

Abstract
Aim
We present the potential false-negative rate of exome sequencing for the detection of pharmacogenomic variants.
Materials & Methods
Depth of coverage of 1928 pharmacogenomically relevant variant positions was ascertained from 62 exome-sequenced samples.
Results
Approximately 14% of the 1928 variant locations examined had inadequate depth of coverage (<20x). The variants with inadequate coverage were predominantly located outside of protein-coding portions and included some clinically relevant variant positions, such as the warfarin VKORC1 variant.
Conclusion
While the use of exome sequencing is becoming more prevalent in fundamental research, clinical trials and clinical use; there is a possibility of false-negative results. The possible quality issues such as false-negative rate should be considered with the use of exome sequencing.
Keywords: exome sequencing, pharmacogenomics, warfarin
Background
One of the overarching goals of the human genome project is the use of personal genomic information for the selection of targeted therapy of diseases. One of the first successes has been in the field of pharmacogenomics (PGx). While many factors can influence drug response in a patient, it has become clear that genetic variation can play an important role. Indeed, diseases ranging from cancer to hypertension have seen an increasing correlation between specific genetic variants and therapeutic success in clinical management. In recent years, several genes with roles in the Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) of drugs have been identified. Specific variants in these genes have been associated with response to specific drugs [1]. For example, variants in the VKORC1 and CYP2C9 have been associated with response to warfarin, an orally prescribed anticoagulant drug [2,3].
Accurate genotyping of variants in these genes is crucial to implementing PGx in a routine clinical setting. Microarray-based approaches have been developed to specifically genotype-known genomic variants in these genes (e.g., AmpliChip CYP450 [4], Roche Molecular Diagnostics, Basel, Switzerland; and DMET Plus [5] Affymetrix, CA, USA). The recent advances in next-generation sequencing (NGS) technologies and specifically exome sequencing have the potential to replace microarray-based protocols to detect these variants. Furthermore, a sequencing-based approach has the ability to detect novel variants not yet characterized, being ‘private’ to an individual or to interrogate additional genes not currently associated with drug response. Despite these benefits, sequencing does come with some limitations. First, the efficiency of exome sequencing relies on the ability of oligonucleotide hybridization probes to capture a target region. Second, any PGx variants not specifically included in the designed target regions will be missed. In contrast, PGx microarrays are specifically designed for each individual variant of interest.
To examine the utility of exome sequencing for PGx, we examined the potential false-negative rate of exome sequencing for the detection of known PGx variants. Using 62 exome-sequenced samples, we explored the coverage of the 1928 PGx variants assayed on the Affymetrix DMET Plus microarray.
Methods
Exome capture methods
Genomic DNA from 62 individuals derived from whole blood was used for all of the exome evaluated. Data from Thomas Jefferson University and the University of Texas Southwestern Medical Center were obtained under separate research protocols approved by their respective Institutional Review Boards. Four different exome capture methods were employed: the TargetSeq (n = 33) (TargetSeq™ Target Enrichment Kit [Life Technologies, CA, USA]); SureSelect v4 (n = 5) (Sure-Select™ Human All Exon Target Enrichment System v4 [Agilent Technologies, CA, USA]), SureSelect v4 + UTR (n = 12) (SureSelect™ Human All Exon Target Enrichment System v4 + UTR [Agilent Technologies]) and, TruSeq (n = 12) (TruSeq™ Exome Enrichment Kit [Illumina, CA, USA]).
Exome sequencing
The TargetSeq and SureSelect v4 libraries were sequenced on a SOLiD 5500xl (Life Technologies) using paired-end 50 and 35 bp read lengths. Sequencing of the Illumina TruSeq exome libraries was performed on a HiSeq 2000 (Illumina) using paired-end 100 bp read lengths.
Sequence read alignment
All sequence reads were mapped to the hg19 reference genome. Exomes sequenced on the SOLiD 5500xl were mapped using the Applied Biosystems LifeScope Genomic Analysis Software v2.5. Each sequence read was allowed to have a maximum of two mismatches. Illumina HiSeq 2000 sequence reads were mapped using the Short Read Mapping Package (SHRiMP2) [6]. The sequence reads were quality-trimmed by cutadapt, which uses read-associated quality values [7]. During mapping, mismatches or insertion–deletion (indels) were allowed that did not comprise more than 4% of a given read’s length. For all sequence mappings, only those reads mapping uniquely to the human genome were maintained.
Variants & calculation of coverage
The 1936 SNPs, indels and copy number variants assayed by the Affymetrix DMET Plus microarray were examined. Six copy number variants examined by the DMET microarray were removed from analysis due to exome sequencing’s inability to accurately capture these variants. For two variants on the microarray that are located at the same genomic positions, but assayed by two distinct probes each, were considered only once. The remaining 1928 variants were maintained for further analysis.
Depth of coverage at each variant position was determined by bedcov in SamTools v1.19 [8], which calculated the number of times a read at each variant location was observed. A minimum depth of coverage of 20x was used as a benchmark for determining adequate coverage of a variant location.
Results
The 1928 genomic variants examined in this study are all included in the Affymetrix DMET Plus microarray assay [9-11]. These variants are from 231 ADMET relevant genes, and are spread throughout different portions of the genes. These variants have varying functional effects and are located within different portions of genes and outside of coding exons (Figure 1A). While 69.5% of the variants are located within protein-coding portions of a gene, the remaining variants are located outside of these regions, including untranslated regions (UTR’s) (12.9%), intronic (10.6%), promoter regions (1.4%) and intergenic regions (5.5%).
Figure 1. Coverage of pharmacogenomic variants by the exome capture kits.
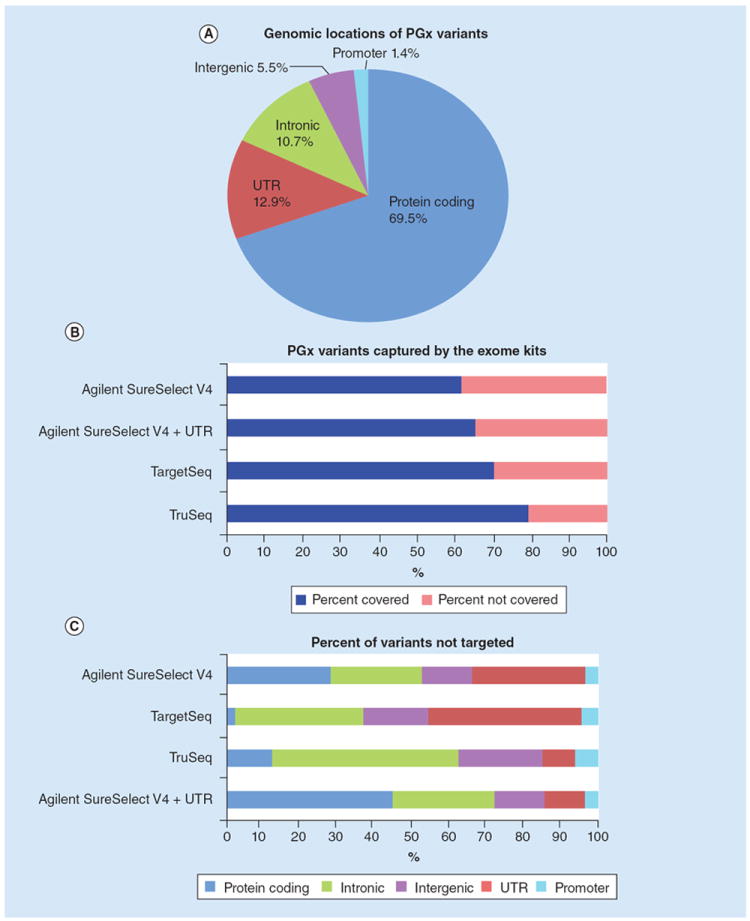
(A) A pie chart showing the genomic distribution of the 1928 PGx variants examined. (B) For each of the four exome capture kits, the ratio of variants targeted versus those not targeted. (C) Of those variants not targeted by the exome capture kits, the distribution of their genomic locations is shown.
PGx: Pharmacogenomics; UTR: Untranslated Regions.
To determine the utility of exome sequencing in capturing these variants, we first examined the percentage of these variants to be specifically targeted by four different exome capture kits (TargetSeq, TruSeq, Agilent SureSelect v4 and Agilent SureSelect v4 + UTRs). Each of the four exome kits examined specifically target more than half of the 1928 variants, but many of the variants are not covered (Figure 1B). The TruSeq capture kit covers the highest proportion of the variants (~80%), while the Agilent SureSelect has the poorest coverage (~60%). Interestingly, the addition of the UTR regions to the Agilent kit allows for an additional ~5% of the variants to be covered. It should be noted that the more recently released version of the Agilent SureSelect kit (version 5) does increase the targeted variants to a level similar to that of the TruSeq kit (data not shown). As exome sequencing is designed to cover protein-coding regions of the genome, it is not surprising that those variants located within the intronic, intergenic and UTR regions make up the highest proportion of variants not targeted by the exome capture kits (Figure 1C).
Despite the varying genomic locations of these variants, the Affymetrix’s targeted use of molecular inversion probes enables efficient genotyping of all the variants. We examined the depth of coverage at these 1928 locations in 62 exome samples to determine whether exome sequencing would have similar success in variant determination at these genomic locations (Supplementary Table 1 gives a summary of the sequence read mapping and coverage obtained across the exome; see online at www.futuremedicine.com/doi/full/10.2217/PME.14.77). As a metric, a minimum coverage of ≥20x at a variant was considered to be sufficient coverage for a reliable genotype call to be made.
Using this criteria, ~30% of the variants in each sample have inadequate depth of coverage (<20x) (Figure 2A). As expected, the variant locations that are present within the protein-coding portions of genes are the most highly covered variants. While ~80% of the variants located within the 5’ and 3’ UTRs are poorly covered, a broader range in coverage is observed compared with other regions (Figure 2A). This heterogeneity in coverage is most likely due to the performance differences between the exome capture kits; some kits have specific hybridization probes targeted toward the UTR regions. In contrast to the variants in coding or UTR regions, those variants located in intronic, promoter and intergenic regions are generally inadequately covered across all samples (Figure 2A; see Supplementary Material 1 for the depth of coverage of each sample at each variant). Thus, a wide range of coverage across these 1928 variant positions is achieved; coverage is highly dependent upon the genomic location of the variant.
Figure 2. Box plots showing coverage of pharmacogenomic variants by exome sequencing.
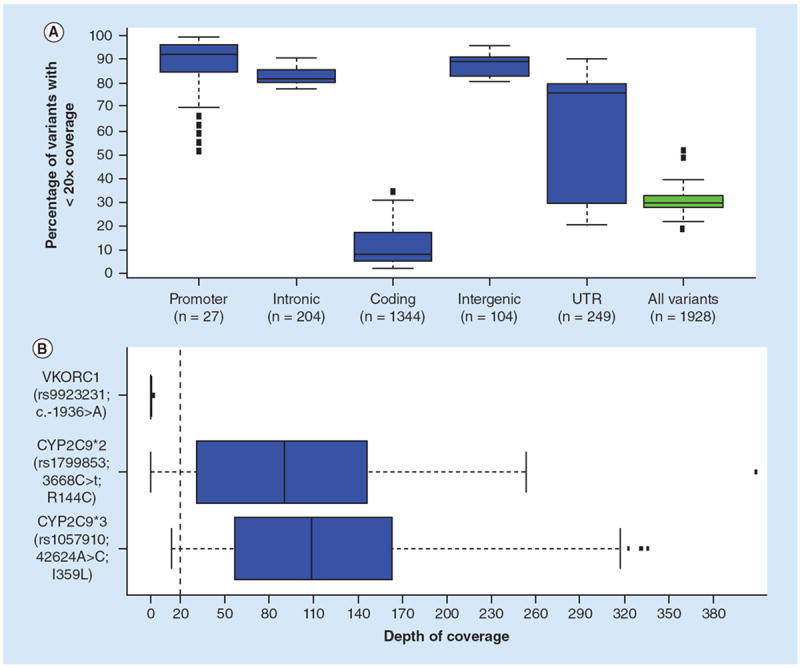
The boxes represent the 25th to 75th quartiles with the median value indicated by the black line for all 62 samples; the whiskers are 1 standard deviation from the interquartile values and the circles represent outliers. (A) Plotted are the distributions of coverage for the variants broken down by genomic region of each variant. Each graph represents all 62 samples and the number of variants at each genomic location is indicated. The y-axis is the percent of variant positions with less than 20x depth of coverage. (B) The depth of coverage for the three variants used for therapeutic dosing of warfarin. The x-axis is the depth of coverage for each of the three variants. The dashed red line indicates a 20x coverage threshold used.
UTR: Untranslated Regions.
Taken together, these results suggest that depending upon the genomic location of the variant, there is a wide range in adequacy of coverage for variant determination using exome sequencing. To determine the effects that this would have on using exome data in determining a person’s susceptibility to a drug response, we focused on variants associated with warfarin response. The warfarin drug label includes CYP2C9 and VKORC1 genotype-based therapeutic dosing guidelines. Based on the specific genotype combinations, different therapeutic doses can be applied [12]. The two CYP2C9 variants (CYP2C9*2 and CYP2C9*3) are both coding non-synonymous SNPs, while the VKORC1 (-16396G>A) is located within the promoter region. We assessed the adequacy of depth of coverage (≥20x) at these three variants within our 62 exome samples, to determine if adequate coverage could be achieved for accurate warfarin dosing (Figure 1B). The two CYP2C9 variants show adequate coverage (>20x) for the majority of the samples analyzed. Inadequate coverage (<20x) was seen in 11 samples for the CYP2C9*2 variant and three samples for the CYP2C9*3, respectively. In comparison, the VKORC1 variant was inadequately covered in all samples analyzed. Based on these results, exome-based determination of warfarin dosing is currently not informative.
It is feasible that despite the fact that the exome kits do not specifically target many of the PGx variants examined here, they may still have indirect capture and sequence data. It is well known that many of the mapped sequence reads do align to ‘off-target’ regions of the genome. As observed in Supplementary Table 1 and Supplementary Figure 1, ~30% of the sequence reads map to ‘off-target’ regions of the genome. Despite the possibility of capturing ‘off-target’ regions, we were still unable to capture those genomic variants not specifically targeted by the exome capture kits.
Discussion
In this study, we examined the utility of exome sequencing for the coverage of known PGx variants. Specifically, we determined if exome sequencing could adequately cover the genomic locations of these variants for reliable genotype determination. Our results show that the genomic location of the variant (coding region or noncoding region) will have an important effect on the reliability of genotypic calls, and subsequent PGx decisions.
The clinical implementation of PGx can lead to improved drug efficacy, reduced adverse effects and improved patient care. However, the ability to integrate PGx is directly associated with the specificity and comprehensiveness of the test. Currently, the Affymetrix DMET Plus assay represents an efficient and specific method to genotype 1936 variants in 231 ADMET-associated genes many of which have known PGx associations. In recent years, NGS has become more prevalent in clinical and research laboratories. NGS and in particular exome-sequencing has many advantages over the use of a microarray. First, instead of relying on assaying only the specific variants in question (the 1936 ADME variants tested here), there is the opportunity to identify and characterize novel variants that may also be actionable. Second, while the 231 ADME genes have been characterized as playing an important role in drug metabolism, other genes of interest may be of clinical importance, and can also be assayed.
Despite the advantages and ever-increasing use of NGS, there is a trade-off. Similar to microarray analysis, exome sequencing will only assess the regions that it is designed to assess. Most of the existing exome methods target the coding regions with varying amounts of noncoding UTR. Although exome is being used for the discovery of novel variants with PGx significance, it should not be assumed that exome testing will adequately assess known PGx variants. Indeed, it is observed here that ~30% of the DMET PGx variants (Figure 1A) lie in intronic and intergenic regions of the genome; these regions generally missed by current exome-sequencing technologies. Additionally, some genes and/or regions generally have poor coverage. For example, the COMT gene (catechol-o-methyltransferase) has an average depth of coverage of 29x across all of the samples, but more than half of the samples examined (34 of 62 exome datasets) had inadequate cover (<20x) (data not shown). An additional limitation of exome sequencing is the current inability to routinely characterize copy number variants. For example copy number variants in the CYP2D6 gene, which are assayed on the Affymetrix DMET Plus microarray, are missed through exome sequencing.
It should be noted that we used a cutoff of 20x sequencing depth to determine if a variant was adequately covered. For the detection of heterozygous germline variants, some laboratories use a minimum coverage of 10–20x. The accuracy of identifying variants is directly related to the depth of coverage, and increased coverage will improve variant calling. Recent papers have examined this [13-15] and for a 90% sensitivity in identifying heterozygous variants, an average depth of coverage between 17 and 36x is needed. Certainly, as the stringency is increased, the number of variants detected will decrease.
While ~30% of the PGx variants examined here are not specifically targeted by the various exome capture kits, it is still possible that these regions may have coverage through “off-target” sequencing. Across the 62 exomes examined here, ~70% of the mapped sequence reads were ‘on-target’ (Supplementary Table 1), leaving 30% of the mapped reads which are located at other regions of the genome. Despite this possibility, the exome sequences did not contain ‘off-target’ reads mapping to the PGx variants, including the VKORC1 variant which is located ~700 bps away from the start of the gene.
Current exome-sequencing methods can miss clinically relevant PGx variants. For example, the VKORC1 promoter variant associated with warfarin response is inadequately covered (Figure 2B). Similarly, examination of the NHBLI Exome Sequencing Project (ESP) Exome Variant Server [16] shows no data for the VKORC1 variant (rs9923231) which is important for warfarin metabolism. In the example of a clinical specimen, unless the deficiency of exome coverage for VKORC1 was specifically noted, the patient report could be falsely negative. Similarly, variants in COMT were poorly covered in the majority of the samples that were exome sequenced. Variants in COMT have been associated with morphine response in acute pain [17], and may affect the levels of levodopa an dentacapone which are used in the Parkinson’s disease [18]. In our datasets, COMT should not be relied upon as a PGx marker. In contrast, it should be noted that many of the well-known genes associated with drug response were adequately covered for genotype analysis: CYP2D6 (tamoxifen) [19,20], CYP2C19 (clopidogrel) [21,22], SLCO1B1 (statins) [23,24] (data not shown).
Conclusion
Our results show that while NGS is becoming increasingly relevant in the clinical setting, there are analytical limitations for the use of current exome data for PGx. The problem is twofold. First, many of the existing exome methods are not designed to return data on noncoding regions. There are many clinically significant PGx variants that are located in noncoding regions. Second, there are regions of coding DNA that are included in the in silico design of the assay, but still do not have adequate sequencing data for interpretation. These two problems create substantial possibilities for false-negative PGx findings when using exome sequencing. Exome methods are a powerful tool for the discovery of new PGx variants that lie in coding sequence. However, until exome methods are redesigned to target noncoding and poorly covered PGx variants, exome sequencing should not be assumed to be a complete analysis of known PGx variants. Additionally, our study addresses only a the basic requirement for sequencing of a targeted region of interest and further studies will be required to address the analytical concordance with SNP arrays and the overall accuracy of based calling by NGS. Furthermore, targeted pharmacogenomic NGS assays such as PGRNseq (National Institutes of Health’s Pharmacogenomics Research Network), which custom captures specific genes with extended targeting of noncoding regions, are a step toward more complete pharmacogenetic analysis [25]. Furthermore, methods such as whole-genome sequencing may become more cost-effective and ameliorate the sequence targeting limitations of exome sequencing.
Future perspective
As our understanding of the complexities of the genome increases, the use of next-generation technologies will become an ever more important and powerful tool for both research and clinical applications. The importance of very rare or ‘private’ variants are becoming apparent; NGS is an ideal method to detect these rare variants. Additionally, the impact of noncoding portions of the genome as highlighted by the Encode project [26] will become more relevant for disease phenotypes. Methods such as whole genome sequencing are ideal for identifying noncoding variants. As the whole genome sequencing cost decreases and bioinformatics methods improve, we will begin to see the increased integration of this method into clinical settings. However, just as exome sequencing has specific limitations and challenges, whole genome sequencing will come with its own set of limitations, such as how to deal with repetitive DNA elements, telomere ends or centromeres. A decade from now, we may be discussing the need to incorporate multiple approaches to adequately examine a disease, such as the genome, transcriptome, epigenome and proteome methodologies. Additionally, for PGx, we may need to address issues of tissue-specific gene regulation or mosaicism. In summary, there is still plenty of work that needs to be done in order to comprehensively address PGx in the clinical setting.
Supplementary Material
Executive summary.
Background
Current assays to interrogate PGx relevant variants are powerful tools to make informed therapeutic decisions.
Microarray assays may miss some novel variants with potential clinical relevance.
Exome sequencing is a powerful tool that is being integrated into clinical and therapeutic use. Many known PGx-associated variants are not specifically included in the exome sequence target design, primarily due to their location in the noncoding portions of the genome.
Results
Some PGx variants do not get adequately covered to make informed variant calls.
~14% of important PGx associated variant positions are poorly covered by exome testing. The VKORC1 warfarin variant (rs9923231) is not captured with current exome capture methods.
Acknowledgments
The work was supported in part by the NIH-NCI Cancer Center Core grant (P30CA56036 to PF) and by TJU Institutional funds (ER).
No writing assistance was utilized in the production of this manuscript.
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Ethical conduct
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
References
Papers of special note have been highlighted as:
of interest
- 1.Pharmg kb. The Pharmacogenomics Knowledgebase. 2014 www.pharmgkb.org.
- 2.Carlson CS, Reider MJ, Nickerson DA, Eberle MA, Kruglyak L. Comment on ‘Discrepancies in dbSNP confirmations rates and allele frequency distributions from varying genotyping error rates and patterns’. Bioinformatics. 2005;21(2):141–143. doi: 10.1093/bioinformatics/bth492. [DOI] [PubMed] [Google Scholar]
- 3.Daly AK, King BP. Pharmacogenetics of oral anticoagulants. Pharmacogenetics. 2003;13(5):247–252. doi: 10.1097/00008571-200305000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Amplichip. CYP450 Test. 2014 http://molecular.roche.com/assays/Pages/AmpliChipCYP450Test.aspx. [PubMed]
- 5.Affymetrix. DMET Plus Solution. 2014 www.affymetrix.com/estore/browse/level_three_category_and_children.jsp?category=35791&categoryIdClicked=35791&expand=true&parent=35923.
- 6.Rumble SM, Lacroute P, Dalca AV, Fiume M, Sidow A, Brudno M. SHRiMP: accurate mapping of short color-space reads. PLoS Comput Biol. 2009;5(5):e1000386. doi: 10.1371/journal.pcbi.1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet journal. 2011;17:10–12. [Google Scholar]
- 8.Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9•.Burmester JK, Sedova M, Shapero MH, Mansfield E. DMET microarray technology for pharmacogenomics-based personalized medicine. Methods Mol Biol. 2010;632:99–124. doi: 10.1007/978-1-60761-663-4_7. Paper describing the DMET platform and its use for pharmacogenomics. [DOI] [PubMed] [Google Scholar]
- 10.Deeken JF, Cormier T, Price DK, et al. A pharmacogenetic study of docetaxel and thalidomide in patients with castration-resistant prostate cancer using the DMET genotyping platform. Pharmacogenomics J. 2010;10(3):191–199. doi: 10.1038/tpj.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sissung TM, English BC, Venzon D, Figg WD, Deeken JF. Clinical pharmacology and pharmacogenetics in a genomics era: the DMET platform. Pharmacogenomics. 2010;11(1):89–103. doi: 10.2217/pgs.09.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson JA, Gong L, Whirl-Carrillo M, et al. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin Pharmacol Ther. 2011;90(4):625–629. doi: 10.1038/clpt.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13•.Asan, Xu Y, Jiang H, et al. Comprehensive comparison of three commercial human whole-exome capture platforms. Genome Biol. 2011;12(9):R95. doi: 10.1186/gb-2011-12-9-r95. Paper compares the performance of different exome capture kits. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meynert AM, Bicknell LS, Hurles ME, Jackson AP, Taylor MS. Quantifying single nucleotide variant detection sensitivity in exome sequencing. BMC Bioinformatics. 2013;14:195. doi: 10.1186/1471-2105-14-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15•.Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med. 2013;369(16):1502–1511. doi: 10.1056/NEJMoa1306555. Describes the use of exome sequencing for clinical applications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.NHLBI. Exome Sequencing Project (ESP) 2014 http://evs.gs.washington.edu/EVS/
- 17.De Gregori M, Garbin G, De Gregori S, et al. Genetic variability at COMT but not at OPRM1 and UGT2B7 loci modulates morphine analgesic response in acute postoperative pain. Eur J Clin Pharmacol. 2013;69(9):1651–1658. doi: 10.1007/s00228-013-1523-7. [DOI] [PubMed] [Google Scholar]
- 18.Agundez JA, Garcia-Martin E, Alonso-Navarro H, Jimenez-Jimenez FJ. Anti-Parkinson’s disease drugs and pharmacogenetic considerations. Expert Opin Drug Metab Toxicol. 2013;9(7):859–874. doi: 10.1517/17425255.2013.789018. [DOI] [PubMed] [Google Scholar]
- 19.Jung JA, Lim HS. Association between CYP2D6 genotypes and the clinical outcomes of adjuvant tamoxifen for breast cancer: a meta-analysis. Pharmacogenomics. 2014;15(1):49–60. doi: 10.2217/pgs.13.221. [DOI] [PubMed] [Google Scholar]
- 20.Martinez De Duenas E, Ochoa Aranda E, Blancas Lopez-Barajas I, et al. Adjusting the dose of tamoxifen in patients with early breast cancer and CYP2D6 poor metabolizer phenotype. Breast. 2014;23(4):400–406. doi: 10.1016/j.breast.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 21.Langaee TY, Zhu HJ, Wang X, et al. The influence of the CYP2C19*10 allele on clopidogrel activation and CYP2C19*2 genotyping. Pharmacogenet Genomics. 2014;24(8):381–386. doi: 10.1097/FPC.0000000000000068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tatarunas V, Jankauskiene L, Kupstyte N, et al. The role of clinical parameters and of CYP2C19 G681 and CYP4F2 G1347A polymorphisms on platelet reactivity during dual antiplatelet therapy. Blood Coagul Fibrinolysis. 2014;25(4):369–374. doi: 10.1097/MBC.0000000000000053. [DOI] [PubMed] [Google Scholar]
- 23.Canestaro WJ, Austin MA, Thummel KE. Genetic factors affecting statin concentrations and subsequent myopathy: a HuGENet systematic review. Genet Med. 2014;16(11):810–819. doi: 10.1038/gim.2014.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Keyser CE, Peters BJ, Becker ML, et al. The SLCO1B1 c.521T>C polymorphism is associated with dose decrease or switching during statin therapy in the Rotterdam Study. Pharmacogenet Genomics. 2014;24(1):43–51. doi: 10.1097/FPC.0000000000000018. [DOI] [PubMed] [Google Scholar]
- 25.Rasmussen-Torvik LJ, Stallings SC, Gordon AS, et al. Design and anticipated outcomes of the eMERGE-PGx project: a multicenter pilot for preemptive pharmacogenomics in electronic health record systems. Clin Pharmacol Ther. 2014;96(4):482–489. doi: 10.1038/clpt.2014.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26•.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. The encode project paper describing the noncoding portions of the genome. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.