

Abstract
Pleomorphic xanthoastrocytoma (PXA) is an uncommon tumor of young adults that typically occurs supratentorially. It is generally considered to be a low-grade, circumscribed tumor that when treated by surgical resection has a relatively favorable outcome. Cases of cerebellar PXA are rare, and those associated with neurofibromatosis type 1 (NF1) are even less common, with only 2 cases reported to date. We present herein a third case of PXA-NF1 with unusual features. A 33-year-old woman presented with a history of headache. Her medical and family history was significant for NF1. Brain MRI revealed a 3.4 cm ill-defined lesion with a gyriform enhancing pattern in the left cerebellum, superficially mimicking Lhermitte-Duclos disease. The patient underwent a gross total resection of the lesion and had an unremarkable postoperative course. While the lesion had histological features typical of “pure” PXA (WHO grade II) it had an unusual growth pattern with thickening of the superficial cerebellar folia and predominant leptomeningeal involvement. No BRAF, IDH-1, or IDH-2 mutation was identified. Three months after surgery, local recurrence was detected, and the patient was treated with radiation therapy. One year after the first surgery, she underwent surgical resection of the recurrent/residual tumor. Histologically, the recurrent tumor showed very similar features to the initially resected tumor, with no anaplastic features. Most cerebellar PXAs have an indolent clinical behavior as do most cerebral PXAs. Whether co-existence of NF1 was a factor in altering the clinical course and biologic behavior of this patient’s tumor is currently unknown.
Keywords: Pleomorphic xanthoastrocytoma (PXA), neurofibromatosis type-1 (NF1), recurrence, cerebellum
Introduction
Pleomorphic xanthoastrocytoma (PXA) is a well-known yet somewhat uncommon tumor with characteristic histopathology that primarily arises in the temporal lobe of children and young adults. It is generally considered to be a low-grade, well-circumscribed tumor and is treated by surgical resection. Although most cases have a favorable prognosis with long-term survival, nearly 20% of patients with PXA, especially those whose tumor was not completely excised, may experience recurrence and/or progression to high-grade glioma.
Neurofibromatosis type 1 (NF1) is an autosomal dominant, multisystem genetic disorder. A variety of nervous system neoplasms are known to occur in association with NF1, and in the central nervous system (CNS), optic nerve glioma (pilocytic astrocytoma) is the most common. Other histological types of gliomas (e.g., diffuse astrocytoma and glioblastoma) can be seen, and other locations in the CNS can also be involved in this disease [1]. Only a few cases of PXA have been described in individuals with NF1 [2-8]. Cases of cerebellar PXA are rare, and those cases associated with NF1 (PXA-NF1) are even rarer, with only 2 cases having been reported to date [6,9]. We present herein a third case of PXA-NF1 with unusual clinicoradiological features. Molecular analysis of this tumor was also performed.
Case report
The patient was a 33-year-old woman who presented with a history of headache for approximately 6 weeks. Her medical and family history was significant for NF1. Brain MRI revealed a 3.4 cm ill-defined lesion with a gyriform enhancing pattern in the left cerebellum on T1-weighted image (WI), bearing a superficial resemblance to Lhermitte-Duclos disease (LDD) (Figure 1). The patient underwent gross total resection of the lesion and had an unremarkable postoperative course.
Figure 1.
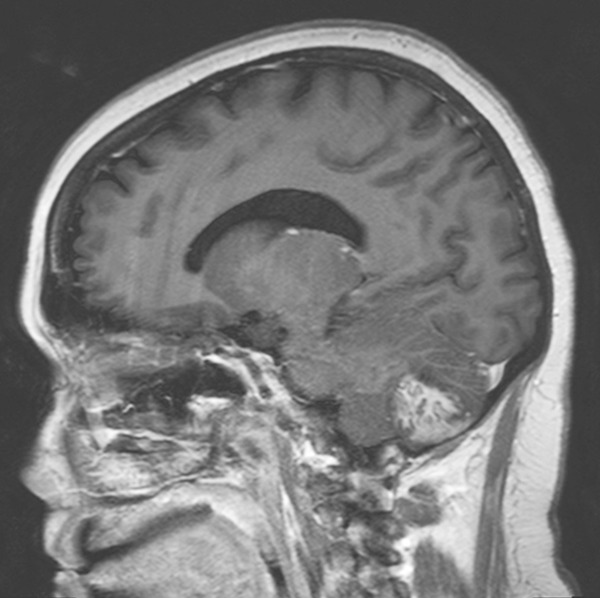
MRI (original tumor) (sagittal T1-weighted image with contrast enhancement): cerebellar tumor with a gyriform enhancing pattern.
Three months after initial resection, local recurrence was detected by MRI, and radiation therapy was initiated. One year after the first surgery, the patient underwent surgical resection of the recurrent/residual tumor and has since been closely monitored.
Pathological findings
The neoplasm from the initial resection had histological features typical of PXA (WHO grade II) characterized by a cellular astrocytic proliferation with bizarre pleomorphic tumor giant cells (some multinucleated), abundant eosinophilic granular bodies (Figure 2), and a typical intercellular reticulin network highlighted by silver impregnation stain. Xanthomatous tumor cells were inconspicuous. No obvious ganglioglioma or other composite tumor components were observed. Of note is that the majority of tumor showed growth within the leptomeninges, with external thickening of the cerebellar folia. No anaplastic features such as significant mitotic activity, microvascular proliferation, or necrosis were seen. Immunohistochemically, the neoplastic cells were diffusely and strongly positive for glial fibrillary acidic protein (GFAP) (Figure 3A). Scattered synaptophysin-positive tumor cells were identified (Figure 3B), and p53 staining was negative. A Ki-67 (MIB-1) labeling index was approximately 5%.
Figure 2.
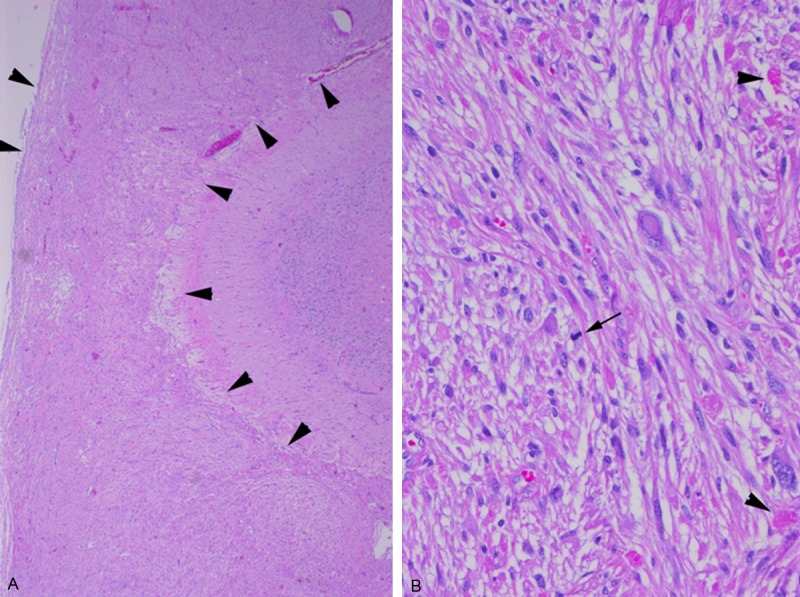
Histologic features of the cerebellar tumor from the initial surgery (H&E stained). A. Low-power view showing markedly-thickened leptomeninges due to extensive involvement of tumor cells (between arrowheads). B. High-power view showing an astrocytic proliferation with bizarre pleomorphic giant cells and scattered eosinophilic granular bodies (arrowheads). Few mitotic figures are identified (arrow).
Figure 3.
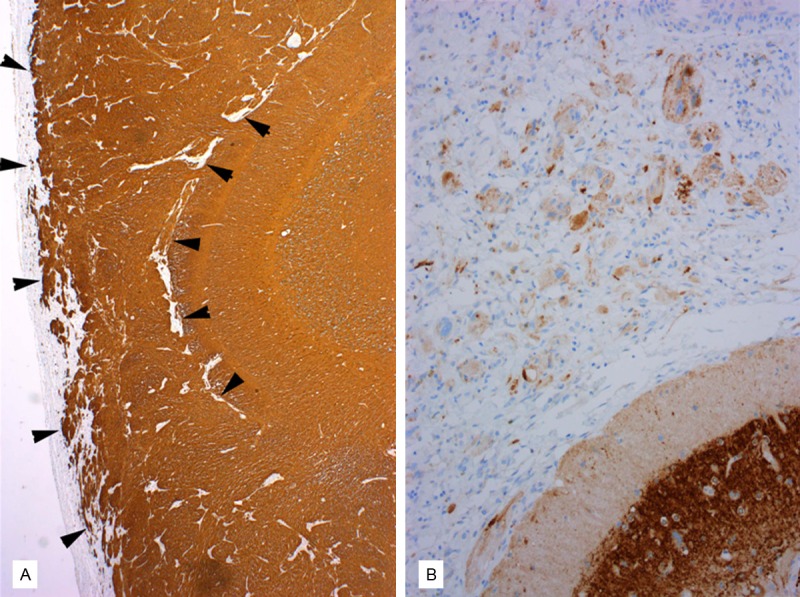
Immunohistochemical findings of the cerebellar tumor from the initial surgery. A. GFAP immunostain is diffusely and strongly positive in the tumor cells within the leptomeninges (between arrowheads). B. Synaptophysin immunostain reveals occasional positive tumor cells.
The lesion from the second resection was histologically very similar to the initially resected tumor, with the additional finding of radiation-related changes. There were no increased mitotic figures or features of anaplastic transformation.
Molecular analysis
Genomic DNA was extracted from the formalin-fixed paraffin-embedded tumor tissue (from the initial resection) following manual microdissection, and tested using the Sequenom MassARRAY OncoCarta Mutation Profiler Panel (San Diego, CA), which evaluates 19 oncogenes for 238 somatic mutations. No mutations, including BRAF V600E, IDH1, or IDH2, were identified.
Discussion
Although very rare, PXAs occurring in the extracerebral compartments have been reported in the literature. These atypical locations include the cerebellum, spinal cord, sellar region, lateral ventricle, and retina [10-14]; among these, the cerebellum is the most frequently reported location [15]. Approximately 20 cases of cerebellar PXA, including composite PXA (PXA-ganglioglioma being most common), have been described in the literature to date, with some interesting clinicopathologic differences from cerebral PXAs [15-18]. Of these reported cases: i) most cases occurred in adults, ranging in age from 3 months to 68 years (versus a younger age profile in cerebral PXAs); ii) radiologically, a higher percentage of cases had a solid enhancing tumor (versus a cyst with enhancing mural nodules); and iii) microscopically, composite histologies (most frequently, PXA-ganglioglioma) were more commonly seen (versus “pure” PXA histology). Given its rarity, awareness of these differences may be useful in diagnosis and differential diagnosis of cerebellar PXA. In contrast, the prognosis of cerebellar PXA appears similar to that of its cerebral counterparts, with an indolent clinical behavior despite some recurrent cases; however, two cases of malignant transformation have been reported [9,15]. Interestingly, one of these two patients had PXA-NF1 (described in detail below) [9].
Individuals with NF1 are predisposed to brain tumors, and the vast majority of these tumors are pilocytic astrocytomas of the optic pathways and brainstem. PXAs rarely occur in association with this disease and are reported to be more commonly located in the cerebrum than in the cerebellum. Only two previous cases of cerebellar PXA-NF1 have been reported to date [6,9]. The first, a 51-year-old woman, presented with worsening ataxia, and an MRI examination demonstrated an atypical infiltrating lesion of the vermis and surrounding cerebellar hemispheres [6]. She subsequently underwent a gross total resection of the tumor and had no recurrence, although details of postoperative follow-up were not provided. The histological features were those of a typical “pure” PXA with a very low proliferation index. The second patient, a 30-year-old woman, presented with headaches and gait disturbance and was found to have two tumors that involved the right occipital lobe and cerebellum [9]. Following surgical resection of both tumors, she experienced multiple recurrences of the cerebellar tumor, which was histologically interpreted as a composite PXA-oligodendroglioma, with progression to a higher-grade lesion that was characterized by significantly increased mitotic figures and microvascular proliferation. She expired three years after the initial presentation. These two previously reported patients had completely opposite outcomes. Our patient had a recurrence three months after the initial gross total resection even though there were no features of malignant transformation. This is earlier than usually reported after gross total resection of PXA. Most cerebellar PXAs have an indolent clinical course, as do most cerebral PXAs; however, our case raises the consideration that NF1 may, by some yet unknown mechanism, alter the clinical course and biologic behavior of these tumors. Further studies are needed to explore this issue, and close follow-up is warranted for this patient.
Recently, BRAF mutations have been detected in up to 70% of PXAs and are now recognized as a genetic hallmark of this tumor. Molecular testing of the current sample did not reveal BRAF, IDH1, or IDH2 mutations. Koelsche et al. recently reported that BRAF V600E mutation is more commonly seen in PXAs located in the temporal lobe than in other locations [19]. To our knowledge, our case is the first cerebellar PXA in which molecular mutation analysis was performed.
Radiologically, this patient’s cerebellar tumor exhibited a gyriform enhancing pattern on T1-weighted MRI, bearing a superficial resemblance to LDD, although LDD is typically hypointense with no enhancement on T1-WI and hyperintense on T2-WI, with parallel linear striation on the surface of the lesions. The gyriform enhancing pattern seen in the current case histologically represents extensive tumor involvement of the leptomeninges, resulting in marked thickening of this structure. We previously reported a similar radiological pattern in a cerebellar ganglio (glio) matous lesion with diffuse leptomeningeal involvement in a child [20].
In summary, this third case of PXA-NF1 with unusual clinicoradiological features raises the consideration that NF-1 may, by a yet unknown mechanism, play a role in altering the clinical course and biologic behavior of cerebellar PXA.
Acknowledgements
Authors would like to thank Kathryn Stockbauer, PhD (Manager, Academic Development at Houston Methodist Hospital) for critical review of this manuscript.
Disclosure of conflict of interest
None.
References
- 1.Guillamo JS, Créange A, Kalifa C, Grill J, Rodriguez D, Doz F, Barbarot S, Zerah M, Sanson M, Bastuji-Garin S, Wolkenstein P Réseau NF France. Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1): a retrospective study of 104 patients. Brain. 2003;126:152–60. doi: 10.1093/brain/awg016. [DOI] [PubMed] [Google Scholar]
- 2.Ozek MM, Sav A, Pamir MN, Oxer AF, Ozek E, Erzen C. Pleomorphic xanthoastrocytoma associated with von Recklinghausen neurofibromatosis. Childs Nerv Syst. 1993;9:39–42. doi: 10.1007/BF00301935. [DOI] [PubMed] [Google Scholar]
- 3.Koeller KK, Henry JM. Armed Forces Institute of Pathology. From the archives of the AFIP: superficial gliomas: radiologie-pathologie correlation. Radiographies. 2001;21:1533–1556. doi: 10.1148/radiographics.21.6.g01nv051533. [DOI] [PubMed] [Google Scholar]
- 4.Kubo O, Sasahara A, Tajika Y, Kawamiira H, Kawabatake H, Takakura K. Pleomorphic xanthoastrocytoma with neurofibromatosis type 1: case report. Noshuyo Byori. 1996;13:79–83. [PubMed] [Google Scholar]
- 5.Ohta S, Ryu H, Miura K. Eighteen-year survival of a patient with malignant pleomorphic xanthoastrocytoma associated with von Recklinghausen neurofibromatosis. Br J Neurosurg. 1999;73:420–422. doi: 10.1080/02688699943583. [DOI] [PubMed] [Google Scholar]
- 6.Naidich MJ, Walker MT, Gottardi-Littelt NR, Han G, Chandler JP. Cerebellar pleomorphic xanthoastrocytoma in a patient with neurofibromatosis type 1. Neuroradiology. 2004;46:825–829. doi: 10.1007/s00234-004-1216-0. [DOI] [PubMed] [Google Scholar]
- 7.Yoshihiro K, Masashi O, Hiroshi R, Kenichi U, Katsiitoshi M. A case of pleomorphic xanthoastrocytoma with malignant findings with complicated neurofibromatosis-1. Bull Fujita Med Society. 1998;22:227–228. [Google Scholar]
- 8.Prayson RA. Pleomorphic xanthoastrocytoma arising in neurofibromatosis type 1. Clin Neuro Pathol. 2012;31:152–154. doi: 10.5414/np300436. [DOI] [PubMed] [Google Scholar]
- 9.Saikali S, Le Strat A, Heckly A, Stock N, Scarabin JM, Hamlat A. Multicentric pleomorphic xanthoastrocytoma in a patient with neurofibromatosis type 1. Case report and review of the literature. J Neuro Surg. 2005;102:376–81. doi: 10.3171/jns.2005.102.2.0376. [DOI] [PubMed] [Google Scholar]
- 10.Arita K, Kurisu K, Tominaga A, Sugiyama K, Sumida M, Hirose T. Intrasellar pleomorphic xanthoastrocytoma: case report. Neurosurgery. 2002;51:1079–1082. doi: 10.1097/00006123-200210000-00042. [DOI] [PubMed] [Google Scholar]
- 11.Bucciero A, De Caro MI, Tedeschi E, De Stefano V, Bianco M, Soricelli A, Vizioli L, Cerillo A. Atypical pleomorphic xanthoastrocytoma. J Neurosurg Sci. 1998;42:153–7. [PubMed] [Google Scholar]
- 12.Fouladi M, Jenkins J, Burger P, Langston J, Merchant T, Heideman R, Thompson S, Sanford A, Kun L, Gajjar A. Pleomorphic xanthoastrocytoma: favorable outcome after complete surgical resection. Neuro Oncol. 2001;3:184–192. doi: 10.1093/neuonc/3.3.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jea A, Ragheb J, Morrison G. Unique presentation of pleomorphic xanthoastrocytoma as a lytic skull lesion in an eight-year-old girl. Pediatr Neuro Surg. 2002;37:254–257. doi: 10.1159/000066217. [DOI] [PubMed] [Google Scholar]
- 14.Yang W, Huang B, Liang C. Pleomorphic xanthoastrocytoma in the lateral ventricle with extensive subarachnoid dissemination: report of a case and review of the literature. Chin Med J. 2012;125:396–399. [PubMed] [Google Scholar]
- 15.Hamlat A, Le Strat A, Guegan Y, Ben-Hassel M, Saikali S. Cerebellar pleomorphic xanthoastrocytoma: case report and literature review. Surg Neurol. 2007;68:89–95. doi: 10.1016/j.surneu.2006.08.064. [DOI] [PubMed] [Google Scholar]
- 16.Yeaney GA, O’Connor SM, Jankowitz BT, Hamilton RL. A 16-year-old male with a cerebellar mass. Brain Pathol. 2009;19:167–70. doi: 10.1111/j.1750-3639.2008.00242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mano Y, Kanamori M, Sonoda Y, Kumabe T, Watanabe M, Tominaga T. [A case report of cerebellar pleomorphic xanthoastrocytoma] . No Shinkei Geka. 2009;37:586–90. [PubMed] [Google Scholar]
- 18.Yu S, He L, Zhuang X, Luo B. Pleomorphic xanthoastrocytoma: MR imaging findings in 19 patients. Acta Radiol. 2011;52:223–8. doi: 10.1258/ar.2010.100221. [DOI] [PubMed] [Google Scholar]
- 19.Koelsche C, Sahm F, Wöhrer A, Jeibmann A, Schittenhelm J, Kohlhof P, Preusser M, Romeike B, Dohmen-Scheufler H, Hartmann C, Mittelbronn M, Becker A, von Deimling A, Capper D. BRAF-mutated pleomorphic xanthoastrocytoma is associated with temporal location, reticulin fiber deposition and CD34 expression. Brain Pathol. 2014;24:221–229. doi: 10.1111/bpa.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takei H, Dauser R, Su J, Chintagumpala M, Bhattacharjee MB, Jones J, Adesina AM. Anaplastic ganglioglioma arising from a Lhermitte-Duclos-like lesion. Case report. J Neurosurg. 2007;107(Suppl):137–42. doi: 10.3171/PED-07/08/137. [DOI] [PubMed] [Google Scholar]