

Abstract
The aim of the present study was to identify differentially expressed (DE) genes in patients with osteoarthritis (OA), and biological processes associated with changes in gene expression that occur in this disease. Using the INMEX (integrative meta-analysis of expression data) software tool, a meta-analysis of publicly available microarray Gene Expression Omnibus (GEO) datasets of OA was performed. Gene ontology (GO) enrichment analysis was performed in order to detect enriched functional attributes based on gene-associated GO terms. Three GEO datasets, containing 137 patients with OA and 52 healthy controls, were included in the meta-analysis. The analysis identified 85 genes that were consistently differentially expressed in OA (30 genes were upregulated and 55 genes were downregulated). The upregulated gene with the lowest P-value (P=5.36E-07) was S-phase kinase-associated protein 2, E3 ubiquitin protein ligase (SKP2). The downregulated gene with the lowest P-value (P=4.42E-09) was Proline rich 5 like (PRR5L). Among the 210 GO terms that were associated with the set of DE genes, the most significant two enrichments were observed in the GO categories of 'Immune response', with a P-value of 0.000129438, and 'Immune effectors process', with a P-value of 0.000288619. The current meta-analysis identified genes that were consistently DE in OA, in addition to biological pathways associated with changes in gene expression that occur during OA, which may provide insight into the molecular mechanisms underlying the pathogenesis of this disease.
Keywords: osteoarthritis, meta-analysis, differentially expressed genes
Introduction
Osteoarthritis (OA) is the most prevalent joint disease and is characterized by an abnormal remodeling of joint tissues, which is predominantly driven by inflammatory mediators within the affected joint (1,2). The pathological changes of OA primarily take place in the articular cartilage, and include cartilage degeneration, matrix degradation and synovial inflammation (3–5). Clinically, features of OA include pain, stiffness, limitation of motion, swelling and deformity (6). Synovial inflammation is hypothesized to be the primary underlying etiology in OA (3). However, the biological mechanisms associated with OA remain to be elucidated.
Microarray, a high-throughput genomics technology, has been developed in order to improve the understanding of complex interactions and networks in disease development (7). Thousands of genes on a genome-wide scale have been measured using microarray technology (8). The successful identification of gene expression signatures that may provide insights into OA pathogenesis and differentiate the diseased state from a healthy state, requires an adequate sample size and heterogeneous datasets (9). Although numerous microarray studies have generated lists of differentially expressed (DE) genes, there are inconsistencies among the results of such studies, due to the limitation of the small sample sizes involved (10).
To overcome these difficulties, meta-analysis has previously been applied to publicly-available genome-wide expression datasets from studies on a number of diseases (11–13). The use of meta-analysis may improve reliability and generalizability, and permit a more precise estimation of gene expression (11). Meta-analyses provide enhanced statistical power, thereby obtaining more robust and reliable gene signatures (7,14–17). Recently, integrative meta-analysis of expression data (INMEX), a new user-friendly microarray meta-analysis tool, has been developed to support meta-analysis of multiple gene expression datasets (18).
In order to overcome the limitations of individual studies, resolve inconsistencies in results, and reduce false-positive or false-negative associations due to random errors, a microarray meta-analysis was performed in the present study. The objective was to identify differentially expressed (DE) genes and biological processes associated with gene expression signature in OA.
Materials and methods
Identification of eligible gene expression datasets of OA
A search of microarray datasets was performed that examined DE genes between OA and healthy controls. The NCBI Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) (19) was used to identify suitable microarray datasets. The keyword 'osteoarthritis' was used for this search. Studies were included in the analysis if they: Were based on gene expression profiling of blood or synovial membrane samples; contained sufficient data to perform a meta-analysis; and included patients diagnosed with OA, based on OA classification criteria (20). The following information was extracted from each of the studies that were selected: GEO accession; sample type; platform; numbers of patients and healthy controls; and gene expression data.
Meta-analysis of microarray datasets
All available OA microarray datasets that met the inclusion criteria were downloaded from the NCBI GEO database. Data tables containing gene expression or relative gene expression values were constructed, with genes/probes in the rows, and samples in the columns. The datasets were uploaded to INMEX (http://www.inmex.ca/INMEX) (18), and the data was subsequently annotated by converting different gene or probe ID to Entrez IDs. For each probe-set, intensity values were log-transformed and/or normalized to zero mean and unit variance, which is the normalization method for high density oligonucleotide array data, as reported by Bolstad et al (21). When all datasets had been uploaded, processed and annotated, a data integrity check was performed prior to the meta-analysis stage.
The random effects model presumes that different studies present substantial diversity, and evaluates between study variance as well as within study sampling error (22,23). The random effects model is used when the between-study heterogeneity is significant (23). The INMEX program was used to conduct statistical analysis (18).
Functional analysis
The functional analysis of INMEX generates new hypotheses by exploiting characteristics of the DE gene lists identified in meta-analysis. A heat map created by 'Pattern extractor' produced gene expression profiles across different datasets/conditions.
In order to examine the functions of the genes in the gene list, gene ontology (GO) enrichment analysis was performed, which detected enriched functional attributes based on gene-associated GO terms, using the hypergeometric test (http://www.geneontology.org/) (24). Functional analysis was performed using the INMEX program (18).
Results
Studies included in the meta-analysis
Three GEO data sets, which met the inclusion criteria, were identified (Table I) (4,25,26). These datasets consisted of two synovial membrane datasets and one blood dataset, and included a total of 137 patients with OA and 52 controls. Selected details of the individual studies are summarized in Table I.
Table I.
Characteristics of the individual studies included in the meta-analysis.
| Study (ref) | GEO accession | Patient number
|
Sample | Platform | |
|---|---|---|---|---|---|
| OA | Control | ||||
| 1 (25) | GSE48556 | 106 | 33 | Blood | Illumina HumanHT-12 V3.0 Expression Beadchip |
| 2 (26) | GSE46750 | 12 | 12 | Synovial membrane | Illumina HumanHT-12 V4.0 Expression Beadchip |
| 3 (4) | GSE32317 | 19 | 7 | Synovial membrane | Affymetrix Human Genome U133 Plus 2.0 Array |
GEO, Gene Expression Omnibus; OA, osteoarthritis; ref, reference.
Meta-analysis of gene expression in OA
A random effects model of effect size (ES) measures was used to integrate gene expression patterns. The present study incorporated between-study heterogeneities across studies, because the estimated Q-value was not in an approximately chi-squared distribution. DE genes with P<0.05 were selected. In the current analysis, 1 'gained' gene and 13402 'lost' genes were identified (Fig. 1). Gained genes are DE genes that were only identified in the meta-analysis (26). The single gained gene exhibited relatively weak but consistent expression profiles across the three different datasets. The large sample size obtained by consisting of the datasets made it possible declare this a DE gene with increased certainty. Lost genes are genes which were identified as DE genes in any of the individual analyses, but not in the meta-analysis. These genes also presented conflicting changes in expression profiles, or large variations across the different studies.
Figure 1.
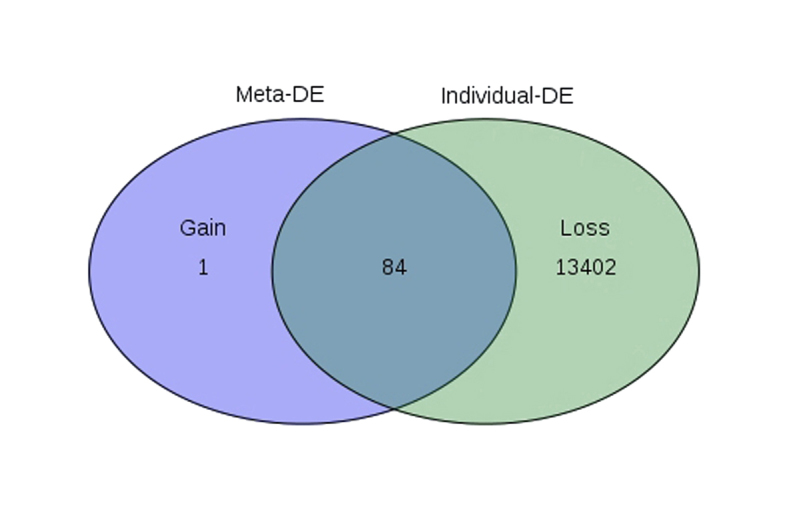
Venn diagram showing the overlap between DE genes identified from the meta-analysis (Meta-DE) and those combined from the individual data analyses (individual-DE). DE, differentially expressed.
Identification of differentially expressed genes in OA
A total of 85 genes were identified, which were consistently differentially expressed in OA. Among these 85 DE genes, 30 were upregulated and 55 were downregulated. A list of the top 20 upregulated and downregulated genes is shown in Table II The upregulated gene with the lowest P-value (P=5.36E-07) was S-phase kinase-associated protein 2, E3 ubiquitin protein ligase (SKP2). The downregulated gene with the lowest P-value (P=4.42E-09) was Proline rich 5 like (PRR5L).
Table II.
Top 20 upregulated and downregulated genes in patients with OA.
| A, Top 20 upregulated genes
| ||||
|---|---|---|---|---|
| Entrez ID | Gene symbol | Combined ES | P-value | Gene name |
| 6502 | SKP2 | −1.1447 | 5.36E-07 | S-phase kinase-associated protein 2, E3 ubiquitin protein ligase |
| 23299 | BICD2 | −1.0632 | 4.24E-06 | Bicaudal D homolog 2 (Drosophila) |
| 8445 | DYRK2 | −0.8747 | 0.000592 | Dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 2 |
| 10116 | FEM1B | −0.9170 | 0.000842 | Fem-1 homolog b (C. elegans) |
| 87 | ACTN1 | −0.8544 | 0.000842 | Actinin, α 1 |
| 147906 | DACT3 | −0.8450 | 0.000867 | Dishevelled-binding antagonist of β-catenin 3 |
| 6627 | SNRPA1 | −0.7981 | 0.002369 | Small nuclear ribonucleoprotein polypeptide A' |
| 84458 | LCOR | −0.7596 | 0.004996 | Ligand dependent nuclear receptor corepressor |
| 55670 | PEX26 | −0.7396 | 0.007656 | Peroxisomal biogenesis factor 26 |
| 284273 | ZADH2 | −0.7241 | 0.009756 | Zinc binding alcohol dehydrogenase domain containing 2 |
| 2983 | GUCY1B3 | −0.7239 | 0.009756 | Guanylate cyclase 1, soluble, β 3 |
| 90550 | MCU | −0.7115 | 0.012319 | Mitochondrial calcium uniporter |
| 359845 | FAM101B | −0.7042 | 0.014203 | Family with sequence similarity 101, member B |
| 158381 | ATP8B5P | −0.7025 | 0.014203 | ATPase, classI, type 8B, member 5, pseudogene |
| 92014 | MCART1 | −0.7010 | 0.014203 | Mitochondrial carrier triple repeat 1 |
| 57456 | KIAA1143 | −0.6989 | 0.014452 | KIAA1143 |
| 51002 | TPRKB | −0.6918 | 0.015972 | TP53RK binding protein |
| 84953 | MICALCL | −0.9119 | 0.015993 | MICAL C-terminal like |
| 80071 | CCDC15 | −0.7894 | 0.018113 | Coiled-coil domain containing 15 |
| 160 | DAB2 | −1.0979 | 0.049846 | Dab, mitogen-responsive phosphoprotein, homolog 2 (Drosophila) |
| B, Top 20 downregulated genes
| ||||
|---|---|---|---|---|
| Entrez ID | Gene symbol | Combined ES | P-value | Gene name |
| 79899 | PRR5L | 1.3235 | 4.42E-09 | Proline rich 5 like |
| 5583 | PRKCH | 1.2343 | 5.08E-08 | Protein kinase C, η |
| 3683 | ITGAL | 1.2126 | 6.62E-08 | Integrin, α L |
| 5051 | PAFAH2 | 1.0668 | 4.24E-06 | Platelet-activating factor acetylhydrolase 2, 40kDa |
| 24144 | TFIP11 | 0.9820 | 4.31E-05 | Tuftelin interacting protein 11 |
| 9595 | CYTIP | 0.9816 | 4.31E-05 | Cytohesin 1 interacting protein |
| 157567 | ANKRD46 | 1.0119 | 7.59E-05 | Ankyrin repeat domain 46 |
| 23294 | ANKS1A | 0.9547 | 7.60E-05 | Ankyrin repeat and sterile α motif domain containing 1A |
| 55272 | IMP3 | 1.2776 | 0.001032 | IMP3, U3 small nucleolar ribonucleoprotein |
| 80213 | TM2D3 | 1.0182 | 0.001425 | TM2 domain containing 3 |
| 474344 | GIMAP6 | 1.1551 | 0.004506 | GTPase, IMAP family member 6 |
| 55303 | GIMAP4 | 1.2787 | 0.008432 | GTPase, IMAP family member 4 |
| 10866 | HCP5 | 1.1647 | 0.001232 | HLA complex P5 (non-protein coding) |
| 54499 | TMCO1 | 1.0355 | 0.014452 | Transmembrane and coiled-coil domains 1 |
| 951 | CD37 | 1.0423 | 0.023921 | CD37 molecule |
| 50650 | ARHGEF3 | 1.0568 | 0.025483 | ρ guanine nucleotide exchange factor (GEF) 3 |
| 56833 | SLAMF8 | 1.0983 | 0.033534 | SLAM family member 8 |
| 65992 | DDRGK1 | 1.4283 | 0.033711 | DDRGK domain containing 1 |
| 112858 | TP53RK | 1.7509 | 0.036334 | TP53 regulating kinase |
| 4236 | MFAP1 | 1.2892 | 0.047164 | Microfibrillar-associated protein 1 |
ES, effect size; OA, osteoarthritis.
Identification of differentially expressed genes in the synovial membrane of patients with OA
An additional meta-analysis was performed on the results from the 2 synovial membrane samples, following exclusion of the third study, which used peripheral blood. A list of the top 20 upregulated and downregulated genes is shown in Table III. The upregulated genes with the lowest P-values (both P=0.004003) were JAZF zinc finger 1 (JAZF1) and Guanine nucleotide binding protein (G-protein), β polypeptide 4 (GNB4), which are involved in coupling membrane receptors to effector proteins, such as ion channels and enzymes (27). The downregulated gene with the largest ES (ES=2.0472; P=0.004003) was multiple inositol-polyphosphate phosphatase 1 (MINPP1). A number of the downregulated genes were related to inflammatory factors (Table III).
Table III.
Top 10 upregulated and downregulated genes in the synovial membrane of patients with OA.
| A, Upregulated genes
| ||||
|---|---|---|---|---|
| Entrez ID | Gene symbol | Combined ES | P-value | Gene name |
| 221895 | JAZF1 | −1.6865 | 0.004003 | JAZF zinc finger 1 |
| 59345 | GNB4 | −1.6774 | 0.004003 | Guanine nucleotide binding protein (G protein), β polypeptide 4 |
| 3070 | HELLS | −1.6193 | 0.005903 | Helicase, lymphoid-specific |
| 9749 | PHACTR2 | −1.5892 | 0.005903 | Phosphatase and actin regulator 2 |
| 9645 | MICAL2 | −1.5726 | 0.006072 | Microtubule associated monooxygenase, calponin and LIM domain containing 2 |
| 10974 | C10orf116 | −1.5715 | 0.006072 | Adipogenesis regulatory factor |
| 283310 | OTOGL | −1.5624 | 0.006072 | Otogelin-like |
| 26230 | TIAM2 | −1.5531 | 0.006563 | T-cell lymphoma invasion and metastasis 2 |
| 51194 | IPO11 | −1.5378 | 0.006822 | Importin 11 |
| 1404 | HAPLN1 | −1.5176 | 0.007348 | Hyaluronan and proteoglycan link protein 1 |
| B, Downregulated genes
| ||||
|---|---|---|---|---|
| Entrez ID | Gene symbol | Combined ES | P-value | Gene name |
| 27242 | TNFRF21 | 1.8378 | 0.003981 | Tumor necrosis factor receptor superfamily, member 21 |
| 84225 | ZMYND15 | 1.8026 | 0.003981 | Zinc finger, MYND-type containing 15 |
| 9562 | MINPP1 | 2.0472 | 0.004003 | Multiple inositol-polyphosphate phosphatase 1 |
| 3600 | IL15 | 1.9554 | 0.004003 | Interleukin 15 |
| 3487 | IGFBP4 | 1.7005 | 0.004003 | Insulin-like growth factor binding protein 4 |
| 54504 | CPVL | 1.6845 | 0.004003 | Carboxypeptidase, vitellogenic-like |
| 9450 | LY86 | 1.6796 | 0.004003 | Lymphocyte antigen 86 |
| 147798 | TMC4 | 1.6305 | 0.004882 | Transmembrane channel-like 4 |
| 834 | CASP1 | 1.5939 | 0.005903 | Caspase 1, apoptosis-related cysteine peptidase |
| 83401 | ELOVL3 | 1.8314 | 0.006822 | ELOVL fatty acid elongase 3 |
ES, effect size; OA, osteoarthritis.
Functional analysis
GO analysis of the DE genes was performed in order to identify the biological processes associated with changes in gene expression in OA. The analysis identified 210 significant enrichments of the DE genes, which were categorized to 10 GO terms (Fig. 2). The two enrichments with the lowest P-values were in the GO category of 'Immune response', with a P-value of 0.000129438, and 'Immune effector process', with a P-value of 0.000288619. Other significant GO categories included 'Regulation of humoral immune response' (P=0.000308832), 'Regulation of immune response' (P=0.00055514) and 'Positive regulation of immune system process' (P=0.00059351; Table IV).
Figure 2.
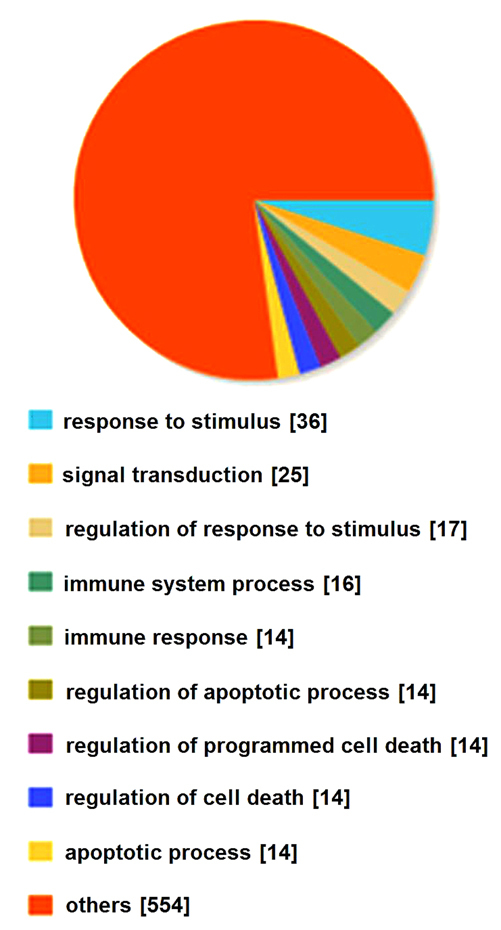
Summary of the enriched GO terms for the list of DE genes in patients with OA compared to controls. GO, gene ontology; DE, differentially expressed; OA, osteoarthritis.
Table IV.
Top 10 enriched GO terms among the DE genes in patients with OA compared with controls.
| GO ID | Term | P-value | Genes |
|---|---|---|---|
| GO:0006955 | Immune response | 0.000129438 | ITGAL, GIMAP5, INPP5D, LAX1, GZMA, IRF1, CST7, SKAP1, CD55, TNFRSF4, CD37, CX3CR1, HLA-C, CXCL3 |
| GO:0002252 | Immune effector process | 0.000288619 | ITGAL, GIMAP5, INPP5D, IRF1, CD55, TNFRSF4, CD37, CX3CR1 |
| GO:0002920 | Regulation of humoral immune response | 0.000308832 | GIMAP5, CD55, CD37 |
| GO:0042981 | Regulation of apoptotic process | 0.000510947 | PRKCH, SKP2, PAFAH2, TMBIM6, GIMAP5; DYRK2, ACTN1, FEM1B, INPP5D, GZMA, IRF1, TNFRSF4, CX3CR1, ARHGEF3 |
| GO:0051250 | Negative regulation of lymphocyte activation | 0.000517654 | GIMAP5, INPP5D, LAX1, IRF1 |
| GO:0045589 | Regulation of regulatory T cell differentiation | 0.000531439 | GIMAP5, IRF1 |
| GO:0043067 | Regulation of programmed cell death | 0.000551712 | PRKCH, SKP2, PAFAH2, TMBIM6, GIMAP5, DYRK2, ACTN1, FEM1B, INPP5D, GZMA, IRF1, TNFRSF4, CX3CR1, ARHGEF3 |
| GO:0050776 | Regulation of immune response | 0.000555143 | PRKCH, ITGAL, GIMAP5, INPP5D, IRF1, SKAP1, CD55, CD37, HLA-C |
| GO:0002684 | Positive regulation of immune system process | 0.000593511 | PRKCH, ITGAL, GIMAP5, INPP5D, IRF1, SKAP1, CD55, TNFRSF4, CD37 |
| GO:0010941 | Regulation of cell death | 0.000708372 | PRKCH, SKP2, PAFAH2, TMBIM6, GIMAP5, DYRK2, ACTN1, FEM1B, INPP5D, GZMA, IRF1, TNFRSF4, CX3CR1, ARHGEF3 |
GO, gene ontology; DE, differentially expressed; OA, osteoarthritis; ITGAL, Integrin, α L; GIMAP5, GTPase, IMAP family member 5; INPP5D, inositol polyphosphate-5-phosphatase; LAX1, lymphocyte transmembrane adaptor 1; GZMA, granzyme A; IRF1, interferon regulatory factor 1; SKAP1, src kinase associated phosphoprotein 1; TNFRSF4, tumor necrosis receptor superfamily, member 4; CX3CR1, CX3C chemokine receptor 1; HLA-C, humna leukocyte antigen C; CXCL3, chemokine (C-X-C motif) ligand 3; PRKCH, protein kinase C, η; SKP2, S-phase kinase-associated protein 2, E3 ubiquitin protein ligase; PAFAH2, platelet-activating factor acetylhydrolase 2, 40kDa; TMBIM6, transmembrane BAX inhibitor motif containing 6; DYRK2, Dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 2; ACTN1, Actinin, α 1; FEM1B, Fem-1 homolog b (C. elegans); ARHGEF3, ρ guanine nucleotide exchange factor (GEF) 3.
Discussion
A number of genes are differentially expressed genes between patients with OA and healthy controls, and it is necessary to identify the genes that may enhance understanding of the molecular and cellular processes, which are involved in the pathogenesis of OA. Although a large quantity of data may be produced using microarray studies, the small sample size of these studies is a significant obstacle to the identification of DE genes. A meta-analysis of multiple microarray datasets increases the sample size, rendering the identification of DE genes more reliable.
In the present study, a meta-analysis was performed using three publicly available GEO datasets in order to identify common biological mechanisms involved in the pathogenesis of OA. The analysis identified 85 genes that were consistently differentially expressed in OA (30 upregulated and 55 downregulated). The upregulated gene with the largest ES was SKP2, which is known to be involved in the inhibition of cell growth and the promotion of apoptosis. Kitagawa concluded that SKP2 controls the p300–p53 signaling pathway in cancer cells (28). Furthermore, this gene encodes a member of the F-box protein family, which is characterized by a ~40 amino acid motif, the F-box (29). The downregulated gene with the lowest P-value was PRR5L, which suppresses mTOR complex 2 (mTORC2)-mediated hydrophobic motif phosphorylation of protein kinase C, but not that of protein kinase B (30). In addition, the PRR5L protein may function to modulate the activity of mTORC2 in a substrate-dependent manner (30). Actinin α 1 (ACTN1), an upregulated gene, encodes an actin-binding protein, which exerts multiple effects in a variety of cell types. ACTN1 may protect osteoclasts from tumor necrosis factor-α (TNF-α); induce apoptosis through increasing the expression of the anti-apoptotic protein, Bcl-2; activate survival signals; and promote Akt phosphorylation and NF-κB activation (31). Although it is currently unclear exactly how these genes contribute to OA, they may be useful as potential biomarkers to facilitate early diagnosis or to monitor the efficacy of treatment in this disease. A number of these genes provide insights into the molecular mechanisms underlying the pathophysiology of OA.
Although osteoarthritis (OA) is understood to be a degradative articular cartilage disease, there is increasing data demonstrating the involvement of the immune system. In recent epidemiological studies involving a large number of patients with OA, an inflammatory synovium has been shown to be involved in increased damage to the cartilage (32) and pain (33). Immune cells, such as T cells, B cells and macrophages, have been identified in the synovial tissue of patients with OA (34–36). Furthermore, immunoglobulins and immune complexes against cartilage components have been detected in the plasma, synovium and cartilage of patients with OA (37), and it has been shown that the synovium is involved in complement activation in OA (38). In the present study, 210 significantly enriched GO terms associated with the DE genes were identified using a meta-analysis. The three enriched terms with the lowest P-values were 'Immune response', 'Immune effector process' and 'Regulation of humoral immune response', which were all involved in the immune system. The identified GO terms may be grouped into a smaller number of categories: 'Response to stimulus', 'Signal transduction', 'Regulation of response to stimulus', 'Immune system process', 'Immune response', 'Regulation of apoptotic process', 'Regulation of programmed cell death', 'Regulation of cell death', 'Apoptotic process' and others. Although it is difficult to identify all the significant functional categories that are expressed differentially in OA, the GO categories identified here, merit further investigation in subsequent studies.
There were certain limitations to the present study, which ought to be considered. Firstly, heterogeneity and confounding factors may have distorted the analysis. Clinical samples may have been heterogeneous with respect to clinical activity, severity or gender. Secondly, there are differences in gene expression between tissues, such as blood and synovial membrane, that were not considered. Although an additional subgroup analysis of the synovial membrane samples was performed, this only included two studies. By contrast, the initial meta-analysis integrated the results obtained from different tissues, which should have enabled detection of the genes that may have been missed in an analysis of two studies only.
In conclusion, the meta-analysis of microarray studies that was performed in the present study, provided an overview of differential gene expression in OA; identifying 85 differential expressed genes (30 upregulated and 55 downregulated genes). Future studies to validate these genes as markers for the diagnosis and response to biological therapy for OA may provide further insight into their involvement in the development and progression of OA.
Acknowledgments
This study was supported by the Key Scientific and Technological Innovation Special Projects of Shaanxi '13115' of China (grant no. 2009ZDKG-79) and the National Natural Science Foundation of China (grant no. 81472924).
References
- 1.Aigner T, Rose J, Martin J, Buckwalter J. Aging theories of primary osteoarthritis: From epidemiology to molecular biology. Rejuvenation Res. 2004;7:134–145. doi: 10.1089/1549168041552964. [DOI] [PubMed] [Google Scholar]
- 2.Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012;64:1697–1707. doi: 10.1002/art.34453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benito MJ, Veale DJ, FitzGerald O, van den Berg WB, Bresnihan B. Synovial tissue inflammation in early and late osteoarthritis. Ann Rheum Dis. 2005;64:1263–1267. doi: 10.1136/ard.2004.025270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma CH, Lv Q, Cao Y, et al. Genes relevant with osteoarthritis by comparison gene expression profiles of synovial membrane of osteoarthritis patients at different stages. Eur Rev Med Pharmacol Sci. 2014;18:431–439. [PubMed] [Google Scholar]
- 5.Duan C, Guo X, Zhang XD, et al. Comparative analysis of gene expression profiles between primary knee osteoarthritis and an osteoarthritis endemic to Northwestern China, Kashin-Beck disease. Arthritis Rheum. 2010;62:771–780. doi: 10.1002/art.27282. [DOI] [PubMed] [Google Scholar]
- 6.Sellam J, Berenbaum F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat Rev Rheumatol. 2010;6:625–635. doi: 10.1038/nrrheum.2010.159. [DOI] [PubMed] [Google Scholar]
- 7.Choi SJ, Rho YH, Ji JD, Song GG, Lee YH. Genome scan meta-analysis of rheumatoid arthritis. Rheumatology (Oxford) 2006;45:166–170. doi: 10.1093/rheumatology/kei128. [DOI] [PubMed] [Google Scholar]
- 8.Golub TR, Slonim DK, Tamayo P, et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 9.Ramasamy A, Mondry A, Holmes CC, Altman DG. Key issues in conducting a meta-analysis of gene expression microarray datasets. PLoS Med. 2008;5:e184. doi: 10.1371/journal.pmed.0050184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siddiqui AS, Delaney AD, Schnerch A, Griffith OL, Jones SJ, Marra MA. Sequence biases in large scale gene expression profiling data. Nucleic Acids Res. 2006;34:e83. doi: 10.1093/nar/gkl404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griffith OL, Melck A, Jones SJ, Wiseman SM. Meta-analysis and meta-review of thyroid cancer gene expression profiling studies identifies important diagnostic biomarkers. J Clin Oncol. 2006;24:5043–5051. doi: 10.1200/JCO.2006.06.7330. [DOI] [PubMed] [Google Scholar]
- 12.Rung J, Brazma A. Reuse of public genome-wide gene expression data. Nat Rev Genet. 2013;14:89–99. doi: 10.1038/nrg3394. [DOI] [PubMed] [Google Scholar]
- 13.Cahan P, Rovegno F, Mooney D, Newman JC, St Laurent G, III, McCaffrey TA. Meta-analysis of microarray results: Challenges, opportunities, and recommendations for standardization. Gene. 2007;401:12–18. doi: 10.1016/j.gene.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee YH, Nath SK. Systemic lupus erythematosus susceptibility loci defined by genome scan meta-analysis. Hum Genet. 2005;118:434–443. doi: 10.1007/s00439-005-0073-1. [DOI] [PubMed] [Google Scholar]
- 15.Lee YH, Witte T, Momot T, et al. The mannose-binding lectin gene polymorphisms and systemic lupus erythematosus: Two case-control studies and a meta-analysis. Arthritis Rheum. 2005;52:3966–3974. doi: 10.1002/art.21484. [DOI] [PubMed] [Google Scholar]
- 16.Lee YH, Rho YH, Choi SJ, Ji JD, Song GG. Ankylosing spondylitis susceptibility loci defined by genome-search meta-analysis. J Hum Genet. 2005;50:453–459. doi: 10.1007/s10038-005-0277-1. [DOI] [PubMed] [Google Scholar]
- 17.Lee YH, Rho YH, Choi SJ, Ji JD, Song GG. Meta-analysis of genome-wide linkage studies for bone mineral density. J Hum Genet. 2006;51:480–486. doi: 10.1007/s10038-006-0390-9. [DOI] [PubMed] [Google Scholar]
- 18.Xia J, Fjell CD, Mayer ML, Pena OM, Wishart DS, Hancock RE. INMEX - a web-based tool for integrative meta-analysis of expression data. Nucleic Acids Res. 2013;41:W63–W70. doi: 10.1093/nar/gkt338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrett T, Troup DB, Wilhite SE, et al. NCBI GEO: Archive for functional genomics data sets - 10 years on. Nucleic Acids Res. 2011;39:D1005–D1010. doi: 10.1093/nar/gkq1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altman R, Asch E, Bloch D, et al. Development of criteria for the classification and reporting of osteoarthritis. Classification of osteoarthritis of the knee. Diagnostic and Therapeutic Criteria Committee of the American Rheumatism Association. Arthritis Rheum. 1986;29:1039–1049. doi: 10.1002/art.1780290816. [DOI] [PubMed] [Google Scholar]
- 21.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 22.Choi JK, Yu U, Kim S, Yoo OJ. Combining multiple microarray studies and modeling interstudy variation. Bioinformatics. 2003;19(Suppl 1):i84–i90. doi: 10.1093/bioinformatics/btg1010. [DOI] [PubMed] [Google Scholar]
- 23.DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986;7:177–188. doi: 10.1016/0197-2456(86)90046-2. [DOI] [PubMed] [Google Scholar]
- 24.Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23:257–258. doi: 10.1093/bioinformatics/btl567. [DOI] [PubMed] [Google Scholar]
- 25.Ramos YF, Bos SD, Lakenberg N, et al. Genes expressed in blood link osteoarthritis with apoptotic pathways. Ann Rheum Dis. 2013;73:1844–1853. doi: 10.1136/annrheumdis-2013-203405. [DOI] [PubMed] [Google Scholar]
- 26.Lambert C, Dubuc JE, Montell E, et al. Gene expression pattern of cells from inflamed and normal areas of osteoarthritis synovial membrane. Arthritis Rheumatol. 2014;66:960–968. doi: 10.1002/art.38315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruiz-Velasco V, Ikeda SR, Puhl HL. Cloning, tissue distribution, and functional expression of the human G protein beta 4-subunit. Physiol Genomics. 2002;8:41–50. doi: 10.1152/physiolgenomics.00085.2001. [DOI] [PubMed] [Google Scholar]
- 28.Kitagawa M, Lee SH, McCormick F. Skp2 suppresses p53-dependent apoptosis by inhibiting p300. Mol Cell. 2008;29:217–231. doi: 10.1016/j.molcel.2007.11.036. [DOI] [PubMed] [Google Scholar]
- 29.Bai C, Sen P, Hofmann K, et al. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996;86:263–274. doi: 10.1016/S0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- 30.Gan X, Wang J, Wang C, et al. PRR5L degradation promotes mTORC2-mediated PKC-δ phosphorylation and cell migration downstream of Gα12. Nat Cell Biol. 2012;14:686–696. doi: 10.1038/ncb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Triplett JW, Pavalko FM. Disruption of alpha-actinin-integrin interactions at focal adhesions renders osteoblasts susceptible to apoptosis. Am J Physiol Cell Physiol. 2006;291:C909–C921. doi: 10.1152/ajpcell.00113.2006. [DOI] [PubMed] [Google Scholar]
- 32.Ayral X, Pickering EH, Woodworth TG, Mackillop N, Dougados M. Synovitis: A potential predictive factor of structural progression of medial tibiofemoral knee osteoarthritis - results of a 1 year longitudinal arthroscopic study in 422 patients. Osteoarthritis Cartilage. 2005;13:361–367. doi: 10.1016/j.joca.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 33.Hill CL, Hunter DJ, Niu J, et al. Synovitis detected on magnetic resonance imaging and its relation to pain and cartilage loss in knee osteoarthritis. Ann Rheum Dis. 2007;66:1599–1603. doi: 10.1136/ard.2006.067470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Revell PA, Mayston V, Lalor P, Mapp P. The synovial membrane in osteoarthritis: A histological study including the characterisation of the cellular infiltrate present in inflammatory osteoarthritis using monoclonal antibodies. Ann Rheum Dis. 1988;47:300–307. doi: 10.1136/ard.47.4.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sakkas LI, Scanzello C, Johanson N, et al. T cells and T-cell cytokine transcripts in the synovial membrane in patients with osteoarthritis. Clin Diagn Lab Immunol. 1998;5:430–437. doi: 10.1128/cdli.5.4.430-437.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakamura H, Yoshino S, Kato T, Tsuruha J, Nishioka K. T-cell mediated inflammatory pathway in osteoarthritis. Osteoarthritis Cartilage. 1999;7:401–402. doi: 10.1053/joca.1998.0224. [DOI] [PubMed] [Google Scholar]
- 37.Jasin HE. Immune mediated cartilage destruction. Scand J Rheumatol Suppl. 1988;76:111–116. doi: 10.3109/03009748809102960. [DOI] [PubMed] [Google Scholar]
- 38.Wang Q, Rozelle AL, Lepus CM, et al. Identification of a central role for complement in osteoarthritis. Nat Med. 2011;17:1674–1679. doi: 10.1038/nm.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]