

Abstract
The aim of the present study was to explore the toxic effects and underlying mechanisms of nickel ions during therapeutic nickel-based alloy-treatment in congenital heart disease by investigating the metal-induced cytotoxicity to the human monocyte-derived macrophage cell line THP-1. THP-1 cells were treated with NiCl2·6H2O (25, 50, 100, 200, 400 and 800 µM) for 24, 48 and 72 h, respectively. MTT was applied to detect THP-1 cell proliferation following NiCl2 treatment. Apoptosis of THP-1 cells was quantified using flow cytometry. Illumina sequencing was used for screening the associated genes, whose mRNA expression levels were further confirmed by quantitative real-time polymerase chain reaction. High concentrations of nickel ions had a significant suppressive effect on cell proliferation at the three concentrations investigated (200, 400 and 800 µM). Treatment with nickel ions (25–400 µM) for 48 h reduced cell viability in a dose-dependent manner. The mRNA expression levels of RELB, FIGF, SPI-1, CXCL16 and CRLF2 were significantly increased following nickel treatment. The results of the present study suggested that nickel ions exert toxic effects on THP-1 cell growth, which may indicate toxicity of the nickel ion during treatment of congenital heart disease. The identification of genes modified by the toxic effects of nickel on THP-1 cells (EPOR, RELB, FIGF, SPI-1, TGF-β1, CXCL16 and CRLF2) may aid in the development of interventional measures for the treatment/prevention of nickel ion-associated toxic effects during the treatment of congenital heart disease.
Keywords: toxicity, nickel ion, THP-1 cell, Illumina sequencing
Introduction
Congenital heart disease is the most common type of congenital malformation (1). Only certain types of congenital heart disease are able to recover naturally, and others present with increased severity and frequency of complications with increasing age (2). Interventional therapy is one of the therapeutic methods used for congenital heart disease (2). Blocking cardiac defects with metal occluders of different shapes is a commonly used surgical alternative, with the nickel-chromium (Ni-Cr) alloy occluder being most widely used in the clinical setting (3).
The clinical application of the Ni-Cr alloy occluder is a result of advances in medical technology and material sciences, and that of a process of continuous research and improvement. Use of the Ni-Cr alloy occluder avoids the risks and trauma of open-heart surgery, allowing short hospital stays and fast recovery for patients (4). However, long-term nickel-based alloy implantation has been increasingly documented to be associated with biological effects, including cytotoxicity and genotoxicity, eliciting tissue inflammation and potential sensitization, thus raising questions over its bio-safety (5). In previous experiments following-up children receiving an atrial septal defect occluder implantation, it was observed that the nickel concentration in the blood was significantly increased 24 h and one month subsequent to surgery (Zhang et al; unpublished results). Other studies have also reported that the Ni-Cr alloy occluder implantation results in the release of the nickel ion into the chambers of the heart, which may result in cell damage due to local and systemic toxicity (6) and lead to headaches, dyspnea and other unpleasant symptoms (7,8).
Being the 'first line of defense', macrophages are essential determinants of implant biocompatibility (9). Pro-inflammatory studies have observed that during the first six weeks following implantation, macrophages interact heavily with the prosthetic device (10). Therefore, the present study aimed to investigate the cytotoxicity of nickel ions on THP-1, a human monocytic cell line derived from the peripheral blood, and to explore the underlying mechanism of the toxicity of the nickel alloy in the heart during its application for congenital heart disease. The present study additionally aimed to provide further insights for physicians and patients on the clinical use and adverse effects of nickel alloy implantation in congenital heart disease and various additional associated diseases.
Materials and methods
Materials
The human monoblastic leukemia cell line THP-1 was obtained from the American Type Culture Collection (Manassas, VA, USA). Cell culture reagents RPMI 1640 medium and fetal bovine serum (FBS) were from Gibco-BRL (Invitrogen Life Technologies, Carlsbad, CA, USA). NiCl2·6H2O, MTT, Hoechst 33342 and propidium iodide (PI) were obtained from Sigma-Aldrich (St. Louis, MO, USA). TRIzol reagent was obtained from Invitrogen Life Technologies. All reagents were of analytical grade.
Cell culture and drug treatment
THP-1 cells were cultured in RPMI 1640 medium supplemented with 10% FBS in a humidified atmosphere of 5% CO2 and 95% air at 37°C. Cells were serum-starved for 48 h prior to treatment with NiCl2·6H2O (25, 50, 100, 200, 400 or 800 µM) for 24, 48 or 72 h, respectively.
Cell growth assay
The MTT assay and cell-counting method were applied to detect THP-1 cell proliferation subsequent to receiving different treatment regimens. The apoptosis of THP-1 in response to nickel ion challenge was detected using flow cytometry (FCM). For the MTT assay, THP-1 cells were seeded in 96-well plates at a density of 4×105 cells/well and then exposed to various concentrations of NiCl2·6H2O for the different time periods indicated (25, 50, 100, 200, 400 and 800 µM for 24, 48 and 72 h). At the end of the treatments, the plates were centrifuged at 2,000 × g for 5 min and the supernatants were discarded. Then cells were further incubated with RPMI 1640 (200 µl/well) supplemented with 0.2% MTT (20 µl/well) for 4 h at 37°C. Following MTT incubation, 100 µl 100% dimethyl sulfoxide was added to dissolve the formazan crystals. Viable cells were counted by reading the absorbance at 570 nm using a 96-well plate reader (Multiskan MK3; Thermo Fisher Scientific, Waltham, MA, USA).
Cytotoxicity analysis
Cells were plated on six-well plates and were allowed to adhere. Following different treatments for 48 h, cells from each group (5×105 cells/group re-suspended in 0.5 ml ice-cold RPMI 1640) were transferred into a clean centrifuge tube and incubated with 1.25 µl Hoechst 33342 for 15 min at room temperature in the dark to stain the nuclei of all cells. Subsequently, the plates were centrifuged at 2,000 × g for 5 min and the supernatants were discarded. Cells were then re-suspended in 0.5 ml ice-cold RPMI 1640 prior to the addition of 10 µl PI (5 µg/ml) for staining of the dead cells. The samples were kept on ice in the dark and immediately analyzed using a FACSCalibur Cell flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). Cytotoxicity was expressed as the percentage of dead cells (PI-positive) relative to the total number of cells.
RNA-sequencing assay
THP-1 cells were cultured with different concentrations of NiCl2·6H2O (25, 50 or 100 µM) for 1 h following starvation for 48 h. Total RNA was prepared by the acid phenol method using TRIzol reagent in accordance with the manufacturer's instructions. Areas containing THP-1 RNAs were sent to Beijing Genomics Institution (Shenzhen, China) for Illumina sequencing.
Pathway analysis of differentially expressed genes
Pathways were constructed using the Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/) database using the Web-based GEne SeT Analysis Toolkit (http://bioinfo.vanderbilt.edu/webgestalt/) to identify differential networks and core regulators involved in the nickel-induced cytotoxic response. The analysis completed was limited to categories with P<0.05.
Quantitative polymerase chain reaction (qPCR)
PCR was performed using the total RNA (1 µg) prepared for the RNA-sequencing assay with the primers listed in Table I under the following conditions: 40 cycles of 94°C for 2 min, 94°C for 30 sec, 58°C for 30 sec and 72°C for 20 sec. RNA expression levels were detected by fluorescent qPCR in the presence of SYBR Green on a Bio-Rad iCycler (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Table I.
Primer sequences for quantitative polymerase chain reaction.
| Genes | Sequence |
|---|---|
| EPOR | Forward: GGGCAACTACAGCTTCTCCT |
| Reverse: ATGGCATGGACTGTGGTCAT | |
| RELB | Forward: TGATCCACATGGAATCGAGA |
| Reverse: CAGGAAGGGATATGGAAGCA | |
| FIGF2 | Forward: ATGGACCAGTGAAGCGATCAT |
| Reverse: GTTCCTCCAAACTAGAAGCAGC | |
| SPI-1 | Forward: GAAAGGTGGGTGAAAGGACCA |
| Reverse: TGTTGGACTCCTTTGGGCAG | |
| TGF-β1 | Forward: TACAGCACGGTATGCAAGCC |
| Reverse: GCAACCGATCTAGCTCACAGAG | |
| CXCL16 | Forward: GACATGCTTACTCGGGGATTG |
| Reverse: GGACAGTGATCCTACTGGGAG | |
| CRLF2 | Forward: AGTGACGGTGACGTGTTCTG |
| Reverse: CTATGGTGACGTTGCAGGTATT | |
| GAPDH | Forward: TGTTCGTCATGGGTGTGAAC |
| Reverse: ATGGCATGGACTGTGGTCAT |
Statistical analysis
Values are expressed as the mean ± standard deviation. The independent-samples t-test was used to compare two samples and one-way analysis of variance was used for multiple comparisons. P<0.05 was considered to indicate a statistically significant difference.
Results
Nickel ions inhibit THP-1 cell growth
The effect of nickel ions on the proliferation of THP-1 was investigated. Cells were treated with NiCl2·6H2O (25, 50, 100, 200, 400 and 800 µM) for 24, 48 and 72 h, and an MTT assay was conducted in order to examine the growth-inhibitory effect of the treatments. As presented in Fig. 1, high concentrations of nickel chloride significantly suppressed cell proliferation at the three concentrations used (200, 400 and 800 µM; P<0.001). Cell proliferation was completely abolished following incubation of cells with nickel chloride at these high concentrations. Lower concentrations of NiCl2·6H2O (25–100 µM) inhibited cell proliferation in a time- and dose-dependent manner as compared with that in the control group.
Figure 1.
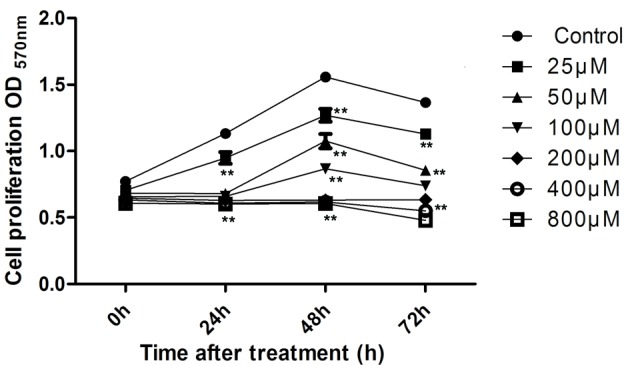
Effect of nickel ion treatment on cell proliferation. The OD of MTT-formazan was determined to assess cell proliferation at 0, 24, 48 and 72 h after treatment with NiCl2·6H2O at concentrations of 25, 50, 100, 200, 400 and 800 µM (**P<0.001, compared with the control). OD, optical density.
Nickel ions exert toxic effects on THP-1 cells
To investigate the toxicity of the nickel ion on THP-1 cells, the viability of THP-1 cells treated with nickel chloride was assessed via FCM. As illustrated in Fig. 2, treatment with nickel chloride (25–400 µM) for 48 h reduced cell viability in a dose-dependent manner. The apoptotic rates of the 25, 50, 100, 200 and 400 µM treatment groups were 14.4, 31.4, 45.7, 61.9 and 65.7%, respectively. The apoptotic rate of the cells was then examined by Hoechst 33342/PI double staining following the various treatment regimens. In this assay, cytotoxicity of the nickel challenge was indicated as the percentage of dead cells (PI-positive) relative to the total number of cells (Hoechst-positive). The extent of THP-1 cell apoptosis was identified to be proportional to the nickel ion concentration. Incubation of THP-1 for 48 h with low concentrations of nickel (25 and 50 µM) resulted in low apoptotic rates (Fig. 2B and C). By contrast, incubation with high concentrations of nickel ions (>100 µM) was observed to have a negative effect on cell viability as indicated by the increased percentage of dead cells (25.2, 60.7 and 85.7% for 100, 200 and 400 µM nickel, respectively) (Fig. 2D, E and F).
Figure 2.

Analysis of cytotoxicity of nickel ions by flow cytometry. (A) THP-1 cells were treated with (B) 25, (C) 50, (D) 100, (E) 200 and (F) 400 μM nickel ions and were harvested following 48-h treatment. Cytotoxicity is indicated by the percentage of dead cells (PI-positive) relative to the total number of cells. PI, propidium iodide.
Differential gene expression of THP-1 cells following treatment with nickel ions
To elucidate the transcriptional events associated with nickel ion-induced THP-1 cytotoxicity, a high-throughput RNA-sequencing assay was conducted using RNA samples extracted from the nickel-challenged cells. Electrophoresis and ethidium bromide staining indicated the successful extraction of RNA products from treated cells. A total of 1 µg total RNA was then pooled from each treatment group (control, 25, 50 and 100 µM NiCl2·6H2O) for comparison of the gene expression profiles via Illumina sequencing. The differentially expressed genes were then filtered. To characterize the functional consequences of alterations in gene expression associated with nickel cytotoxicity, pathway analysis of DEGs was conducted using the web-based KEGG pathway database. The top four enriched KEGG pathway categories of upregulated genes were cytokine-cytokine receptor interaction, osteoclast differentiation, steroid biosynthesis and the chemokine signaling pathway (Table II). The top four enriched KEGG pathway categories of downregulated genes were olfactory transduction, taste transduction, RNA transport and the mRNA surveillance pathway (Table III).
Table II.
Top four enriched Kyoto Encyclopedia of Genes and Genomes pathway categories of upregulated genes.
| Pathway | P-value | Upregulated genes |
|---|---|---|
| Cytokine-cytokine receptor interaction | 6.77×10−8 | TGFB1, CXCR4, TNFRSF1B, CD70, CCL5, EGFA, VEGFB, TNFRSF10B, TNF, IL10RA, IL3RA, TNFSF9, CSF2RA, EPOR, TNFSF13B, FLT3LG, CD40, IL2RG, IL23A, CSF1, CXCL16, PDGFA, TNFRSF12A, CRLF2, CCL2, CCL24, CTF1, CCL3, FIGF, CCL13, INHBE, LTA, CCL4, CCL21, XCR1, LEP, TNFSF18, CCL3L3, CL3L1, GDF6, CCL4L2, IL17B, GH1, CXCL11, TNFRSF4, INHBA, CCR7, CCR5, CCL26, IFNB1, GDF5, TNFRSF18, CCL8 |
| Osteoclast differentiation | 1.07×10−4 | SPI1, CYBA, TGFB1, NCF4, JUND, RAC1, STAT1, GAB2, NFKBIA, FCGR1A, JUN, PPARG, IKBKG, LILRB5, TNF, NFKB2, LILRA5, RELB, LILRB2, OSCAR, NCF1, SOCS3, CSF1, SOCS1, FCGR1B, BLNK, LILRB3, LILRA6, IFNB1 |
| Steroid biosynthesis | 1.13×10−4 | FDFT1, SQLE, DHCR7, EBP, SDHL, TM7SF2, HSD17B7, CYP51A1, SOAT2 |
| Chemokine signaling pathway | 2.41×10−4 | GNB2, CXCR4, RAC1, STAT1, FOXO3, CCL5, HCK, NFKBIA, NFKBIB, HRAS, IKBKG, SHC2, FGR, ARRB2, NCF1, ADCY2, CXCL16, AC092535.1, CCL2, GRK7, CCL24, CCL3, CCL13, CCL4, CCL21, CXCR1, ADCY8, CCL3L3, GNGT2, CCL3L1, CCL4L2, CXCL11, CCR7, CCR5, CCL26, CCL8 |
Table III.
Top four enriched Kyoto Encyclopedia of Genes and Genomes pathway categories of downregulated genes.
| Pathway | P-value | Downregulated genes |
|---|---|---|
| Olfactory transduction | 2.91×10−4 | PRKACB, CAMK2D, PDC, CNGA4, CNGB1, OR2V1, CYorf17, OR11L1, OR10A2, OR52L1, OR52K1, OR6V1, OR2T33, DAPL1, OR6K3, OR13G1 |
| Taste transduction | 1.22×10−3 | PRKACB, TAS2R10, TAS2R50, TAS2R4, TAS2R46, GRM4, TAS2R43, TAS2R13, TAS2R60, TAS2R3, GNG13, TAS2R39 |
| RNA transport | 1.61×10−3 | EIF4G2, XPO1, TPR, TMEM48, PNN, NUP205, DDX39B, PRMT5, NUP155, NUP160, NCBP1, POP1, NUP50, GEMIN5, NUP107, NUP88, NUP54, NXT2, NXF3, NUPL1, THOC1, SUMO1, EIF4E, NUP43, TRNT1, RPP40, PABPC1L, SUMO4, PABPC4L, NXF2B |
| mRNA surveillance pathway | 2.13×10−3 | GSPT1, NUDT21, PNN, PAPOLA, DDX39B, CPSF3, NCBP1, CSTF3, RNGTT, HBS1L, NXT2, NXF3, PAPOLG, CSTF2T, PPP2R3A, PABPC1L, PABPC4L, PAPOLB, NXF2B |
Confirmation of cytotoxicity-associated genes
The authenticity and reliability of the cytotoxicity-associated genes was then verified by specifically examining the expression levels of ten congenital heart disease-associated genes, including EPOR, As presented in Fig. 3, RELB, FIGF, SPI-1, TGF-β1, CXCL16 and CRLF2 through qPCR analysis. RELB, FIGF, SPI-1, CXCL16 and CRLF2 mRNA expression levels of nickel-treated cells presented were significantly increased compared with those in the control group (P<0.01). TGF-β1 and EPOR mRNA expression levels were also significantly increased following nickel challenge (P<0.05).
Figure 3.

Relative mRNA expression levels of EPOR, RELB, FIGF, SPI-1, CXCL16, TGF-β1 and CRLF2. Values presented in histograms are expressed as the mean ± standard deviation. *P<0.05 and **P<0.01 nickel-treated samples vs. control samples (paired t-test).
Discussion
The present study investigated the cytotoxic effects of nickel ions on THP-1 cells and the possible mechanisms involved. The results suggested that nickel ions exerted a growth-inhibitory effect on THP-1 cells and in particular, it was demonstrated that the toxicity of nickel ions to THP-1 cells may be associated with the differential expression of certain genes, including EPOR, RELB, FIGF, SPI-1, TGF-β1, CXCL16 and CRLF2.
In the present study, it was observed that the proliferation of THP-1 cells gradually increased following stimulation for a short period of time with low concentrations (25–100 µM) of nickel ions, which may be associated with the incompletely compromised ability of the cells to resist and metabolize the insult. However, with the increases in the nickel ion concentration and incubation time, a rapid decline in the number of live cells was observed. The cells began to die when incubated with high concentrations of nickel ions (200–800 µM). Cell viability analysis suggested that the concentration and incubation time with the nickel ions may be two important indicators of the toxic effect of nickel. Thus, correct assessment of nickel ion toxicity requires to comprehensively take the concentration and time period of its use into account.
Multiple studies have examined the association between nickel exposure and blood and heart diseases (11,12). The toxicity of the nickel ion to the heart has become a focus of research in recent years since nickel-associated products are being increasingly used in heart disease therapy (13). Accumulating evidence has demonstrated that nickel exposure negatively regulates cardiac function (14,15). The results from a previous study indicated that heart rate variability (a predictor for arrhythmias, mortality risk and severity of illness) was regulated by nickel-induced oxidative-inflammatory responses (14). Thus, it is suggested that nickel may cause certain cardio-regulatory responses and may induce varying responses during systolic and diastolic phases (15).
Although the phenomenon of nickel-induced cardiotoxicity has raised increasing concerns, only few studies are available on the mechanism of nickel ion-induced toxicity to the heart during its use in congenital heart disease (16). Previous studies have demonstrated that congenital heart disease is regulated and affected by multiple genes and pathways, including EPOR, SPI-1, p38/MAPK and TGF-β1 (17–20). It remains to be fully elucidated whether there is an association between nickel toxicity and congenital heart disease, and if so, what the associated genes and pathways are. In the present study, the expression levels of EPOR, RELB, FIGF, SPI-1, TGF-β1, CXCL16 and CRLF2 were observed to be significantly increased subsequent to nickel ion stimulation in THP-1 cells. Nickel can also generate cytotoxic reactive oxygen species through TGF-β1 activation (20). CRLF2 gene alterations have been observed to correlate with poor prognosis in Japanese BCR-ABL1-negative high-risk B-cell precursor acute lymphoblastic leukemia (21), which suggests that the downregulation of CRLF2 may block the inflammatory effect of nickel ions. FIGF was detected in adult lung and heart tissues and was observed to exhibit mitogenic activity on fibroblasts (22,23). CXCL16 appears to have a role in the homing of CD4(+) T cells in acute and chronic rejection models of heart allotransplantation (24). Nickel ion stimulation exerted differential effects on these heart disease-associated genes, which may indicate that the toxicity mechanism of the nickel ion may be regulated by these genes. Further studies, including loss-of-function analysis by RNA inference and in vivo experiments, are required to fully elucidate the mechanistic significance of these genes and pathways in the observed nickel cytotoxicity.
In conclusion, the present study demonstrated that the toxicity of the nickel ion to THP-1 cells may be controlled by or is associated with certain genes, including EPOR, RELB, FIGF, SPI-1, TGF-β1, CXCL16 and CRLF2. These observations provided insight and advance the understanding of the genetic basis of nickel ion-induced toxicity in its therapeutic use for congenital heart disease.
References
- 1.Stan MN, Ammash NM, Warnes CA, Brennan MD, Thapa P, et al. Body mass index and the development of amiodarone-induced thyrotoxicosis in adults with congenital heart disease - a cohort study. Int J Cardiol. 2013;167:821–826. doi: 10.1016/j.ijcard.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 2.Schranz D, Michel-Behnke I. Advances in interventional and hybrid therapy in neonatal congenital heart disease. Semin Fetal Neonatal Med. 2013;18:311–321. doi: 10.1016/j.siny.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Mori Y, Takahashi K, Nakanishi T. Complications of cardiac catheterization in adults and children with congenital heart disease in the current era. Heart Vessels. 2013;28:352–359. doi: 10.1007/s00380-012-0241-x. [DOI] [PubMed] [Google Scholar]
- 4.Dancea A, Justino H, Martucci G. Catheter intervention for congenital heart disease at risk of circulatory failure. Can J Cardiol. 2013;29:786–795. doi: 10.1016/j.cjca.2013.04.021. [DOI] [PubMed] [Google Scholar]
- 5.Braga M, Quecchia C, Perotta C, Timpini A, Maccarinelli K, et al. Systemic nickel allergy syndrome: Nosologic framework and usefulness of diet regimen for diagnosis. Int J Immunopathol Pharmacol. 2013;26:707–716. doi: 10.1177/039463201302600314. [DOI] [PubMed] [Google Scholar]
- 6.Sutton NJ, Greenberg MA, Menegus MA, Lui G, Pass RH. Caring for the adult with congenital heart disease in an adult catheterization laboratory by pediatric interventionalists - safety and efficacy. Congenit Heart Dis. 2013;8:111–116. doi: 10.1111/chd.12004. [DOI] [PubMed] [Google Scholar]
- 7.Karaś Z, Bładek J. Nickel in the environment and morbid symptoms. Przegl Lek. 2004;61(Suppl 3):55–57. In Polish. [PubMed] [Google Scholar]
- 8.Sunderman FW, Jr, Dingle B, Hopfer SM, Swift T. Acute nickel toxicity in electroplating workers who accidently ingested a solution of nickel sulfate and nickel chloride. Am J Ind Med. 1988;14:257–266. doi: 10.1002/ajim.4700140303. [DOI] [PubMed] [Google Scholar]
- 9.Lavin Y, Merad M. Macrophages: Gatekeepers of tissue integrity. Cancer Immunol Res. 2013;1:201–209. doi: 10.1158/2326-6066.CIR-13-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Röstlund T, Thomsen P, Bjursten LM, Ericson LE. Difference in tissue response to nitrogen-ion-implanted titanium and c.p. titanium in the abdominal wall of the rat. J Biomed Mater Res. 1990;24:847–860. doi: 10.1002/jbm.820240705. [DOI] [PubMed] [Google Scholar]
- 11.Kim Y, Wang X, Zhang XS, Grigoriu S, Page R, et al. Escherichia coli toxin/antitoxin pair MqsR/MqsA regulate toxin CspD. Environ Microbiol. 2010;12:1105–1121. doi: 10.1111/j.1462-2920.2009.02147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hou YP, Gu JY, Shao YF, Song YF, Jing YH, et al. The characteristics of placental transfer and tissue concentrations of nickel in late gestational rats and fetuses. Placenta. 2011;32:277–282. doi: 10.1016/j.placenta.2010.12.021. [DOI] [PubMed] [Google Scholar]
- 13.Ries MW, Kampmann C, Rupprecht HJ, Hintereder G, Hafner G, Mayer J. Nickel release after implantation of the Amplatzer occluder. Am Heart J. 2003;145:737–741. doi: 10.1067/mhj.2003.7. [DOI] [PubMed] [Google Scholar]
- 14.Chuang HC, Hsueh TW, Chang CC, Hwang JS, Chuang KJ, et al. Nickel-regulated heart rate variability: The roles of oxidative stress and inflammation. Toxicol Appl Pharmacol. 2013;266:298–306. doi: 10.1016/j.taap.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 15.Creutzenberg O. Biological interactions and toxicity of nanoma-terials in the respiratory tract and various approaches of aerosol generation for toxicity testing. Arch Toxicol. 2012;86:1117–1122. doi: 10.1007/s00204-012-0833-3. [DOI] [PubMed] [Google Scholar]
- 16.Wertman B, Azarbal B, Riedl M, Tobis J. Adverse events associated with nickel allergy in patients undergoing percu-taneous atrial septal defect or patent foramen ovale closure. J Am Coll Cardiol. 2006;47:1226–1227. doi: 10.1016/j.jacc.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 17.Essafi-Benkhadir K, Refai A, Riahi I, Fattouch S, Karoui H, Essafi M. Quince (Cydonia oblonga Miller) peel polyphenols modulate LPS-induced inflammation in human THP-1-derived macrophages through NF-κB, p38MAPK and Akt inhibition. Biochem Biophys Res Commun. 2012;418:180–185. doi: 10.1016/j.bbrc.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 18.Azakie A, Fineman J, He Y. Differential responses of the right ventricle to abnormal loading conditions in vivo: Possible pathophysiologic mechanisms. J Thorac Cardiovasc Surg. 2013;145:1335–1344. doi: 10.1016/j.jtcvs.2013.01.016. [DOI] [PubMed] [Google Scholar]
- 19.Qin C, Zhou S, Xiao Y, Chen L. Erythropoietin enhances mitochondrial biogenesis in cardiomyocytes exposed to chronic hypoxia through Akt/eNOS signalling pathway. Cell Biol Int. 2014;38:335–342. doi: 10.1002/cbin.10205. [DOI] [PubMed] [Google Scholar]
- 20.Yi X, Li X, Zhou Y, Ren S, Wan W, Feng G, Jiang X. Hepatocyte growth factor regulates the TGF-β1-induced proliferation, differentiation and secretory function of cardiac fibroblasts. Int J Mol Med. 2014;34:381–390. doi: 10.3892/ijmm.2014.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamashita Y, Shimada A, Yamada T, et al. IKZF1 and CRLF2 gene alterations correlate with poor prognosis in Japanese BCR-ABL1-negative high-risk B-cell precursor acute lympho-blastic leukemia. Pediatr Blood Cancer. 2013;60:1587–1592. doi: 10.1002/pbc.24571. [DOI] [PubMed] [Google Scholar]
- 22.Rocchigiani M, Lestingi M, Luddi A, Orlandini M, Franco B, et al. Human FIGF: Cloning, gene structure and mapping to chromosome Xp22.1 between the PIGA and the GRPR genes. Genomics. 1998;47:207–216. doi: 10.1006/geno.1997.5079. [DOI] [PubMed] [Google Scholar]
- 23.Avantaggiato V, Orlandini M, Acampora D, Oliviero S, Simeone A. Embryonic expression pattern of the murine figf gene, a growth factor belonging to platelet-derived growth factor/vascular endothelial growth factor family. Mech Dev. 1998;73:221–224. doi: 10.1016/S0925-4773(98)00049-5. [DOI] [PubMed] [Google Scholar]
- 24.Mitsuhashi N, Wu GD, Zhu H, Kearns-Jonker M, Cramer DV, et al. Rat chemokine CXCL11: Structure, tissue distribution, function and expression in cardiac transplantation models. Mol Cell Biochem. 2007;296:1–9. doi: 10.1007/s11010-005-9010-9. [DOI] [PubMed] [Google Scholar]