

Abstract
Peutz-Jeghers syndrome (PJS) is a rare, inherited autosomal dominant disease characterized by mucocutaneous pigmentation and polyps in the gastrointestinal tract. Here, we report the rare case of a 64-year-old female patient with pigmentation on her lips and extremities for over 63 years and intermittent abdominal pain and, diarrhea for 3 years. The presence of intestinal and colorectal hamartomatous polyps was confirmed. The removal and characterization of her rectal polyp showed it to be a typical hamartomatous polyp with a portion of it being an adenoma with high-grade intramucosal neoplasia. A survey of the patient’s family identified 9 people in the family with Peutz-Jeghers syndrome and three of them have already died from colorectal cancer. This case study serves as an example of how imperative it is to survey the patient about their family history in order to detect early cancerous lesions.
Keywords: Peutz-Jeghers syndrome, endoscopy, hamartoma, cancer
Introduction
Patients diagnosed with Peutz-Jeghers syndrome (PJS) are at an increased risk for developing both gastrointestinal and extra intestinal malignancies; however, most are of gastrointestinal origin. Here, we report a case of PJS-with abundant rectal intramucosal neoplasias and the associated PJS pedigree of the family.
Case report
A 64-year-old female patient presented to our institution with pigmentation on her lips and extremities for over 63 years and intermittent abdominal pain and, diarrhea for 3 years. Three years ago, more than 20 polyps of small intestine and colorectum origin were resected under double-balloon enteroscopy and colonoscopy. Histopathology of the resected polyps was conformed to be multiple hamartomatous polyps. Physical examination of the patient was normal other than the scattered melanin pigments on her lips (Figure 1A) and hands (Figure 1B). The observed pigmentations had definite boundaries, did not fade under pressure, and did not protrude from the skin. Radiographs of the chest appeared normal. Ultrasonography of the pancreas and the liver were also normal. Colonoscopy examination indicated the presence of more than 10 polyps (0.5 cm to 3.5 cm in diameter) scattered from the ascending colon to the rectum. Among them, a flat, sessile polyp lesion (3.0 cm × 3.5 cm) with an uneven surface located in the rectum was removed by colonoscopy (Figure 2). Histopathology of the resected rectal polyp indicated it to be a typical hamartomatous polyp, while a portion of the polyp was shown to be an adenoma with high-grade intramucosal neoplasia (Figure 3).
Figure 1.
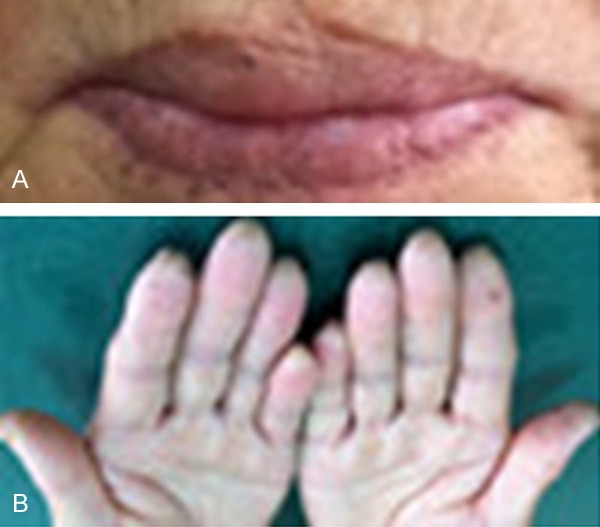
A. Hyperpigmented macules on the patient’s lips. B. Hyperpigmented macules on the patient’s fingers.
Figure 2.
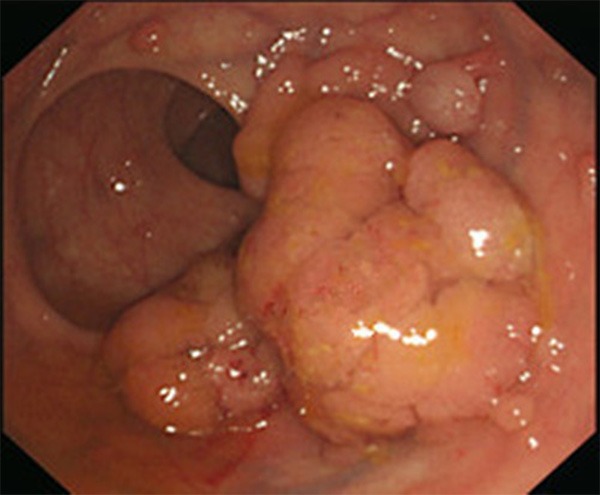
Endoscopic view of the flat, sessile rectal polyp (3.0 cm × 3.5 cm).
Figure 3.
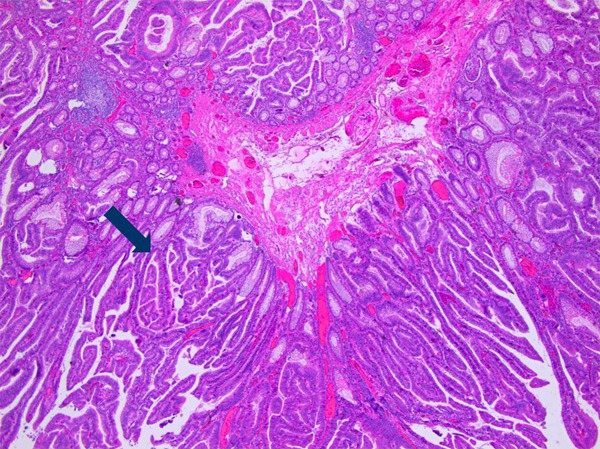
Histopathology image indicating a typical Peutz-Jeghers polyp and an adenoma with high-grade intramucosal neoplasia (HE staining × 100).
Family survey
The family survey included 42 family members from four generations. Nine family members (3 males and 6 females) with mucocutaneous pigmentation and/or multiple hamartomatous polyps were diagnosed with PJS (Figure 4). There were no sexual differences between the family members with PJS, thus indicating the inherited autosomal dominant disorder. Three of the 9 PJS family members, the patient’s mother, sister, and brother died of colorectal cancer.
Figure 4.
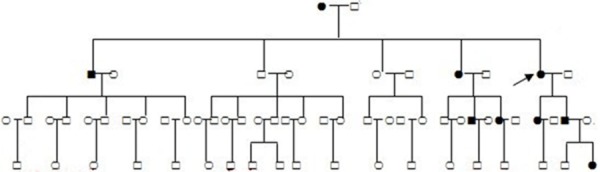
Pedigree of the family with Peutz-Jeghers syndrome (PJS). Circles and squares indicate females and males, respectively, while an arrow indicates the reported patient. Black symbols denote individuals with PJS.
Discussion
PJS is a rare, autosomal dominant inherited disorder with an estimated incidence rate between 1 in 50,000 and 1 in 200,000 people [1]. The disease has a variable penetrance. Some patients will only exhibit mucocutaneous pigmentation which is defined as incomplete PJS, while others will exhibit mucocutaneous pigmentations and hamartomatous polyps in the gastrointestinal tract. According to the established diagnosis criteria for PJS [2-4], if an individual has a family history of PJS in a close relative(s) and presents with a PJ polyp and/or mucocutaneous pigmentation, then a clinical diagnosis of PJS must be determined. In these family patients, those diagnosis criteria were met, so they were diagnosed with PJS. Germline mutations in the serine threonine kinase 11 gene (STK11 or LKB1 gene) cause PJS [5]. STK 11 is a tumor suppressor gene and is localized on chromosome 19p13.3. Determination of the STK 11 mutation detection is useful for PJS diagnosis, but the type and/or site of the STK 11 mutation does not influence a patient’s risk of developing cancer [6]. The most common malignancy associated with PJS is colorectal cancer, followed by breast, small bowel, gastric, and pancreatic cancers. The lifetime risk for any cancer varies between 37 and 93%, with relative cancer risks ranging from 9.9 to 18 in comparison with general population, with lifetime cumulative cancer risks up to 93% [7]. Among PJS patients aged 27- to 71-years-old, Giardiello et al. [8] found an overall risk of developing colorectal cancer to be 39%. Mechanisms of PJS-associated carcinogenesis remain unclear and controversial. Some hypothesize the hamartoma-adenoma-carcinoma pathway is responsible [9], while others think the PJ polyps may actually represent a form of abnormal mucosal prolapse [10] and do not have malignant potential. In this case, we found a hamartoma and an adenoma co-existing in the same polyp, which supports the hamartoma-adenoma-carcinoma pathway hypothesis and the notion that malignant tumors are associated with hamartomas. Therefore, hamartomatous polyps should be considered as cancer precursors and should be removed [11]. Cancer surveillance may detect early malignancies and thereby improve outcome. In this case, the patient’s rectal polyp was associated with a high-grade intramucosal neoplasia, which was then resected using colonoscopy, which ultimately saved her life. Endoscopy now appears to be an essential method for screening for gastrointestinal malignancies in PJS patients. The polypectomy of polyps in more than 1-1.5 cm in diameter has been recommended [7]. Double-balloon endoscopy (DBE) enables endoscopic resection of small-bowel polyps. Using DBE to remove small-bowel polyps in PJS patients is considered safe and effective [12]. In this case, intestinal polyps were safely removed using DBE 3 years ago.
In conclusion, PJS is associated with an increased risk for developing both gastrointestinal and extra intestinal malignancies; therefore, it is necessary to regularly survey patients about their family history in order to identify patients that are at-risk for developing PJS-associated malignancies. Identifying patients at-risk for developing PJS-associated malignancies earlier will ultimately help to save lives.
Disclosure of conflict of interest
None.
References
- 1.Giardiello FM, Trimbath JD. Peutz-Jeghers syndrome and management recommendations. Clin Gastroenterol Hepatol. 2006;4:408–415. doi: 10.1016/j.cgh.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 2.Aaltonen LA. Hereditary intestinal cancer. Semin Cancer Biol. 2000;10:289–298. doi: 10.1006/scbi.2000.0148. [DOI] [PubMed] [Google Scholar]
- 3.Aretz S, Stienen D, Uhlhaas S, Loff S, Back W, Pagenstecher C, McLeod DR, Graham GE, Mangold E, Santer R, Propping P, Friedl W. High proportion of large genomic STK11 deletions in Peutz-Jeghers syndrome. Hum Mutat. 2005;26:513–519. doi: 10.1002/humu.20253. [DOI] [PubMed] [Google Scholar]
- 4.Beggs AD, Latchford AR, Vasen HF, Moslein G, Alonso A, Aretz S, Bertario L, Blanco I, Bülow S, Burn J, Capella G, Colas C, Friedl W, Møller P, Hes FJ, Järvinen H, Mecklin JP, Nagengast FM, Parc Y, Phillips RK, Hyer W, Ponz de Leon M, Renkonen-Sinisalo L, Sampson JR, Stormorken A, Tejpar S, Thomas HJ, Wijnen JT, Clark SK, Hodgson SV. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59:975–986. doi: 10.1136/gut.2009.198499. [DOI] [PubMed] [Google Scholar]
- 5.Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Höglund P, Järvinen H, Kristo P, Pelin K, Ridanpää M, Salovaara R, Toro T, Bodmer W, Olschwang S, Olsen AS, Stratton MR, de la Chapelle A, Aaltonen LA. Aserine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 6.Hearle N, Schumacher V, Menko FH, Olschwang S, Boardman LA, Gille JJ, Keller JJ, Westerman AM, Scott RJ, Lim W, Trimbath JD, Giardiello FM, Gruber SB, Offerhaus GJ, de Rooij FW, Wilson JH, Hansmann A, Möslein G, Royer-Pokora B, Vogel T, Phillips RK, Spigelman AD, Houlston RS. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res. 2006;12:3209–3215. doi: 10.1158/1078-0432.CCR-06-0083. [DOI] [PubMed] [Google Scholar]
- 7.van Lier MG, Wagner A, Mathus-Vliegen EM, Kuipers EJ, Steyerberg EW, van Leerdam ME. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol. 2010;105:1258–1264. doi: 10.1038/ajg.2009.725. [DOI] [PubMed] [Google Scholar]
- 8.Giardiello FM, Brensinger JD, Tersmette AC, Goodman SN, Petersen GM, Booker SV, Cruz-Correa M, Offerhaus JA. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119:1447–1453. doi: 10.1053/gast.2000.20228. [DOI] [PubMed] [Google Scholar]
- 9.Bosman FT. The hamartoma-adenoma-carcinoma sequence. J Pathol. 1999;188:1–2. doi: 10.1002/(SICI)1096-9896(199905)188:1<1::AID-PATH327>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 10.Jansen M, de Leng WW, Baas AF, Myoshi H, Mathus-Vliegen L, Taketo MM, Clevers H, Giardiello FM, Offerhaus GJ. Mucosal prolapse in the pathogenesis of Peutz-Jeghers polyposis. Gut. 2006;55:1–5. doi: 10.1136/gut.2005.069062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sinha N, Chatterjee U, Sarkar S. Jejunal carcinoma in a patient with Peutz-Jeghers syndrome. Can J Surg. 2009;52:299–300. [PMC free article] [PubMed] [Google Scholar]
- 12.Sakamoto H, Yamamoto H, Hayashi Y, Yano T, Miyata T, Nishimura N, Shinhata H, Sato H, Sunada K, Sugano K. Nonsurgical management of small-bowel polyps in Peutz-Jeghers syndrome with extensive polypectomy by using double-ballon endoscopy. Gastrointest Endosc. 2011;74:328–333. doi: 10.1016/j.gie.2011.04.001. [DOI] [PubMed] [Google Scholar]