

Abstract
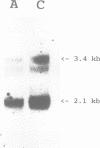
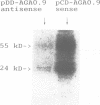
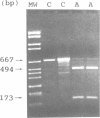
We have isolated a 2.1 kb cDNA which encodes human aspartylglucosaminidase (AGA, E.C. 3.5.1.26). The activity of this lysosomal enzyme is deficient in aspartylglucosaminuria (AGU), a recessively inherited lysosomal accumulation disease resulting in severe mental retardation. The polypeptide chain deduced from the AGA cDNA consists of 346 amino acids, has two potential N-glycosylation sites and 11 cysteine residues. Transient expression of this cDNA in COS-1 cells resulted in increased expression of immunoprecipitable AGA protein. Direct sequencing of amplified AGA cDNA from an AGU patient revealed a G----C transition resulting in the substitution of cysteine 163 with serine. This mutation was subsequently found in all the 20 analyzed Finnish AGU patients, in the heterozygous form in all 53 carriers and in none of 67 control individuals, suggesting that it represents the major AGU causing mutation enriched in this isolated population. Since the mutation produces a change in the predicted flexibility of the AGA polypeptide chain and removes an intramolecular S-S bridge, it most probably explains the deficient enzyme activity found in cells and tissues of AGU patients.
Full text
PDF








Images in this article


Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Alwine J. C., Kemp D. J., Stark G. R. Method for detection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridization with DNA probes. Proc Natl Acad Sci U S A. 1977 Dec;74(12):5350–5354. doi: 10.1073/pnas.74.12.5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronson N. N., Jr, Kuranda M. J. Lysosomal degradation of Asn-linked glycoproteins. FASEB J. 1989 Dec;3(14):2615–2622. doi: 10.1096/fasebj.3.14.2531691. [DOI] [PubMed] [Google Scholar]
- Aula P., Astrin K. H., Francke U., Desnick R. J. Assignment of the structural gene encoding human aspartylglucosaminidase to the long arm of chromosome 4 (4q21----4qter). Am J Hum Genet. 1984 Nov;36(6):1215–1224. [PMC free article] [PubMed] [Google Scholar]
- Aula P., Raivio K., Autio S. Enzymatic diagnosis and carrier detection of aspartylglucosaminuria using blood samples. Pediatr Res. 1976 Jun;10(6):625–629. doi: 10.1203/00006450-197606000-00012. [DOI] [PubMed] [Google Scholar]
- Aula P., Renlund M., Raivio K. O., Koskela S. L. Screening of inherited oligosaccharidurias among mentally retarded patients in northern Finland. J Ment Defic Res. 1986 Dec;30(Pt 4):365–368. doi: 10.1111/j.1365-2788.1986.tb01332.x. [DOI] [PubMed] [Google Scholar]
- Baumann M., Peltonen L., Aula P., Kalkkinen N. Isolation of a human hepatic 60 kDa aspartylglucosaminidase consisting of three non-identical polypeptides. Biochem J. 1989 Aug 15;262(1):189–194. doi: 10.1042/bj2620189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton W. D., Davis R. W. Screening lambdagt recombinant clones by hybridization to single plaques in situ. Science. 1977 Apr 8;196(4286):180–182. doi: 10.1126/science.322279. [DOI] [PubMed] [Google Scholar]
- Borud O., Torp K. H. Letter: Aspartylglycosaminuria in Northern Norway. Lancet. 1976 May 15;1(7968):1082–1083. doi: 10.1016/s0140-6736(76)92266-2. [DOI] [PubMed] [Google Scholar]
- Chirgwin J. M., Przybyla A. E., MacDonald R. J., Rutter W. J. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979 Nov 27;18(24):5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- Christomanou H., Aignesberger A., Linke R. P. Immunochemical characterization of two activator proteins stimulating enzymic sphingomyelin degradation in vitro. Absence of one of them in a human Gaucher disease variant. Biol Chem Hoppe Seyler. 1986 Sep;367(9):879–890. doi: 10.1515/bchm3.1986.367.2.879. [DOI] [PubMed] [Google Scholar]
- Conzelmann E., Sandhoff K. Purification and characterization of an activator protein for the degradation of glycolipids GM2 and GA2 by hexosaminidase A. Hoppe Seylers Z Physiol Chem. 1979 Dec;360(12):1837–1849. doi: 10.1515/bchm2.1979.360.2.1837. [DOI] [PubMed] [Google Scholar]
- D'Azzo A., Hoogeveen A., Reuser A. J., Robinson D., Galjaard H. Molecular defect in combined beta-galactosidase and neuraminidase deficiency in man. Proc Natl Acad Sci U S A. 1982 Aug;79(15):4535–4539. doi: 10.1073/pnas.79.15.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denhardt D. T. A membrane-filter technique for the detection of complementary DNA. Biochem Biophys Res Commun. 1966 Jun 13;23(5):641–646. doi: 10.1016/0006-291x(66)90447-5. [DOI] [PubMed] [Google Scholar]
- Felgner P. L., Gadek T. R., Holm M., Roman R., Chan H. W., Wenz M., Northrop J. P., Ringold G. M., Danielsen M. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc Natl Acad Sci U S A. 1987 Nov;84(21):7413–7417. doi: 10.1073/pnas.84.21.7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehler J., Sewell A. C., Becker C., Hartmann J., Spranger J. Clinical and biochemical delineation of aspartyl-glycosaminuria as observed in two members of an Italian family. Helv Paediatr Acta. 1981;36(2):179–189. [PubMed] [Google Scholar]
- Grön K., Aula P., Peltonen L. Linkage of aspartylglucosaminuria (AGU) to marker loci on the long arm of chromosome 4. Hum Genet. 1990 Jul;85(2):233–236. doi: 10.1007/BF00193202. [DOI] [PubMed] [Google Scholar]
- Hreidarsson S., Thomas G. H., Valle D. L., Stevenson R. E., Taylor H., McCarty J., Coker S. B., Green W. R. Aspartylglucosaminuria in the United States. Clin Genet. 1983 Jun;23(6):427–435. doi: 10.1111/j.1399-0004.1983.tb01977.x. [DOI] [PubMed] [Google Scholar]
- Inui K., Emmett M., Wenger D. A. Immunological evidence for deficiency in an activator protein for sulfatide sulfatase in a variant form of metachromatic leukodystrophy. Proc Natl Acad Sci U S A. 1983 May;80(10):3074–3077. doi: 10.1073/pnas.80.10.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. An analysis of 5'-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 1987 Oct 26;15(20):8125–8148. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel L. M., Smith K. D., Boyer S. H., Borgaonkar D. S., Wachtel S. S., Miller O. J., Breg W. R., Jones H. W., Jr, Rary J. M. Analysis of human Y-chromosome-specific reiterated DNA in chromosome variants. Proc Natl Acad Sci U S A. 1977 Mar;74(3):1245–1249. doi: 10.1073/pnas.74.3.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J., Doolittle R. F. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982 May 5;157(1):105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Laemmli U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970 Aug 15;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lathe R. Synthetic oligonucleotide probes deduced from amino acid sequence data. Theoretical and practical considerations. J Mol Biol. 1985 May 5;183(1):1–12. doi: 10.1016/0022-2836(85)90276-1. [DOI] [PubMed] [Google Scholar]
- Maury C. P. Accumulation of glycoprotein-derived metabolites in neural and visceral tissue in aspartylglycosaminuria. J Lab Clin Med. 1980 Nov;96(5):838–844. [PubMed] [Google Scholar]
- McGovern M. M., Aula P., Desnick R. J. Purification and properties of human hepatic aspartylglucosaminidase. J Biol Chem. 1983 Sep 10;258(17):10743–10747. [PubMed] [Google Scholar]
- Mullis K. B., Faloona F. A. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol. 1987;155:335–350. doi: 10.1016/0076-6879(87)55023-6. [DOI] [PubMed] [Google Scholar]
- Okayama H., Berg P. A cDNA cloning vector that permits expression of cDNA inserts in mammalian cells. Mol Cell Biol. 1983 Feb;3(2):280–289. doi: 10.1128/mcb.3.2.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollitt R. J., Jenner F. A., Merskey H. Aspartylglycosaminuria. An inborn error of metabolism associated with mental defect. Lancet. 1968 Aug 3;2(7562):253–255. doi: 10.1016/s0140-6736(68)92355-6. [DOI] [PubMed] [Google Scholar]
- Sanger F., Nicklen S., Coulson A. R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977 Dec;74(12):5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southern E. M. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol. 1975 Nov 5;98(3):503–517. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- Tollersrud O. K., Aronson N. N., Jr Purification and characterization of rat liver glycosylasparaginase. Biochem J. 1989 May 15;260(1):101–108. doi: 10.1042/bj2600101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G. A new method for predicting signal sequence cleavage sites. Nucleic Acids Res. 1986 Jun 11;14(11):4683–4690. doi: 10.1093/nar/14.11.4683. [DOI] [PMC free article] [PubMed] [Google Scholar]