BALF [5]. These data support that CXCL12 may play a role to recruit inflammatory cells in asthmatic condition; however, the exact underlying mechanisms are yet to be fully addressed.
Matrix metalloproteinases (MMPs) are a large family of zinc- and calcium-dependent endopeptides that degrade most components of the extracellular matrix with distinct specificities for substrates, a crucial process during the course of asthma development including airway inflammation, angiogenesis and airway remodeling [6,7]. Among all MMPs, MMP-9 is the major proteinase that has been recognized closely related to asthmatic pathologies. Particularly, mice deficient in MMP-9 manifest decreased airway inflammation and peribronchial mononuclear cell infiltration, and therefore, these mice are resistant to allergen-induced airway hyperresponsiveness (AHR) as compared with wild-type (WT) animals [8]. Similarly, higher levels of MMP-9 are found in the sputum and BALF of stable asthmatic patients [9,10], and severe asthmatic patients also manifest higher MMP-9 activity [11]. Therefore, MMP-9 is also found to be involved in the disease process of bronchial asthma.
Although both experimental and clinical data support that CXCL12 and MMP-9 are implicated in the pathogenesis of asthma, their relationship during the course of asthmatic responses, however, remains unknown. We thus hypothesize that CXCL12 enhances bronchial epithelial cells expression of MMP9, which then promotes allergic inflammatory response during the course of asthmatics. Asthmatic mice induced by OVA and bronchial epithelial 16HBE cell line were employed to test the above hypothesis.
Materials and methods
Reagents
Ovalbumin was purchased from Sigma (St. Louis, MO, USA). Recombinant human CXCL12 and IL-13 were purchased from Peprotech (Rocky Hill, USA). AMD3100, a specific CXCR4 antagonist [12,13] was purchased from Cayman chemical company (Ann Arbor, MI, USA). PD98059, a specific ERK inhibitor, was obtained from Enzo Life Sciences (Shanghai, China). Antibodies against MMP-9 and CXCR4 were purchased from Abcam (Cambridge, MA, USA), while anti-ERK1/2 antibody was obtained from Cell Signaling (Danvers, MA, USA).
Mice
Female BALB/c mice were purchased from Laboratory Animal Center of Hubei Province, Wuhan, China. All mice were housed in an SPF facility in microisolator cages supplied with autoclaved food and acidified water with a 12/12 h light/dark cycle. The studies involving mice were done according to a protocol reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the Tongji Hospital.
Generation of asthmatic model and treatment
To generate an asthmatic model, mice were sensitized and challenged with OVA as previously reported [14]. Briefly, mice were immunized intraperitoneally on days 0, 7 and 14 with 100 μg OVA plus 1 mg alum in 200 μL saline. The mice were then challenged by intranasal administration of 1 mg of OVA in 50 μL saline on days 21, 22 and 23. AMD3100 was freshly dissolved in saline and then administered intraperitoneally at the dose of 10 mg/kg on day 20.
Collection of bronchoalveolar lavage fluid (BALF) and histological analysis
Mice were sacrificed 24 h after last OVA or saline challenge. The lungs were lavaged 3 times with 0.8 ml of saline, and the collected cells were centrifuged and subjected to Wright-Giemsa staining. Culture supernatants were collected for assay of MMP-9 activity. Left lungs were isolated and fixed in 4% paraformaldehyde, and then embedded in paraffin. The blocks were sectioned at 5 μm thickness and then subjected to H&E staining as previously reported [14].
Cell culture and treatment
The human bronchial epithelial cell line (16HBE) was obtained from Fuxiang Biotechnology Co. Ltd (Shanghai, China). 16HBE cells were maintained in DMEM medium supplement with 10% fetal bovine serum, 100 U/ml penicillin and 100ug/ml streptomycin at 37°C in a humidified atmosphere containing 5% CO2. The cells were plated onto six-well plates. Subconfluent cell monolayers were first deprived of serum for 24 h, and then stimulated with CXCL12 in the absence or presence of IL-13 at indicated time. Culture medium was replaced every 24 h, and PD98059 was added into the culture 30 min before stimulation.
Immunostaining
HBE cells were fixed, permeabilized and probed with a CXCR4 antibody, followed by staining with a secondary antibody conjugated with FITC, and nuclei were stained with PI using the established techniques [14].
Western blotting
Whole cell lysates were prepared after 72 h of stimulation from 16HBE cells using RIPA lysis buffer with protease inhibitors (Beyotime, China). The loaded proteins (20 µg) were separated by 10% SDS-PAGE, and then transferred onto PVDF membranes. After blocking with 5% milk, the membranes were probed with antibodies against MMP-9 or ERK1/2, followed by incubation with an HRP-conjugated secondary antibody. These reactive bands were developed using an ECL chemiluminescence detection kit as instructed (Thermo Pierce, USA).
Zymographic analysis
To assay MMP-9 activity, 16 μL of BALF or culture supernatant were used for the analysis. BALF or culture supernatant were mixed with SDS-PAGE loading buffer (lacking reducing agents), and then fractionated by SDS-polyacrylamide gel electrophoresis at 4°C on 10% gels containing 0.1% gelatin. The gels were next washed in 2.5% Triton X-100 for 1h to promote the recovery of protease activity before incubation for 42 h at 37°C in a reaction buffer containing 50 mM Tris-HCl (pH 7.5), 5 mM CaCl2 and 1% Triton X-100. Subsequently, the gels were stained with brilliant blue. After washes, MMP-9 was detected as transparent bands on the blue background of a Coomassie blue-stained gel.
Statistical analysis
All data were presented as mean ± SEM. Statistical differences were assessed by one-way analysis of variance (ANOVA) followed by the Tukey’s multiple comparison test or the unpaired Student’s t-test. In all cases, P < 0.05 was considered with statistically significance.
Results
Administration of AMD3100 provides protection for mice against OVA-induced asthma
Given that AMD3100 acts as a CXCR4 antagonist, we first sought to demonstrate its role in OVA-induced inflammatory infiltration in the lung. It was noted that AMD3100 administration significantly reduced total cell counts and eosinophil counts in the BALF after OVA sensitization and challenge (Figure 1A). Histological analysis of lung sections further confirmed these observations (Figure 1B).
Figure 1.

Blockade of CXCL12/CXCR4 signaling attenuates OVA-induced asthmatic responses along with suppressed MMP-9 expression. BALB/c mice (n = 5) were intraperitoneally administered AMD3100 (10 mg/kg) on the day before OVA challenge. BALF and lungs were collected 24 h after OVA last challenge. A. Cell counts in the BALF for macrophages (Mac), eosinophils (Eos), lymphocytes (Lymph) and neutrophils (Neu). Saline, normal control mice treated with saline only; OVA, OVA-sensitized/challenged mice; OVA+AMD3100; OVA-sensitized/challenged mice along with AMD3100 treatment. *, P < 0.05 as compared with Saline group; #, P < 0.05 as compared with OVA group. B. Histological analysis of lung sections. Images for H&E stained sections were taken under × 200 magnification. Three mice were analyzed for each study group. C. Zymographic results for MMP-9 expressions. Consistent results were obtained for all mice (n = 5) analyzed in each group.
We next examined the impact of AMD3100 on MMP-9 expression, in which we assayed MMP-9 activity in the BALF between control and experimental mice. As expected, OVA-challenged mice demonstrated significantly elevated MMP-9 activity. In sharp contrast, treatment of mice with AMD3100 (10 mg/kg) attenuated MMP-9 activity by almost 2-fold (Figure 1C). Together, our data indicate that administration of AMD3100 provides protection for mice against OVA-induced asthma.
CXCL12/CXCR4 signaling induces bronchial epithelial cells expression of MMP-9
Given the role of bronchial epithelial cells played in the pathogenesis of asthma, we next conducted studies with focus on epithelial cells to dissect the mechanisms underlying the implication of CXCL12/CXCR4 signaling in asthmatic mechanism. We first examined CXCR4 expression in human bronchial epithelial cells, in which 16HBE cells were used for the study. Immunostaining of 16HBE cells revealed high levels of CXCR4 expression (Figure 2). We further noted that CXCR4 is constitutively expressed in bronchial epithelial cells.
Figure 2.
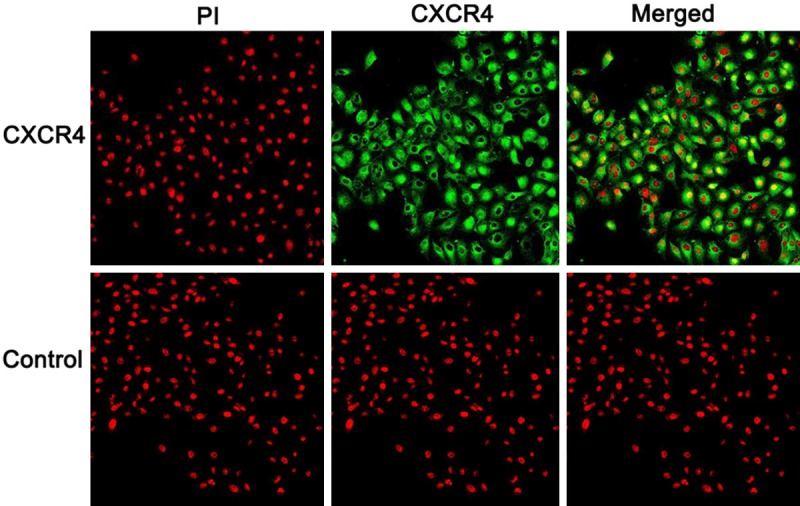
Results for immunostaining of CXCR4 in bronchial epithelial cells. CXCR4 in 16HBE cells were first probed with a rabbit derived mAb and then stained a green fluorescent labeled anti-rabbit IgG (green). Nuclei were stained in red by PI (original magnification × 400).
We next sought to address the impact of CXCL12/CXCR4 signaling on the induction of MMP-9 expression in bronchial epithelial cells. We assumed that MMP-9 is downstream of CXCL12/CXCR4 signaling, we thus first stimulated 16HBE cells with recombinant CXCL12, and then examined MMP-9 synthesis. We first conducted pilot studies to optimize the CXCL12 dose, and through which 200 ng/ml of CXCL12 was noted to be the most optimal dose for our purpose. Interestingly, CXCL12 time-dependently induced high levels of MMP-9 expression as manifested by Western blot analysis (Figure 3A). Of which, a significant increase for MMP-9 expression in response to CXCL12 stimulation was noted within the first 24 h, and the maximal response was achieved around 6 h stimulation. To further confirm these results, we conducted zymographic analysis of MMP-9 protein levels, and similar results were obtained as shown in Figure 3B. Collectively, our data support that CXCL12/CXCR4 signaling enhances asthma by inducing MMP-9 expression in bronchial epithelial cells.
Figure 3.

CXCL12/CXCR4 synergizes with IL-13 to enhance epithelial MMP-9 expression. A. CXCL12 time-dependently induced epithelial cells expression of MMP-9. 16HBE cells were cultured in serum-free medium at 37°C for 24 h and then stimulated with CXCL12 (200 ng/ml) as indicated times. B. Gelatin zymographic results for conditioned media collected from CXCL12 treated 16HBE cells. C. Western blot analysis of epithelial MMP-9 expression after CXCL12 and/or IL-13 stimulation. 16HBE cells were treated for 24 h with 10 ng/ml IL-13, 200 ng/ml CXCL12, 10 ng/ml IL-13 and 200 ng/ml CXCL12, CXCL12 and saline, CXCL12 and AMD3100, respectively. The cells were then harvested for Western blot analysis of MMP-9 expression. D. A bar graphic figure showing the results of 5 independent experiments conducted. *, P < 0.05 as compared with Control group; #, P < 0.05 as compared with CXCL12 group.
CXCL12/CXCR4 signaling synergizes with IL-13 to induce MMP-9 expression
Previous studies including ours also demonstrated a critical role for IL-13 in asthmatic pathogenesis [15-17]. We thus further assumed that CXCL12/CXCR4 signaling synergizes with IL-13 to promote MMP-9 expression. It was noted that IL-13 possesses almost similar potency to induce MMP-9 expression in 16HBE cells, while stimulation of 16HBE cells with combined CXCL12 and IL-13 induced much higher levels of MMP-9 expression as compared with that of IL-13 or CXCL12 alone (Figure 3C and 3D), indicating a synergistic effect between CXCL12 and IL-13 in terms of induction of MMP-9 expression in bronchial epithelial cells. More importantly, addition of AMD3100, a specific small-molecule CXCR4 antagonist, completely attenuated the effect of CXCL12 stimulation on 16HBE cells (Figure 3C, 3D), which further confirmed that the enhanced MMP-9 expression was caused by CXCL12/CXCR4 signaling.
CXCL12/CXCR4 signaling induces MMP-9 expression via ERK1/2 pathway
To further address the molecular mechanisms by which CXCL12/CXCR4 signaling induces MMP-9 expression, we examined ERK1/2 activity. For this purpose, 16HBE cell lysates after CXCL12 stimulation were prepared at indicated time points, and then subjected to Western blot analysis of total ERK1/2 and activated (phosphorylated) ERK1/2. Interestingly, CXCL12 stimulation significantly enhanced the protein levels for both total and activated ERK1/2 (p-ERK1/2). Specifically, the increase of total ERK1/2 and pERK1/2 could be noted 15min after CXCL12 stimulation, and the increase reached a peak 45 min after CXCL12 stimulation (Figure 4A). To confirm the involvement of ERK1/2 pathway in CXCL12/CXCR4 induced MMP-9 expression, we then treated 16HBE cells with CXCL12 in the presence of PD98059, a specific ERK1/2 kinase inhibitor. As expected, addition of PD98059 diminished the stimulatory effect of CXCL12 on 16HBE cells (Figure 4B). Collectively, our data support that CXCL12/CXCR4 signaling induces MMP-9 expression through ERK1/2 pathway.
Figure 4.
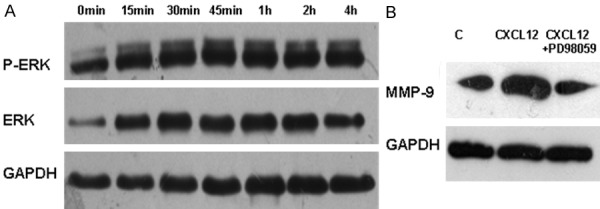
CXCL12/CXCR4 signaling induces ERK1/2 expression and activation. 16HBE cells were incubated with 200 ng/ml CXCL12 as the indicated time and then subjected to Western blot analysis of ERK1/2 expression and activation. A. CXCL12 potently increased total ERK1/2 protein levels and pERK1/2 levels. The results are a representative of 3 independent experiments conducted. B. Blockade of ERK1/2 signaling by PD98059 abolished the stimulatory effect of CXCL12 on epithelial MMP-9 expression. 16HBE cells were pretreated with PD98059 (50 μM) for 30 min before CXCL12 stimulation. Addition of PD98059 completely diminished CXCL12 induced MMP-9 expression in 16HBE cells.
Discussion
We demonstrated evidence supporting that CXCL12/CXCR4 signaling implicates in OVA-induced lung allergic inflammation by inducing MMP-9 expression in bronchial epithelial cells. Of which, blockade of CXCL12/CXCR4 signaling by AMD3100 completely attenuated CXCL12 induced MMP-9 expression in 16HBE cells. We also noted CXCL12/CXCR4 synergizes with IL-13 to promote bronchial epithelial cells expression of MMP-9. Our mechanistic studies further revealed that CXCL12/CXCR4 mediates MMP-9 expression by inducing ERK1/2 expression and activation. Together, those data would be important to understand the mechanisms for CXCL12/CXCR4 signaling in the pathogenesis of asthmatic responses.
Although the CXCL12/CXCR4 signaling has long been recognized to play a crucial role in asthmatic inflammation, the exact underlying mechanisms are yet to be fully addressed. Other than CXCL12/CXCR4 signaling, MMP-9 has also been suggested to play a crucial role in bronchial inflammation and airway remodeling [18-21]. In the present report, we sought to address that MMP-9 is a downstream molecule of CXCL12/CXCR4 signaling. Previous study showed that the expression of CXCL12 and CXCR4 increased in asthmatic mice model [22], and our results further demonstrate the role of CXCL12/CXCR4 signaling in OVA-induced asthmatics. We next treated OVA-challenged mice with AMD3100, and found blockade of CXCL12/CXCR4 signaling significantly attenuated MMP-9 expression. Histological studies and analysis of BALF further confirmed these results. To further address that MMP-9 is downstream of CXCL12/CXCR4 signaling, we then conducted studies in 16HBE cells. In line with studies in animals, CXCL12 was noted time-dependently to induce epithelial MMP-9 expression, as manifested by Western blot analysis and zymographic studies. Together, these data provided convincing evidence indicating that CXCL12 stimulates bronchial epithelial cells expressing and releasing of MMP-9.
IL-13 is a pleiotropic cytokine that has been suggested to play a critical role in the pathogenesis of asthma. Indeed, IL-13 showed similar potency to stimulate HBE16 cells expression of MMP-9. More interestingly, when 16HBE cells stimulated with combination of CXCL12 and IL-13, the expression of MMP-9 was significantly higher than that of CXCL12 or IL-13 alone. As expected, addition of AMD3100 completely diminished the stimulatory effect of CXCL12 on epithelial MMP-9 expression. These data suggest a synergistic effect between CXCL12/CXCR4 and IL-13 signaling on epithelial MMP-9 expression.
ERK1/2 can be induced and activated by a variety of cytokines such as IL-4 [23], IL-13 [24] and IL-17 [25], and a greater level of ERK1/2 activity was observed in the asthmatic mice model [26], suggested that ERK1/2 pathway may play an essential role in asthmatic inflammation. Given that MMP-9 expression has been suggested to be regulated by ERK pathway in a number of cell types [27-29], we thus conducted studies in 16HBE cells to demonstrate whether CXCL12/CXCR4 regulates epithelial MMP-9 expression through ERK1/2 pathway. Interestingly, CXCL12 not only promoted ERK1/2 activation, but also enhanced ERK1/2 expression. More importantly, stimulation of 16HBE cells with CXCL12 in the presence of PD98059, a specific ERK1/2 inhibitor, almost completely abolished the stimulatory effect of CXCL12 on epithelial MMP-9 expression. Altogether, we demonstrated evidence indicating that CXCL12/CXCR4 regulates epithelial MMP-9 expression through ERK1/2 pathway. Nonetheless, it is worthy of note that additional pathways other than ERK1/2 may be involved in CXCL12/CXCR4 induced MMP-9 expression in epithelial cells, additional studies would be necessary to fully address this question.
In summary, we demonstrated evidence for the first time that CXCL12/CXCR4 signaling implicates in asthmatic pathology through induction of epithelial MMP-9 expression, which involves induction of ERK1/2 expression and activation. These studies would bring novel insight into the understanding of mechanisms underlying the implication of CXCL12/CXCR4 signaling in asthmatic responses during the course of disease development.
Acknowledgements
None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript. This work was supported by the National Natural Science Foundation of China (No. 81170021 and No. 30900647).
Disclosure of conflict of interest
None.
References
- 1.Cinamon G, Shinder V, Alon R. Shear forces promote lymphocyte migration across vascular endothelium bearing apical chemokines. Nat Immunol. 2001;2:515–22. doi: 10.1038/88710. [DOI] [PubMed] [Google Scholar]
- 2.Nagase H, Miyamasu M, Yamaguchi M, Fujisawa T, Ohta K, Yamamoto K, Morita Y, Hirai K. Expression of CXCR4 in eosinophils: functional analyses and cytokine-mediated regulation. J Immunol. 2000;164:5935–43. doi: 10.4049/jimmunol.164.11.5935. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalo JA, Lloyd CM, Peled A, Delaney T, Coyle AJ, Gutierrez-Ramos JC. Critical involvement of the chemotactic axis CXCR4/stromal cell-derived factor-1 alpha in the inflammatory component of allergic airway disease. J Immunol. 2000;165:499–508. doi: 10.4049/jimmunol.165.1.499. [DOI] [PubMed] [Google Scholar]
- 4.Lukacs NW, Berlin A, Schols D, Skerlj RT, Bridger GJ. AMD3100, a CxCR4 antagonist, attenuates allergic lung inflammation and airway hyperreactivity. Am J Pathol. 2002;160:1353–60. doi: 10.1016/S0002-9440(10)62562-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Negrete-García MC, Velazquez JR, Popoca-Coyotl A, Montes-Vizuet AR, Juárez-Carvajal E, Teran LM. Chemokine (C-X-C motif) ligand 12/stromal cell-derived factor-1 is associated with leukocyte recruitment in asthma. Chest. 2010;138:100–6. doi: 10.1378/chest.09-2104. [DOI] [PubMed] [Google Scholar]
- 6.Nagase H, Woessner JF Jr. Matrix metalloproteinases. J Biol Chem. 1999;274:21491–4. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- 7.Kelly EA, Jarjour NN. Role of matrix metalloproteinases in asthma. Curr Opin Pulm Med. 2003;9:28–33. doi: 10.1097/00063198-200301000-00005. [DOI] [PubMed] [Google Scholar]
- 8.Cataldo DD, Tournoy KG, Vermaelen K, Munaut C, Foidart JM, Louis R, Noël A, Pauwels RA. Matrix metalloproteinase-9 deficiency impairs cellular infiltration and bronchial hyperresponsiveness during allergen-induced airway inflammation. Am J Pathol. 2002;161:491–8. doi: 10.1016/S0002-9440(10)64205-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vignola AM, Riccobono L, Mirabella A, Profita M, Chanez P, Bellia V, Mautino G, D’accardi P, Bousquet J, Bonsignore G. Sputum metalloproteinase-9/tissue inhibitor of metalloproteinase-1 ratio correlates with airflow obstruction in asthma and chronic bronchitis. Am J Respir Crit Care Med. 1998;158:1945–50. doi: 10.1164/ajrccm.158.6.9803014. [DOI] [PubMed] [Google Scholar]
- 10.Mautino G, Oliver N, Chanez P, Bousquet J, Capony F. Increased release of matrix metalloproteinase-9 in bronchoalveolar lavage fluid and by alveolar macrophages of asthmatics. Am J Respir Cell Mol Biol. 1997;17:583–91. doi: 10.1165/ajrcmb.17.5.2562. [DOI] [PubMed] [Google Scholar]
- 11.Bossé M, Chakir J, Rouabhia M, Boulet LP, Audette M, Laviolette M. Serum matrix metalloproteinase-9: Tissue inhibitor of metalloproteinase-1 ratio correlates with steroid responsiveness in moderate to severe asthma. Am J Respir Crit Care Med. 1999;159:596–602. doi: 10.1164/ajrccm.159.2.9802045. [DOI] [PubMed] [Google Scholar]
- 12.Donzella GA, Schols D, Lin SW, Esté JA, Nagashima KA, Maddon PJ, Allaway GP, Sakmar TP, Henson G, De Clercq E, Moore JP. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat Med. 1998;4:72–7. doi: 10.1038/nm0198-072. [DOI] [PubMed] [Google Scholar]
- 13.Schols D, Esté JA, Henson G, De Clercq E. Bicyclams, a class of potent anti-HIV agents, are targeted at the HIV coreceptor fusin/CXCR-4. Antiviral Res. 1997;35:147–56. doi: 10.1016/s0166-3542(97)00025-9. [DOI] [PubMed] [Google Scholar]
- 14.Cheng S, Chen H, Wang A, Bunjhoo H, Cao Y, Xie J, Xu Y, Xiong W. Exclusion of IL-21 in the pathogenesis of OVA-induced asthma in mice. Int J Clin Exp Med. 2014;7:3202–3208. [PMC free article] [PubMed] [Google Scholar]
- 15.Ingram JL, Kraft M. IL-13 in asthma and allergic disease: asthma phenotypes and targeted therapies. J Allergy Clin Immunol. 2012;130:829–42. doi: 10.1016/j.jaci.2012.06.034. quiz 843-4. [DOI] [PubMed] [Google Scholar]
- 16.Mitchell J, Dimov V, Townley RG. IL-13 and the IL-13 receptor as therapeutic targets for asthma and allergic disease. Curr Opin Investig Drugs. 2010;11:527–34. [PubMed] [Google Scholar]
- 17.Martinez FD, Vercelli D. Asthma. Lancet. 2013;382:1360–72. doi: 10.1016/S0140-6736(13)61536-6. [DOI] [PubMed] [Google Scholar]
- 18.Ueno-Iio T, Shibakura M, Iio K, Tanimoto Y, Kanehiro A, Tanimoto M, Kataoka M. Effect of fudosteine, a cysteine derivative, on airway hyperresponsiveness, inflammation, and remodeling in a murine model of asthma. Life Sci. 2013;92:1015–23. doi: 10.1016/j.lfs.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 19.Vliagoftis H, Schwingshackl A, Milne CD, Duszyk M, Hollenberg MD, Wallace JL, Befus AD, Moqbel R. Proteinase-activated receptor-2-mediated matrix metalloproteinase-9 release from airway epithelial cells. J Allergy Clin Immunol. 2000;106:537–45. doi: 10.1067/mai.2000.109058. [DOI] [PubMed] [Google Scholar]
- 20.Lee KS, Min KH, Kim SR, Park SJ, Park HS, Jin GY, Lee YC. Vascular endothelial growth factor modulates matrix metalloproteinase-9 expression in asthma. Am J Respir Crit Care Med. 2006;174:161–70. doi: 10.1164/rccm.200510-1558OC. [DOI] [PubMed] [Google Scholar]
- 21.Liu CP, Hsieh CH, Wu BN, Yeh JL, Dai ZK, Chai CY, Chen IJ. Inhaled KMUP-1 Prevents Allergic Pulmonary Vascular Inflammation and Remodeling via NO and Suppressed MMP-9 and ICAM-1/VCAM-1. Inflamm Allergy Drug Targets. 2012;11:251–61. doi: 10.2174/187152812800958960. [DOI] [PubMed] [Google Scholar]
- 22.Luan B, Huang XJ, Qiao JY. Expression of stromal cell derived factor-1 and CXC chemokine receptor 4 and the effects of budesonide on their expression in mice with asthma. Zhongguo Dang Dai Er Ke Za Zhi. 2010;12:215–8. [PubMed] [Google Scholar]
- 23.Chiu PR, Lee WT, Chu YT, Lee MS, Jong YJ, Hung CH. Effect of the Chinese herb extract osthol on IL-4-induced eotaxin expression in BEAS-2B cells. Pediatr Neonatol. 2008;49:135–40. doi: 10.1016/S1875-9572(08)60028-5. [DOI] [PubMed] [Google Scholar]
- 24.Ip WK, Wong CK, Lam CW. Interleukin (IL)-4 and IL-13 up-regulate monocyte chemoattractant protein-1 expression in human bronchial epithelial cells: involvement of p38 mitogen-activated protein kinase, extracellular signal-regulated kinase 1/2 and Janus kinase-2 but not c-Jun NH2-terminal kinase 1/2 signalling pathways. Clin Exp Immunol. 2006;145:162–72. doi: 10.1111/j.1365-2249.2006.03085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laan M, Lötvall J, Chung KF, Lindén A. IL-17-induced cytokine release in human bronchial epithelial cells in vitro: role of mitogen-activated protein (MAP) kinases. Br J Pharmacol. 2001;133:200–6. doi: 10.1038/sj.bjp.0704063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar A, Lnu S, Malya R, Barron D, Moore J, Corry DB, Boriek AM. Mechanical stretch activates nuclear factor-kappaB, activator protein-1, and mitogen-activated protein kinases in lung parenchyma: implications in asthma. FASEB J. 2003;17:1800–11. doi: 10.1096/fj.02-1148com. [DOI] [PubMed] [Google Scholar]
- 27.Lee EJ, Lee SJ, Kim S, Cho SC, Choi YH, Kim WJ, Moon SK. Interleukin-5 enhances the migration and invasion of bladder cancer cells via ERK1/2-mediated MMP-9/NF-kappaB/AP-1 pathway: involvement of the p21WAF1 expression. Cell Signal. 2013;25:2025–38. doi: 10.1016/j.cellsig.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 28.Lee SJ, Cho SC, Lee EJ, Kim S, Lee SB, Lim JH, Choi YH, Kim WJ, Moon SK. Interleukin-20 promotes migration of bladder cancer cells through extracellular signal-regulated kinase (ERK)-mediated MMP-9 protein expression leading to nuclear factor (NF-kappaB) activation by inducing the up-regulation of p21 (WAF1) protein expression. J Biol Chem. 2013;288:5539–52. doi: 10.1074/jbc.M112.410233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma L, Lan F, Zheng Z, Xie F, Wang L, Liu W, Han J, Zheng F, Xie Y, Huang Q. Epidermal growth factor (EGF) and interleukin (IL)-1beta synergistically promote ERK1/2-mediated invasive breast ductal cancer cell migration and invasion. Mol Cancer. 2012;11:79. doi: 10.1186/1476-4598-11-79. [DOI] [PMC free article] [PubMed] [Google Scholar]