

Abstract
Although previous reports purpored that the unique magnetic resonance imaging (MRI) features of Lhermitte-Duclos disease (LDD) obviates the need for biopsy, we have made a misdiagnosis of LDD which has an indistinguishable imaging appearance. We present a patient who suffered from a normal cerebellum with arachnoid vascular malformation that had imaging characteristics which were indistinguishable from LDD before operation. This atypical imaging appearance, which could potentially be confused with LDD, may lead to misdiagnosis and inappropriate treatment in the absence of tissue sampling. Thus, this finding suggests that in those patients where images are highly suggestive of LDD but lack other manifestations of Cowden syndrome, biopsy is required and advanced imaging with magnetic resonance spectroscopy (MRS) should be strongly considered.
Keywords: Lhermitte-Duclos disease, Cowden syndrome, magnetic resonance imaging, misdiagnosis
Introduction
Lhermitte-Duclos disease (LDD), also known as dysplastic gangliocytoma of the cerebellum, is an extremely rare pathological entity. The disease is characterized by a cerebellar mass composed of enlarged cerebellar folia containing abnormal ganglion cells. More than 200 cases have been reported in the literature, but the precise incidence is still unknown [1]. Clinically, LDD usually presents in young and middle-aged adults, rarely in children, commonly with a symptom of intracranial hypertension and cerebellar dysfunction [2]. Magnetic resonance (MR) imaging is the appropriate technique allowing preoperative diagnosis with the characteristic striated pattern of exaggerated folia appearance on T2-weighted images [3]. The characteristic histopathological hallmarks of the lesion include the destruction of the normal laminar cytoarchitecture of the cerebellar cortex with abundant dysplastic and hypertrophic neurons in the internal granular cell layer, absence of the Purkinje cell layer, and hypertrophy of the molecular layer [3].
Recent advances in imaging techniques especially in MR imaging have helped us to understand the pathological and metabolic changes preoperatively and advance appropriate surgical treatment. However, the imaging diagnosis may sometimes lead to misdiagnosis. Therefore pathological diagnosis technique is the golden standard. Here, we present a case of LDD and a case of non-LDD, which have the similar MR imaging characters, but different pathological results.
Materials and methods
A case of LDD and a case of non-LDD were retrieved in the department of Neurosurgery, Beijing Tiantan Hospital. All tissues were fixed in 10% neutral buffered formalin, routinely processed, and embedded in paraffin. Hematoxylin-eosin stained sections were reviewed independently by two experienced pathologists.
Results
Case 1
A 40-year-old woman was accidentally found to have a posterior fossa mass on CT during a post-traumatic physical examination. Her past medical history revealed an operation of breast cancer resection. No neurologic or systemic symptoms were found, and no family history of tumor was noted. MRI showed a mass in the right cerebellar hemisphere with compression of the fourth ventricle. The mass was hypointense on T1-weighted images, hyperintense on T2-weighted images and showed a pathognomonic ‘tiger-striped’ appearance (Figure 1A, 1B). There was a very subtle enhancement along the folia on post-contrast T1-weighted images (Figure 1C). These findings strongly indicated LDD. Subtotal surgical excision was carried out via the right paramedian suboccipital approach. At surgery, the mass presented with an abnormal surface consisting of thickened, mildly soft and elastic folia. Margins of the mass were not easy to define, although its color was rather pale compared to normal tissue. After surgery, histopathological examination showed loss of Purkinje cells, replacement of granular cells, hypertrophy of dysplastic ganglion cells and expanded abnormal molecular layers. (Figure 2A). LDD was diagnosed based on these findings. Postoperative course was uneventful and the patient was discharged 10 days post-operation with normal neurologic functions. The patient remained symptom-free at a 18-month follow-up examination.
Figure 1.
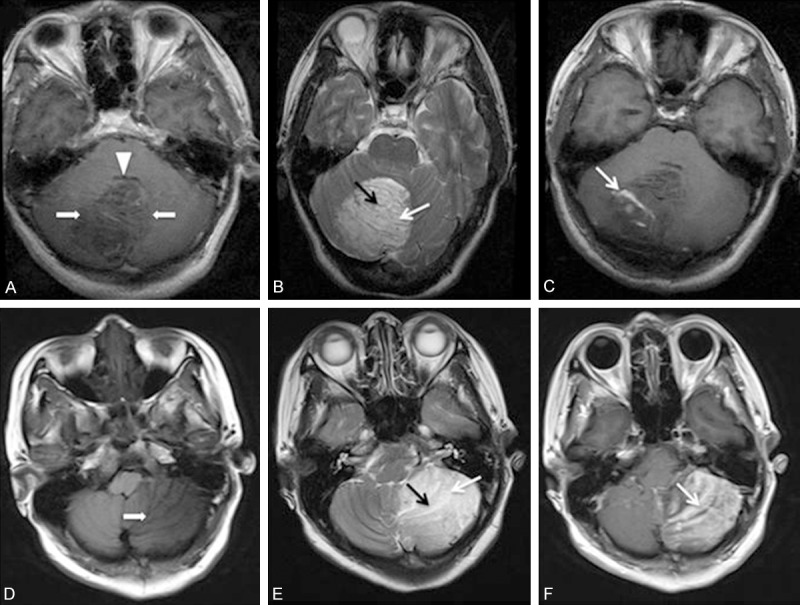
A. Axial T1-weighted MR image in the first patient shows the typical striated appearance, with hypo- and isointense bands. Note the abnormal hypointense area (arrows) in the region of the right cerebellar hemisphere. The fourth ventricle (arrowhead) is compressed. B. T2-weighted axial image in the first patient shows the right cerebellar lesion with the characteristic ‘tiger striped’ appearance. The inner hyperintense (white arrow) and the outer iso- to hypointense intense (black arrow) portions of the abnormal folia are to be noted. C. Contrast-enhanced T1-weighted axial image in the first patient shows the ‘vascular pattern’ of enhancement between the folia (white arrows). D. T1-weighted axial MR image in the second patient shows hypointense-to-isointense left cerebellar lesion with linear striations. Note the abnormal hypointense area (arrows) in the region of the left cerebellar hemisphere. E. T2-weighted axial image in the second patient shows the left cerebellar lesion with the characteristic ‘tiger striped’ appearance. The inner hyperintense (white arrow) and the outer iso- to hypointense intense (black arrow) portions of the abnormal folia are to be noted. F. Contrast-enhanced T1-weighted axial image in the second patient shows obvious heterogeneous enhancement of the linear striations (white arrows) interspersed within the hypodense linear areas.
Figure 2.
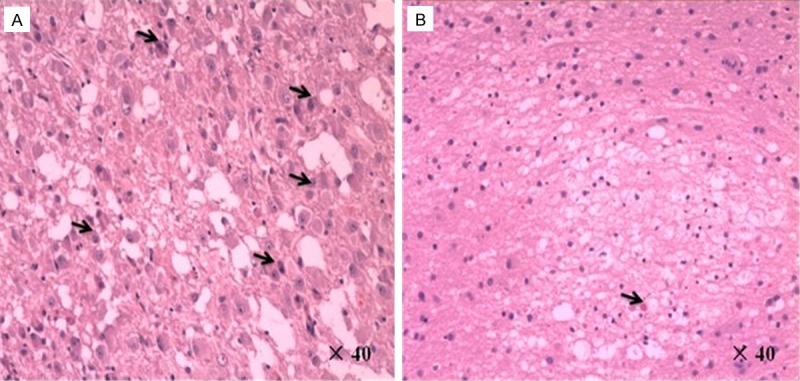
A. High-power photomicrograph in the first patient showed loss of Purkinje cells, replacement of granular cells, hypertrophy of dysplastic ganglion cells with prominent nucleoli (black arrows) and expanded abnormal molecular layers. (Hematoxylin-eosin stain; original magnification, × 40). B. High-power photomicrograph in the second patient showed normal cerebellum with arachnoid vascular malformation (black arrow) and partial loss of Purkinje cells. (Hematoxylin-eosin stain; original magnification, × 40).
Case 2
A 42-year-old woman presented with a 2-month history of intermittent and paroxysmal posterior headaches and progressive gait disturbance was admitted. Upon examination, the patient had a dysdiadochokinesia, papilledema and truncal ataxia. MRI showed the hypointense-to-isointense left cerebellar lesion with linear striations on T1-weighted imaging (Figure 1D). The lesion was hyperintense on T2-weighted imaging with areas of linear hypointensity streaks running throughout the lesion. The parallel linear striations on the surface of the lesion were characteristic of the “tiger striping” or the “striated cerebellum” effect of LDD (Figure 1E). Contrast-enhanced T1-weighted axial MRI showed obvious heterogeneous enhancement of the linear striations interspersed within the hypodense linear areas (Figure 1F). A left paramedian suboccipital craniectomy revealed a yellowish gray tumor of soft texture with a defined plane of cleavage from the surface of cerebellum. Subtotal removal of the tumor was achieved. Histopathological examination revealed normal cerebellum with arachnoid vascular malformation and partial loss of Purkinje cells (Figure 2B). The postoperative course was unremarkable. The patient remained symptom-free at a 12-month follow-up examination.
Discussion
LDD is a very rare, progressive pathological entity with an unclear fundamental nature and pathogenesis. Histopathologically, LDD is characterized by replacement of the granular cell layer and Purkinje cells by scattered dysplastic ganglion cells, which results in global thickening of the cerebellar folia [4]. Recently, LDD has been shown to be a sporadic, isolated disease or associated with Cowden’s syndrome (CS) characterized by multiple hamartomas and high risk of thyroid, endometrial and breast malignancies [5,6]. Currently LDD and Cowden syndromeare considered a single phakomatosis with autosomal dominant inheritance, which is caused by a mutation of phosphatase and tensin homolog (PTEN) suppressor oncogene [6-8].
Most cases of LDD are documented to occur in adults in the third and fourth decades without gender predilection. The typical symptoms with LDD are headaches, cerebellar dysfunction, occlusive hydrocephalus and cranial nerve palsies due to progressive mass effect in the posterior fossa. However, the clinical presentation of most posterior fossa pathologies is similar; and many diseases share a similar MRI appearance except LDD. It is considered so unique that some authors have proposed that the diagnosis of LDD can be definitively made based on the MRI features [3,9,10]. The characteristic appearance of LDD on MRI is that a non-enhancing cerebellar lesion, hypointense on T1-weighted images and hyperintense on T2-weighted images with a ‘tiger-striped’ appearance. With the characteristic MR imaging of LDD, we have diagnosed the disease before treatment.
However, the imaging diagnosis may sometimes lead to misdiagnosis. Douglas-Akinwande AC et al. [11] reported a single case that a medulloblastoma mimicked LDD on MRI and CT. In our study, we also found that a left cerebellar lesion was misdiagnosed as LDD preoperatively, and the pathological diagnosis was a normal tissue of cerebellum with arachnoid vascular malformation. Recent studies reported that in challenging cases, 1H MR spectroscopy with typical normal or low Cho/Cr, PWI showing elevated cerebral blood volume (CBV) despite lack of robust contrast enhancement and ADC map of normal diffusion pattern could establish the diagnosis of LDD [12]. Therefore, MRI alone is insufficient to make the diagnosis of LDD, although MRS may provide additional information histopathologic confirmation is required.
Except for the fact that the diagnosis of LDD was overlooked, the long follow-up period of this case is noteworthy. Naoya Hashimoto et al. provided new insights on the natural history of the disease. They revealed that the tumor doubling time (Tdt) was about 42 months in MRI-based volumetry, which seems compatible with a tumor of benign nature. When compared with asymptomatic meningiomas, mean Tdt of 93.6 months, this finding seems to suggest that LDD grows more rapidly than meningiomas [13]. In addition, it was somewhat surprising that the tumor has shown a linear growth pattern, although they expected an exponential growth like other cancers or neuro-glial tumors. It may represent that this tumor grows very slowly and we are just looking at a part of the very long-term natural history [13].
In 1991, Padberg et al. were credited for the recognition of the coexistence of LDD and a multiple hamartoma-neoplasia syndrome, also called Cowden syndrome (CS) [14]. CS is an autosomal dominant hereditary cancer syndrome, characterized by mucocutaneous lesions and other systemic hamartomas associated with a high incidence of breast, thyroid and genitourinary malignancies [5,6]. In 2000, Robinson and Cohen found a clinical coexistence of LDD and CS on reviewing the files of five patients, previously diagnosed of dysplastic cerebellar gangliocytoma [1]. All patients had other hamartomas and malignancies attributable to the Cowden syndrome complex. From their findings the authors considered that this association between the two disorders might represent a true “neurocutaneous syndrome”, based on a hereditary aberration of a tumor suppressor gene [1]. Thus, Robinson and Cohen defined a new phakomatosis named “Cowden and Lhermitte-Duclos disease complex (COLD)” and concluded that detection of either one of these two disorders should prompt a thorough search for the other. Recognition of this association has direct clinical relevance, as long-term follow up of patients with LDD may lead to early diagnosis of cancer. In our reports, we also found the patient of LDD had a diagnosis of breast cancer previously, and the other patient of LDD-misdiagnosis didn’t have CS-associated symptoms and signs.
In symptomatic patients, surgical excision can improve the symptoms rapidly, and appears to be the only effective treatment. But, the management of asymptomatic patients with LDD is controversial and needs prudent decision making. Both the progressive nature and recurrence after incomplete resection can lead to poor prognosis. On the other hand, the lesion usually blends into normal cerebellar tissue and is difficult to distinguish from healthy cerebellar tissue, which may result in extensive resection and cause neurological deficits such as cerebellar mutism. In addition, the patients with LDD may lead to early diagnosis of cancer, and the histopathologic confirmation is required [15]. Thus, better prognosis can be achieved from surgical intervention in the early stage of LDD.
In conclusion, MRI alone is insufficient to make the diagnosis of LDD, the histopathologic confirmation is required. If the preoperation imaging is highly suggestive of LDD, detailed medical history and clinical examination are conducive to diagnosis of LDD or CS. Furthermore, long-term follow up of patients with LDD may lead to early diagnosis of cancer.
Consent
Written informed consents were obtained from the patients for publication of this case report and any accompanying images. A copy of the written consent is available for review by the editor-in-chief of this journal.
Acknowledgements
This study was supported by National Key Technology Research and Development Program of the Ministry of Science and Technology of China (No. 2013BAI09B03) and Beijing Institute for Brain Disorders (No. BIBD-PXM2013_014226_07_000084).
Disclosure of conflict of interest
None.
References
- 1.Robinson S, Cohen AR. Cowden disease and Lhermitte-Duclos disease: characterization of a new phakomatosis. Neurosurgery. 2000;46:371–383. doi: 10.1097/00006123-200002000-00021. [DOI] [PubMed] [Google Scholar]
- 2.Nowak DA, Trost HA, Porr A, Stolzle A, Lumenta CB. Lhermitte-Duclos disease (Dysplastic gangliocytoma of the cerebellum) Clin Neurol Neurosurg. 2001;103:105–110. doi: 10.1016/s0303-8467(01)00124-x. [DOI] [PubMed] [Google Scholar]
- 3.Thomas B, Krishnamoorthy T, Radhakrishnan VV, Kesavadas C. Advanced MR imaging in Lhermitte-Duclos disease: moving closer to pathology and pathophysiology. Neuroradiology. 2007;49:733–738. doi: 10.1007/s00234-007-0241-1. [DOI] [PubMed] [Google Scholar]
- 4.Nowak DA, Trost HA. Lhermitte-Duclos disease (dysplastic cerebellar gangliocytoma): a malformation, hamartoma or neoplasm? Acta Neurol Scand. 2002;105:137–145. doi: 10.1034/j.1600-0404.2002.1r127.x. [DOI] [PubMed] [Google Scholar]
- 5.Afshar-Oromieh A, Linhart H, Podlesek D, Schrempf W, Schackert G, Krex D. Postoperative cerebellar mutism in adult patients with Lhermitte-Duclos disease. Neurosurg Rev. 2010;33:401–408. doi: 10.1007/s10143-010-0278-1. [DOI] [PubMed] [Google Scholar]
- 6.Govindan A, Premkumar S, Alapatt JP. Lhermitte-Duclos disease (dysplastic gangliocytoma of the cerebellum) as a component of Cowden syndrome. Indian J Pathol Microbiol. 2012;55:107–108. doi: 10.4103/0377-4929.94875. [DOI] [PubMed] [Google Scholar]
- 7.Pilarski R, Eng C. Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumor syndrome. J Med Genet. 2004;41:323–326. doi: 10.1136/jmg.2004.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sutphen R, Diamond TM, Minton SE, Peacocke M, Tsou HC, Root AW. Severe Lhermitte-Duclos disease with unique germline mutation of PTEN. Am J Med Genet. 1999;82:290–293. doi: 10.1002/(sici)1096-8628(19990212)82:4<290::aid-ajmg3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 9.Nair P, Pal L, Jaiswal AK, Behari S. Lhermitte-Duclos disease associated with dysembryoplastic neuroepithelial tumor differentiation with characteristic magnetic resonance appearance of “tiger striping”. World Neurosurg. 2011;75:699–703. doi: 10.1016/j.wneu.2010.10.046. [DOI] [PubMed] [Google Scholar]
- 10.Carlson JJ, Milburn JM, Barre GM. Lhermitte-Duclos disease: case report. J Neuroimaging. 2006;16:157–162. doi: 10.1111/j.1552-6569.2006.00020.x. [DOI] [PubMed] [Google Scholar]
- 11.Douglas-Akinwande AC, Payner TD, Hattab EM. Medulloblastoma mimicking Lhermitte-Duclos disease on MRI and CT. Clin Neurol Neurosurg. 2009;111:536–539. doi: 10.1016/j.clineuro.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 12.Cianfoni A, Wintermark M, Piludu F, D’Alessandris QG, Lauriola L, Visocchi M, Colosimo C. Morphological and functional MR imaging of Lhermitte-Duclos disease with pathology correlate. J Neuroradiol. 2008;35:297–300. doi: 10.1016/j.neurad.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 13.Goto Y, Hashimoto N, Okita Y, Goto T, Rabo C, Hirayama H, Horikawa Y, Kinoshita M, Kagawa N, Yoshimine T. A surgically treated case of Lhermitte-Duclos disease with a precise natural history and high uptake of FDG on PET. J Neurooncol. 2010;97:445–450. doi: 10.1007/s11060-009-0042-y. [DOI] [PubMed] [Google Scholar]
- 14.Padberg GW, Schot JD, Vielvoye GJ, Bots GT, de Beer FC. Lhermitte-Duclos disease and Cowden disease: a single phakomatosis. Ann Neurol. 1991;29:517–523. doi: 10.1002/ana.410290511. [DOI] [PubMed] [Google Scholar]
- 15.Kumar R, Vaid VK, Kalra SK. Lhermitte-Duclos disease. Childs Nerv Syst. 2007;23:729–732. doi: 10.1007/s00381-006-0271-8. [DOI] [PubMed] [Google Scholar]