

Abstract
Aggregation of proteins to fiber-like aggregates often involves a transformation of native monomers to β-sheet-rich oligomers. This general observation underestimates the importance of α-helical segments in the aggregation cascade. Here, using a combination of experimental techniques and accelerated molecular dynamics simulations, we investigate the aggregation of a 43-residue, apolipoprotein A-I mimetic peptide and its E21Q and D26N mutants. Our study indicates a strong propensity of helical segments not to adopt cross-β fibrils. The helix-turn-helix monomeric conformation of the peptides is preserved in the mature fibrils. Furthermore, we reveal opposite effects of mutations on and near the turn region in the self-assembly of these peptides. We show that the E21-R24 salt bridge is a major contributor to helix-turn-helix folding, subsequently leading to abundant fibril formation. On the other hand, the K19-D26 interaction is not required to fold the native helix-turn-helix. However, removal of the charged D26 residue decreases the stability of helix-turn-helix monomer, and consequently reduces aggregation. Finally, we provide a more refined assembly model for the helix-turn-helix peptides from apolipoprotein A-I based on the parallel stacking of helix-turn-helix dimers.
Keywords: peptide, fibril, α-helix, apolipoprotein A-I, conformation state, non-amyloid aggregation, ion-mobility mass spectrometry, accelerated molecular dynamics
INTRODUCTION
Protein self-assembly into fibrils has raised important questions into the nature of secondary structures in the aggregation cascade. It has been shown extensively that many fatal neurodegenerative diseases (e.g. Alzheimer’s, Parkinson’s, Creutzfeldt–Jacob’s and amyotrophic lateral sclerosis) correlate strongly to the fibrillization of intrinsically disordered proteins such as amyloid-β (Aβ), α-synuclein, prion and Tau.1–3 Moreover, many peptides and proteins, including biologically active and de novo fragments, have been found to form amyloid fibrils in vitro.4–5 This process typically involves structural transitions into β-sheets, with a loss of globular native states and any associated α-helicity.6–8 Despite the dominant role of the cross-β-sheet in mature fibrils,9–11 compelling evidence points to an intermediate role for the α-helix in the cascade.12–15 Many aggregating proteins exhibit α-helical character when associated with membrane.15–17 However, the crowded environment in cells makes the study of in vivo aggregation challenging. In order to directly evaluate the role of the α-helix in fibril formation, previous studies have been focused on inducing α-helical peptides to self-assemble into fibrils in solution by novel peptide design.18–20 Several model systems have been successfully developed based on apolipoprotein A-I (apo A-I) and its mimetic peptides.19–20
Apo A-I is the major structural component of HDL, a heterogeneous combination of lipids and proteins used to shuttle lipids in the body.21 Helical intermediates have been described in the fibrillogenesis of the apolipoprotein E protein.22–23 Explanted fibrils of the apo A-I variant L174S from cardiac tissue show both cross-β and residual helical structure.24 Fiber diffraction data indicate that the α-helices are oriented perpendicular to the main axis of the fibril, similar to the wt peptide fibrils. More recently, the apo A-I variant L178H has been shown to form fibrils with exclusively α-helical structure.25 The L178H variant is the first report of a full-length variant of apo A-I that forms α-helical fibrils. The presence of α-helical character in physiological apo A-I fibrils indicates that this secondary structure is important in protein aggregation and disease, and suggests that our work on apo A-I mimetic peptides may inform this growing area of exploration.
The helix-turn-helix motif is a key structural motif in apo A-I. Residues 44–243 of apo A-I contain ten tandem, amphiphilic α-helices, many of which are connected by proline residues. A number of short, amphiphilic, α-helical peptides have been shown to recapitulate many of the physical properties of the entire 243 residue apolipoprotein. For example, the 18-residue, amphiphilic α-helical peptide developed by Anantharamaiah and colleagues (peptide 18A shown in Scheme 1)26 binds to lipid, activates the enzyme lecithin-cholesterol acyltransferase (LCAT), and has other physical properties typical of the full-length protein.
Scheme 1.

The primary sequences of the peptides studied. The turn regions are labeled in red with the mutations shown in green.
An early work by Fezoui et al., in which one 18A segment was used to construct the N-terminal helix of a 38-residue helix-turn-helix, showed that the peptide undergoes α-helix/β-sheet transition leading to cross-β fibrils.19 On the other hand, a previous work by Lazar et al. involved the synthesis of a fibril-forming, 43-residue helix-turn-helix peptide with two segments of peptide 18A connected by a putative turn sequence from apo A-I (the wt peptide shown in Scheme 1).20 Unlike Fezoui et al.’s design, circular dichroism (CD) of the 43-residue peptide in solution and CD and FTIR of fibril films showed significant α-helical character. Synchrotron X-ray fiber diffraction on a magnetically aligned sample of the peptide confirmed the α-helical character in the fibrils and indicated that the helical axes are oriented perpendicular to the fibril axis.
The 43-mer peptide (henceforth referred to as the wt peptide) forms α-helical fibrils instantaneously upon dissolution at concentrations greater than 1 μM.20 It was thought that the high propensity of the wt peptide to form fibrils may be due to electrostatic interactions in the turn region. The turn region contains four charged residues placed so that, at pH close to neutrality, oppositely charged groups could interact with one another; Glu (E) and Arg (R) could interact (E21-R24) and Lys (K) could interact with the Asp (D) (K19-D26) which would stabilize the helix-turn-helix. We made modifications in the turn region of the wt peptide to abolish these potential electrostatic interactions and determine if these interactions play an important role in fibril formation. E21Q eliminates one of the potential salt bridges in the turn region while D26N eliminates the other. The sequences of the 18A, wt, E21Q and D26N peptides are shown in Scheme 1.
The current work explores the propensities of peptides 18A, E21Q and D26N to form α-helical fibrils at the molecular level. Transmission electron microscopy (EM), CD, intrinsic tryptophan fluorescence, ion-mobility mass spectrometry (IM-MS) and computational modeling were used to explore the fibrillogenesis potential of these peptides.
MATERIALS AND METHODS
Peptide synthesis and purification
The wt peptide was purchased from Midwest Biotech and the E21Q and D26N peptides were purchased from Aapptec. Peptide 18A was synthesized using standard 9-fluorenylmethoxycarbonyl (Fmoc) and HBTU/HOBt manual solid-phase synthesis. The peptide was acetylated with acetic anhydride and amidated with a Fmoc-Rink Amide resin (Anaspec). The peptide was cleaved from the resin using 94% TFA, 5% of triisopropylsilane and 1% phenol for 2 h at 295K. Crude peptides were purified by RP-HPLC on a semi-preparative C18 column (Phenomenex) using gradients of water (0.1% v/v TFA) and acetonitrile (0.1% v/v TFA). Peptide purity was greater than 91% by analytical RP-HPLC. The molecular masses of the peptides were verified by ESI mass spectrometry.
Determination of peptide concentrations
Peptide concentrations were calculated using the extinction coefficients for the tryptophan and tyrosine residues. The concentration of peptide 18A was measured using ε280 = 7,300 M−1 cm−1, and the concentrations of the wt, E21Q and D26N peptides were measured using ε280= 14,600 M−1 cm−1 (Thermo Electron Corporation Nicolet Evolution 300).
Fibril sample preparation
Fibril assays were performed by incubating the wt, E21Q, D26N and 18A peptides at 50 μM in 50 mM ammonium acetate buffer, pH 7.0. The samples were incubated at 310K and contained 0.01% NaN3 to prevent microbial growth. To determine if a higher temperature promotes fibril formation in the E21Q peptide, 50 μM peptide was heated for 16 hours at 343K in 50 mM ammonium acetate buffer, pH 7.0, 0.01% NaN3.
Transmission Electron microscopy
Aliquots of fibril samples (5 μL) were applied to a glow-discharged, 400-mesh, carbon-coated support film and stained with 1% uranyl acetate. Images of fibrils at magnifications of 21,000x and 137,200x were recorded using a FEI Tecnai F30 STEM Microscope operated at 300 kV and equipped with a Gatan CCD digital micrograph.
Far-UV Circular Dichroism
CD data were collected on an Applied Photophysics Chirascan CD spectrometer. The CD spectra of freshly dissolved peptides were measured in 10 mM ammonium acetate, pH 7.0. The wt, E21Q and D26N peptides were measured at 1 μM, and the 18A peptide at 2 μM. A concentration of 1 μM was chosen for the helix-turn-helix peptides because the wt peptide forms fibrils at concentrations greater than 1 μM.20 The CD measurements were collected at 1 nm intervals from 265 to 190 nm with a 1 s averaging time and a 1 nm bandwidth. All spectra were collected in a 1 cm quartz cuvette at 298K. Three scans were collected, and the data were averaged. A buffer baseline was subtracted from the averaged data and the curves were base-line corrected. The α-helical content of the peptides was estimated from the mean residue ellipticities (MRE) at 222 nm.27
Intrinsic Tryptophan Fluorescence
Samples were prepared to explore the aggregation behavior of the wt, E21Q and D26N peptides. 1 mL samples were prepared containing 50 μM peptide in 20 mM ammonium acetate buffer, pH 7.0, 0.01% NaN3. To minimize tyrosine fluorescence, the excitation wavelength was set at 295 nm and the tryptophan emission was followed from 310 to 440 nm. The excitation and emission bandpasses were set to 2 nm and 8 nm, respectively. A quartz 1 cm cuvette containing 0.4 mL of 20 mM ammonium acetate buffer, pH 7.0, and a 20 μL aliquot of the peptide solution, was mixed in the cuvette and the fluorescence was measured (Aminco Bowman Series Luminescence Spectrometer). The peptide samples were incubated at 310K in between the time points. A background was also taken and subtracted from the peptide scan.
Ion-mobility Mass Spectrometry
Sample Preparation
Prior to introduction into the mass spectrometer, samples were freshly prepared from peptide powder to a final concentration of 50 μM in 20 mM ammonium acetate buffer (pH = 7.0).
Instrumentation and Method
A majority of IM-MS experiments were performed using a lab-built mass spectrometer (Instrument I). This instrument consists of a nanospray source, an ion funnel, a 2-m long drift cell, an exit funnel and a quadrupole mass filter.28 The high resolution in ion mobility offers arrival time separation among different conformations and oligomeric species of the same mass to charge (m/z) ratio.
A second instrument (Instrument II) was also utilized for injection energy studies of some ATDs at higher m/z values. Details about this instrument were previously described.29 The major difference is that the drift-cell is shorter (~4.8 cm) and thus the instrument has a lower ion-mobility resolution.
In an ion-mobility experiment, arrival time distributions (ATDs) can be measured at different pressure to voltage (P/V) ratios, allowing for the determination of a reduced mobility K0 and subsequently a collisional cross section σ, a quantity independent from instrumental parameters (see Equation 1).30 The experimental cross sections can be compared with those of the theoretical structures obtained from simulations.
| (Eq. 1) |
Here, T is the temperature of the buffer gas (295–300K), N0 is the number density of helium at standard temperature and pressure, e is the total charge of the ion, μ is the reduced mass of the He-ion collision system, and kB is the Boltzmann factor. The relative abundance of a single species can be approximated using the kinetic theory of gases. A measured ATD peak is fitted with single or multiple Gaussian peaks using peak heights and exact arrival times as variables. Each Gaussian peak indicates a single species.31
Molecular Dynamics Simulations
Molecular Dynamics (MD) provides a means to computationally investigate a variety of events within time scales on the order of tens to thousands of nanoseconds at an atomistic level.32 Since the first publication on computer simulations of protein recognition,33 the field has grown tremendously and holds great promise to unravel complex processes including protein structure predictions and interactions. Quantitative analyses of three-dimensional structure, and kinetic and thermodynamic phenomena, are vital steps toward the understanding of driving forces behind folding and unfolding pathways. In the most basic form of MD, referred to as conventional MD (cMD) or “brute force” dynamics, the biological systems of interest are often trapped in a few local minima of a complex potential energy landscape. Such long interstate lag times lead to insufficient sampling of folding or interaction events. Furthermore, many relevant processes (not limited to biology) occur in much longer timescales (i.e., milliseconds to seconds), which are intractable for any traditional MD techniques. All of these processes are assumed to have a long-time dynamics, in which there are few infrequent jumps among different states which highly depend on the lag time.34 In order to overcome the nonergodic nature of system states obtained from cMD, and to increase the accuracy of structure prediction, new innovative MD methodologies have been developed.34–37
Accelerated MD (aMD) was proposed by Voter and co-workers under the name Hyperdynamics,38 in which a designed bias potential is adaptively utilized to fill the basins other than the transition states between the basins. The technique has quickly matured and has been applied to the simulations of biological events in the subsequent work done by Hamelberg, McCammon and many others.37, 39 aMD allows more rapid sampling of conformations within a rough energy surface by modifying the potential energy surface when V(r) is below a pre-defined energy level E. Details about this method can be found in the Supporting Information section S1 or elsewhere.39
Temperature-based Replica Exchange Molecular Dynamics (T-REMD) offer a means to sample conformation distributions and extract equilibrium thermodynamic functions, taking advantage of the decrease in escape time from local minima at elevated temperatures.40 In T-REMD, multiple copies are simulated at different temperatures and adjacent replicas are exchanged periodically with a probability defined by the corresponding temperatures and energies of the copies (for a review, see Morriss-Andrews, A. et al41).
Structures obtained from the aMD and T-REMD simulations were subjected to a short minimization in the gas phase to mimic the dehydration process occurring inside the mass spectrometer. All theoretical collision cross sections of the wt, E21Q and D26N monomers and their model structures were computed using both the trajectory method (TJ) available from the Mobcal package42–43 and the PSA method (http://luschka.bic.ucsb.edu:8080/WebPSA/).44–45 For 18A systems, the Projection Superposition Approximation (PSA) method44–45 was employed because the TJ method can overestimate the cross sections for small systems.46–47
Implicit solvent aMD simulations and in-vacuo T-REMD simulations
Method and Force field Validation
Duan and co-workers folded several small helix bundles using the Amber0348 force field in combination with the Onufriev, Bashford, and Case solvation model.49–51 Since the wt and its mutants are highly helical according to CD spectra (see Lazar et al.20 and next sections), we used the same procedure to fold all of these peptides. All implicit solvent simulations were performed using the AMBER12 package.52
To validate the choice of force field and the implicit solvation model, aMD simulations were performed for the 18A peptide, whose NMR structure is known (PDB code 2FQ5,53 see Section S1 in the Supporting Information for simulation parameters and other details). The correct native states (backbone RMSD of 1 Å) were reached for this system, with multiple folding and unfolding events sampled as seen in Figure 1. It is noted that 18A alternatively takes on either a full helix or partially folded helical conformations (see Supporting Information Figure S1). aMD samples a much wider range of conformations, making it an inexpensive method for probing the native folded states of the peptides.
Figure 1.

Backbone RMSD of structures obtained from cMD and aMD simulations of the 18A peptide. The reference structure was obtained from PDB code 2FQ5 (shown in blue).
To simulate the wt, E21Q and D26N peptides, several hundreds of nanoseconds to few-microseconds long aMD trajectories (the longest trajectory was 1.65-μs) were performed at 300K, with the dual boost potentials chosen according to a procedure described in Pierce et al.39 (see Supporting Information for the values of E(dih), E(tot), α(dih) and α(total) used in each simulation) and randomized initial velocities. In all simulations, the starting structures were extended conformations built using tleap and minimized using the sander module of the AMBER12 package.52
In vacuum T-REMD simulations40 were performed to generate solvent-free structures for the wt starting with 32 identical replicas (T = 285.00, 298.97, 313.48, 328.55, 344.20, 360.46, 377.33, 394.87, 413.09, 432.00, 451.65, 472.06, 493.27, 515.29, 538.18, 561.95, 586.64, 612.29, 638.93, 666.58, 695.34, 725.22, 756.26, 788.63, 822.14, 856.98, 893.15, 930.73, 969.77, 1010.30, 1052.44, 1096.20 K). Extremely high temperatures were used to overcome slow structural relaxation in the gas phase.54 The production run is 350-ns with the last 200-ns subjected to analysis. The theoretical cross sections of these structures offer the lower-bound for experimental cross sections of the monomers. Chirality restraints were applied for all residues, and dihedral restraints were used to prevent trans to cis isomerization for all residues except proline. At the end, all structures are clustered based on the backbone root-mean-squared deviation (RMSD) using the Daura algorithm55 with a cut-off value of 0.2 nm to identify the major families of structures.
Explicit solvent T-REMD and standard MD simulations
Standard MD simulations in explicit solvent were performed to examine the stability of different dimer models using the GROMACS 4.6.3 package56–58 and the Amber03 force field48 in combination with the TIP3P water model.59 The presence of explicit water and counterions is necessary to capture intermolecular electrostatic interactions between two monomer units. Initial structures were constructed from the wt helix-turn-helix monomers obtained from the aMD simulation, then solvated in a cubic box of side 6.5 nm by 8647 water molecules and neutralized by 2 Cl− counterions. The whole systems were minimized and then subjected to a 3-ns equilibration in NPT ensemble (T = 300K and P = 1 bar) to optimize box size. The production run includes two independent 50-ns trajectories in NVT ensemble for different initial velocities. The LINCS algorithm60 was employed to constrain bonds between heavy atoms and hydrogen, and the SETTLE algorithm61 was used for water molecules. These constraints allowed an integration time step of 2.0 fs. The electrostatic and dispersion forces were computed with a real space cutoff of 1.2 nm, and the particle mesh Ewald method was used to treat long-range electrostatics.62 The temperature was maintained by the Nose-Hoover thermostat.63 The temperature and pressure coupling constants were 0.1 ps and 1.0 ps, respectively. The equations of motion were integrated accordingly to the leap-frog algorithm. The stability of the final structure is determined on the basis of the secondary structure of each monomer unit and the center-of-mass distance between the two monomers.
Robetta Full-chain Protein Structure Prediction
The monomeric structures of the wt, E21Q and D26N peptides were also predicted using the Robetta server (http://robetta.bakerlab.org/).64–65 Robetta provided automated structure predictions using a comparative modeling method. Five probable structures were generated for each peptide.
RESULTS
Formation of Fibrils and Appearance by EM
The wt peptide has been shown previously to form α-helical fibrils in 10 mM sodium phosphate buffer, pH 7.6.20 For the current study, the peptides were incubated in ammonium acetate buffer, the buffer employed in the IM-MS experiments. The wt, E21Q, D26N and 18A peptides were incubated at a concentration of 50 mM in 50 mM ammonium acetate buffer, pH 7.0. The wt peptide formed a pellet (13,200 rpm) within an hour of incubation at 310K. EM revealed the presence of fibrils (Figure 2A,B). The E21Q and the D26N peptides were used to investigate the role of the potential electrostatic interactions in the turn region in fibril formation. The E21Q peptide did not form fibrils after a month of incubation at 310K in 50 mM ammonium acetate buffer, pH 7.0, indicating that the glutamic acid at position 21 is essential for fibril formation (Figure 2C,D). The D26N peptide, however, did form fibrils after incubation at 310K, indicating that the aspartic acid at position 26 is not a key contributor in fibril formation (Figure 2E,F). It is of interest to note that unlike the wt peptide, a pellet (13,200 rpm) was not observed after incubation of D26N for several days, but EM revealed the presence of fibrils and intrinsic tryptophan fluorescence experiments support fibril formation (see Supporting Information Figure S1). EM and fluorescence both show that the D26N mutation reduces the aggregation propensity of the wt peptide. The EM images show fewer fibrils for the D26N mutant. The 18A peptide did not show fibrils after a month of incubation at 310K (Figure 2G,H).
Figure 2.

Electron micrographs of the precipitates formed by the wt, E21Q, D26N and 18A peptides. Precipitates stained with 1% uranyl acetate are shown magnified at 21,000x and 137,200x as follows: wt (A and B), E21Q (C and D), D26N (E and F), and 18A (G and H). Panels A, C, E and G, scale bar = 200 nm. Panels B, D, F and H, scale bar = 50 nm.
The fibrillogenesis potentials of the wt, E21Q and D26N peptides have also been explored in 10 mM sodium phosphate buffer, pH 7.4, after incubation at 310K (data not shown). Both the wt and the D26N peptides show a strong aggregation propensity under these conditions, with a large number of fibrils observed by EM. This indicates that there is a strong solvent effect, with ammonium acetate showing fewer fibrils than samples incubated in sodium phosphate buffer. The E21Q peptide does not show fibrils in 10 mM phosphate buffer, pH 7.4, similar to the results obtained in 10 mM ammonium acetate buffer, pH 7.0 (Figure 2C,D).
Far-UV Circular Dichroism
Far-UV CD data were collected on freshly dissolved peptide samples in 10 mM ammonium acetate, pH 7.0. Ammonium acetate buffer gives a high absorbance at low wavelengths (190–200 nm). Consequently, the Far-UV CD data are reported from 200 – 260 nm. The CD spectra of the wt, E21Q and D26N peptides show minima at 208 and 222 nm, consistent with significant α-helical character (Figure 3). The helical content of the peptides was estimated from their mean residue ellipticities (MRE) at 222 nm.27 The helical contents of the wt, E21Q, D26N and 18A peptides in 10 mM ammonium acetate buffer, pH 7.0, are 45, 43, 49, and 31%, respectively. The percent α-helix for the wt peptide in ammonium acetate buffer (45%) is slightly less than the value reported previously in 10 mM sodium phosphate buffer, pH 7.6 (52%).20 The percent α-helix for peptide 18A (31%) is similar to the value given in the literature (34%) for this peptide in PBS.66 In addition, the zero crossover point (wavelength at which MRE is zero) in the CD spectrum of 18A in PBS is 197 nm.67 A similar result is seen in the current study with a zero crossover point below 200 nm in ammonium acetate buffer. The exact value is not reported due to the high absorbance of the ammonium acetate buffer below 200 nm.
Figure 3.

CD spectra of the wt (●), E21Q (■), D26N (◆) and 18A (▲) peptides in 10 mM ammonium acetate buffer, pH 7.0. For clarity, every third point is shown.
Molecular Dynamics Simulations
The data for the two mutant peptides were collected and analyzed from two independent aMD trajectories with different dihedral boost potentials, unlike the wt peptide which required only one aMD trajectory.
wt
The 1.00 μs-long aMD trajectory reaches the predicted helix-turn-helix structure with an average radius of gyration (Rg) of 11.6 nm (i.e., helix-turn-helix, which is referred to as a native (N) state, see Supporting Information Figure S3 for the orientations and positions of different residue types). However, the turn region appears to be shorter than what was proposed by Lazar et al.20 The folding is initiated by a quick hydrophobic collapse, followed by the rearrangement of the C-terminal 18A fragment into a helix. The rate determining step for the formation of this helix is a transition from a partial to a full helix, which is also observed in other helical peptides. The folding of the second helix at the N-terminus is another rate determining step, which requires the breaking of non-native hydrophobic interactions between the hydrophobic faces created by the first helix and the hydrophobic residues of the remaining helix (see Supporting Information Figure S4 and further discussion later). When the helix-turn-helix is formed, a majority of the turn region excluding Pro22 is part of the two helices. The interlocking hydrophobic seams, while contributing to the stability of the helix-turn-helix, allow a small degree of twisting and fraying, as previously described in CD experiments.20 In addition to the folded helix-turn-helix, the trajectories also sample a considerable population of helical bundles (Rg < 11 nm), which are likely the intermediates of the folding process (see Figure 4A and Supporting Information Figure S5).
Figure 4.

Normalized histograms of Rg values of the structures obtained from independent aMD trajectories. (A) wt, (B) E21Q and (C) D26N. Clusters of final structures are shown in the insets, with the corresponding Rg value. The helix bundle (left) and helix-turn-helix structure (right) of the E21Q mutant are shown in panel B. (D) Secondary structure propensity of each residue (Turn, π-helix, α-helix, 310-helix, anti-parallel β-sheet, parallel β-sheet) obtained from aMD trajectories of the wt and D26N mutant using the same low boost potentials.
E21Q
The trajectory started with a lower dihedral boost does not reach the helix-turn-helix within the time scale of 1.65 μs. From this trajectory, a family of helix bundles are sampled, composed of a C-terminal fully-folded helix and a partially folded N-terminal helix with a small turn at V10-A11, referred to as a P state. Since the turn region randomizes into a coil due to the lack of E21-R24 salt-bridge, it is unable to position the N-terminal fragment to support native hydrophobic interactions and electrostatic contacts among charged residues in the helix-turn-helix within the timescale of the simulation. On the other hand, the 0.85-μs trajectory, with a higher boost potential, reaches the helix-turn-helix structure (Rg = 11.5 nm). The simulation suggests that the mutation does not completely abolish the formation of these folded structures, but can limit or shift the populations from a native N state toward partially folded P state structures. As seen in Figure 4B, the two trajectories sample as least two distinct, non-interconverting populations of structures. Since the Rg distributions are narrow and relatively unimodal for both trajectories, the simulations qualitatively suggest that the helix bundles and the helix-turn-helix are stable conformations of the E21Q peptide within the timescale of the simulations.
D26N
The peptide was sampled using two aMD trajectories at 1.40 and 0.85-μs. Both of these trajectories sample helix-turn-helix structures. Figure 4C shows bimodal distributions of Rg, similar to that of the wt, suggesting that the P state helix bundles and N state helix-turn-helix are sampled in both trajectories and internally converted, with the latter being favored. Helix-turn-helix structures are the final conformations in both cases. The turn region in the helix-turn-helix folded by the first trajectory is shorter than that folded by the second trajectory, leading to a small difference in average cross sections.
Figure 4D compares the folding of the wt and D26N trajectories. In the wt trajectory, after the helix-turn-helix is reached, there are minimal fluctuations in secondary structure of the two residues in the two helices. On the other hand, the residues of the D26N mutant continue to alter between α-helix and turn structures, leading to the instability of the native state.
The aMD simulation results are in good agreement with the Robetta structure predictions (see Supporting Information Figure S6). Both methods suggest that all three peptides can adopt helix-turn-helix structures. There are partially folded structures populated by all three peptides. There are also two structures populated by the two mutants, in which there are no significant interactions between the two helicies of the helix-turn-helix. These two structures will be discussed later in light of the experimental results.
Ion-mobility Mass Spectrometry
18A
The ESI-q mass spectrum of 18A shows two major peaks at 748 and 1121 m/z, corresponding to oligomer to charge ratios (n/z) of +1/3 and +1/2, respectively (see Supporting Information Figure S7). Ion-mobility experiments reveal two monomer species of charge state z = +3 with cross sections of σexp = 435 and 457 Å2 (Figure S7B). The theoretical cross section of the 18A helix obtained from aMD simulation is σPSA = 463 Å2 (see the inset of Figure 1). Here, the PSA method is employed because the trajectory method can overestimate the cross sections for small systems.46–47 The structure with the smaller experimental cross section is likely the imperfect helix with a small turn near the Val10-Ala11 hydrophobic pair (σPSA = 436 Å2, see Figure S2). The ATD at 1121 m/z shows the presence of two species, a monomer with z = +2 and a dimer with z = +4 (Figure S7C). The doubly charged monomer has a cross section of σexp = 410 Å2, similar to that of a helix-turn-helix (σPSA = 412 Å2, see Supporting Information Figure S2). The feature at shorter arrival time is assigned to a dimer (σexp = 659 Å2), due to the difference in arrival time between the two features. The data suggest that the 18A monomer motif is amenable to dimer formation. However, larger oligomers are not observed.
Three possible pathways can occur leading to an 18A dimer. In the first pathway, the two monomers would interact through hydrophobic interactions and expose charged residues to solvent similar to those between the two helices of the wt helix-turn-helix. However, unlike the wt peptide where these two 18A segments are connected by a turn, the entropic penalty for a pair of independent helices to associate is predicted to be much higher. Furthermore, the experimental cross section of the 18A dimer (σexp = 659 Å2) is much smaller than the hypothetical dimer composed of two 18A helices (see Supporting Information Figure S3, panel A; σPSA = 705 Å2). The second mechanism is that 18A undergoes a conformational transition from α-helix to β-sheet, from which the two molecules dimerize through the means of hydrogen bonding interactions. However, 18A is an amphipathic peptide known for retaining helical structure.26, 66, 68 In 18A, blocks composed of two consecutive oppositely charged residues are often separated by two to three bulky, hydrophobic amino acids. This pattern stabilizes helical structures better than β-sheets which favor a hydrophobic-polar repeating pattern.69 Further, the CD spectrum of 18A shows no β-sheet signature (Figure 3). The last possibility suggests random electrostatic interactions among charged residues and hydrophobic collapse among others, leading to a dimer with less defined secondary structures and an oligomer that is unable to grow further. The last mechanism appears to be the most probable, since it is consistent with the formation of amorphous, non-fibrillar aggregates observed in the TEM images of incubated 18A peptide samples (Figure 2G,H). It also suggests a more globular dimer with a cross section compatible with the experimental cross section (a representative model structure is shown in Figure S3, panel C; σPSA = 669 Å2). Finally, the data on the 18A peptide suggest a vital role of the turn in positioning the two 18A helices to support fibrillization.
wt, E21Q and D26N
In Figure 5, the ESI-quadrupole mass spectra of the wt, E21Q and D26N peptides show the presence of mainly monomer species with charge states z = +5 and +4 (1044 and 1305 m/z, respectively) in which z = +4 is dominant. (The molecular weights of these peptides differ by only one amu.) The insets in Figure 5 show the ATDs of the major peak (1305 m/z; n/z = 1/4) of the wt, E21Q and D26N mutants. The overall ATDs are very similar in the number of features, suggesting that the peptides can adopt similar conformations but their distributions are different. Each ATD is composed of at least five main peaks; three with cross sections less than 820 Å2 and the remaining two with larger cross sections. The shorter arrival time ATD peaks of E21Q are dominant and the two other features better resolved than those of the wt and D26N mutant. On the other hand, the wt and D26N mutant have the later-arrival time features dominant, but less resolved.
Figure 5.

nano-ESI-q-mass spectra of (A) wt, (B) E21Q and (C) D26N. Major peaks are annotated with oligomer to charge ratios (n/z). Representative ATDs of n/z = +1/4 for the (A) wt, (B) E21Q and (C) D26N peptides are shown in the insets. Each feature is fitted to a Gaussian distribution and labeled with oligomer size, n/z and experimental cross section.
The cross section of the shortest arrival time feature in the z = +4 ATDs (σexp = 741 Å2) is larger than those of the most populated clusters obtained from in-vacuo T-REMD simulation (σTJ = 716 Å2, see Supporting Information Figure S8), suggesting that all of the features are dehydrated solution-phase monomers. The major features at long arrival times in all ATDs (i.e. the native helix-turn-helix N states in Figure 5 insets) have cross sections in good agreement with those of the theoretical “native” helix-turn-helix structures (Figure 4). Side chain rearrangements and a small degree of fraying between the two helices give rise to a broad distribution and a large range of cross sections also observed in the simulation. The remaining structures with cross sections smaller than 820 Å2 are helix bundles or other metastable, more compact structures (i.e. the partially folded P state).
The n/z = +1/3 peak at 1740 m/z is also present but very minor. Dimer populations are also detected at n/z = +2/5 in some cases. The intensities of the peaks were too low for IM-MS measurements by Instrument I, but the ATDs could be obtained from Instrument II (see Figure S10).
Injection energy studies for the wt and mutants
n/z = 1/4
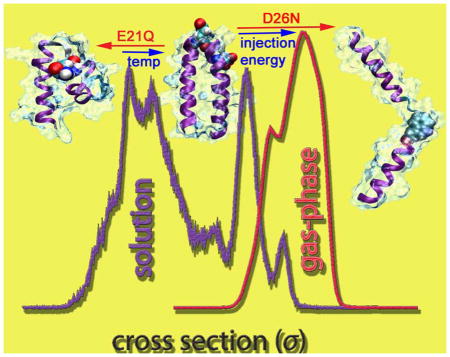
The ATDs of n/z = 1/4 (1305 m/z) for both the wt and its mutants obtained from Instrument II show only extended monomers (σexp = 831–847 Å2 and 968–1012 Å2) with no compact, metastable structures (Figure 6). At high injection energies (IE; > 75V), the molecules are energized leading to a shift in distribution to favor the most extended structure. This change is more profound in the case of the mutants. While the shortest time feature is the native N state, the more extended feature is a “gas-phase” helix-turn-helix A state with less interacting surface between the two helices (see Figure 7). Due to the construction of instrument II, the ions generated from ESI are often energized before entering the drift cell, that can lead to the partial loss of native hydrophobic contacts in folded species. In solution, the native helix-turn-helix motif favors an interlocking hydrophobic seam as seen in Figure 4 and exposes charged residues to the solvent. However, once the solvent evaporates and the peptides are subjected to an injection enery at the drift cell entrance, the hydrophobic surface between the two helices partially unzips and the two helices can translate more freely. This gives rise to the solvent free A states with larger cross sections. Hence low injection energies favor the solution like N states and higher injection energies favor the solvent free A states. The native state of the wt is more stable than both of the mutants, as shown by the ratios between N states and A states at IE = 100V.
Figure 6.

Injection energy studies using Instrument II for peaks at 1305 m/z (n/z = 1/4) and 1740 m/z (n/z = 1/3). Each feature is annotated with oligomer size (M = monomer, D = dimer), n/z ratio and an experimental cross section.
Figure 7.

Model structures for P, N and A states. The P and N states are obtained from aMD simulations, the A states from Robetta predictions. The A1 and A2 are structures in which the hydrophobic surface between the two helices is disrupted. The cross section of each structure is shown.
n/z = 1/3
The ATDs of n/z = 1/3 (1740 m/z) of the wt and the two mutants show two resolved features at low injection energies (< 40 V) (see Figure 6). The feature at shorter times is assigned to the z = +6 dimer for all peptides based on its cross section. As the injection energy is increased, the hydrophobic interface between the two monomers making up the dimer decreases, leading to a reduction in intensity of the shorter arrival time feature and eventually its total disapprerance at high IE. The remaining peak at longer arrival time is a compact monomer (σexp = 747–761 Å2). The dimer of the D26N mutant is more stable than that of the wt and E21Q mutant, as shown by the ATDs obtained at IE = 40V. At this IE, the monomer is the dominant species for the wt and E21Q mutant whereas the dimer is dominant for the D26N mutant. The feature labeled with an asterisk in the ATDs of the D26N mutant does not correspond to a large oligomer (i.e. tetramer), since its intensity remained unchanged at high injection energies.
DISCUSSION
The E21Q mutation favors a partially folded P state over the native N state. The native state of the E21Q monomer and dimer is also less stable than that of the wt
Far-UV CD spectra (Figure 3) of the three peptides show α-helical character, which is consistent with the theoretical modelling and IM-MS experimental results. aMD simulations (Figure 4) and ion-mobility data obtained from the high resolution ion mobility experiments (Figure 5) reveal that all three peptides can fold into the helix-turn-helix N states. They also show the presence of metastable, partially folded P states with smaller cross sections. The population of P states in the E21Q mutant is higher than in the wt because there is a higher barrier for structural conversion between P and N states of the E21Q mutant compared to the wt, as suggested by aMD boost potentials. We incubated the E21Q peptide for 16 hours at 343K and observed fibrils by EM (data not shown), indicating that the N-state many be formed at higher temperature.
Under high injection voltages and harsher experimental conditions (Figure 6), the P states disappear and a new structure appears which we term a solvent free A state. In the A states, the helix-turn-helix has lost significant hydrophobic contact between the two helices resulting in free movement of the helices and larger cross sections than the N states. Figure 7 presents the model structures for P, N and A states together with their theoretical cross sections obtained from the TJ42–43 and PSA methods.44–45 The A states were obtained from the Robetta structure predictions (see Figure S6).
Injection energy studies also indicate that the E21Q dimer is less stable than those of the wt and D26N mutant. This finding is consistent with the fact that EM results (Figure 2) indicate E21Q has significantly weaker aggregation propensity than the other two peptides.
The formation of an E21-R24 salt bridge is a major contributor to helix-turn-helix folding, while K19-D26 has an opposite effect.
A detailed analysis (see Figure S9) of distances between E/Q21 and R24 indicates that the folding of the helix-turn-helix monomer requires the formation of a stable E21-R24 salt-bridge. When this salt-bridge is formed, all trajectories sample the helix-turn-helix (e.g. traj1 of the wt, traj2 of the E21Q mutant, and both trajectories of the D26N mutant). When the salt-bridge is broken, the trajectories sample other structures (e.g. traj1 of the E21Q mutant, see also Supporting Information Figure S5). Furthermore, it is clear from the simulation that the E21-R24 distance is better preserved than the Q21-R24 distance. As a result, the helix-turn-helix N states are favored by the wt and D26N mutant, whereas there is a strong competition between the N state and partially folded P states as mentioned above.
On the other hand, the K19-D26 distances (Figure S9, right panels), and this range of distances does not specifically correspond to the helix-turn-helix monomer. In fact, the large distances between the two residues imply that the K19-D26 salt bridge does not exist in the stable helix-turn-helix monomer. The only time that this salt bridge exists is in the early stage, but the K19-D26 distance quickly increases when the helix-turn-helix starts to form. This suggests that the K19-D26 salt bridge has a negative effect on the folding since this salt bridge must be severed to form the helix-turn-helix. It is also supported by the trajectories of D26N, in which the K19-N26 distances do not change significantly from the beginning to the end of the trajectory, but the peptide is able to fold into a helix-turn-helix similar to the wt. Thus we conclude that K19-D26 interactions are not required for the folding of helix-turn-helix. However, the D26N mutation removes a charge on the second helix segment, which reduces the overall stability of the helix-turn-helix monomer (as suggested by IM-MS that the monomer of D26N is less stable that that of the wt; Figure 6 and by aMD; Figure 4D), and consequently decrease aggregation propensity as shown the EM and fluorescence data.
Parallel stacking of helix-turn-helix monomers to form a dimer is the most probable pathway for fibril formation
The IM-MS and aMD data show that the significant differences between the non-aggregating E21Q mutant and the fibril-forming wt peptide are the population and stability of the helix-turn-helix N state. We speculate that this state is essential for fibril formation, especially since the far-UV CD spectra shows no β-sheet character (Figure 3) and the fibrils were previously shown to be non-amyloid.20 We construct some structures based on the models shown in Figure 8 and compare their cross sections with experimental cross sections of the z = +5 and +6 dimers (see Figure 6 and Figure S10). These structures are built based on the helix-turn-helix monomers obtained from the aMD simulation of the wt and subjected to short minimization. Because the native helix-turn-helix creates a hydrophobic surface between the two helices and two hydrophilic surfaces on the outside (see Supporting Information Figure S4), there are four possible arrangements for the dimer in which the helix-turn-helix N state is preserved. The first two models have the two monomers stabilized by charged surfaces formed by two inter-molecular helices running parallel or anti-parallel to each other (see Figure 8, models A and B). However, the cross sections of these model structures are significantly larger than the experimental values (the models have cross sections σav = 1499 ± 32 Å2 and 1468 ± 27 Å2; see Figure 7 and Figure S10, whereas experimental values are from 1214–1248 Å2; Figure 8). In the last two models, the two monomers interact face-to-back through hydrophobic surfaces, in which the two helix-turn-helix monomers are stacked on top of each other; either parallel or anti-parallel (Figure 8, models C and D). The structures have cross sections similar to the experimental cross sections of both dimers z = +5 and +6 (model C: σav = 1282 ± 17 Å2 and model D: σav = 1286 ± 26 Å2). The native N state creates a strong hydrophobic core in the middle of the two helices, allowing another helix-turn-helix to stack below (or above) it so that the whole system can further elongate. This hydrophobic surface is vital to the aggregation of these helix-turn-helix peptides. The results from standard explicit MD simulations of the wt dimers suggest that parallel stacking is more stable than antiparallel stacking (see Figure S11 for the center-of-mass distances between the two monomers as a function of simulation time). Therefore, we suggest that parallel stacking of two monomers on top of each other, rather than elongation through side-by-side interactions in other directions, initiates the fibril formation process.
Figure 8.

Four dimer models showing possible helix-turn-helix arrangements to form a dimer. Each helix is shown as a rectangle with the hydrophobic face color coded in green and the hydrophilic face exposed to solution in red. In models A and B, two monomers interact through hydrophilic surfaces in an anti-parallel and a parallel face-to-back manner, respectively. The face-to-back implies the top surface of a monomer is interacting with the back surface of another monomer. In models C and D, two monomers are stacked on top of each other, and interact through hydrophobic surfaces in the middle of the helix-turn-helix monomers.
Next, we compare our proposed pathway with previously published X-ray fiber diffraction data from Lazar et al.20 In the monomer within the dimer obtained from MD simulations, the average center-of-mass intramolecular distance between backbone Cα atoms of the two helices inside the helix-turn-helix is approximately 10 Å. The 10 Å distance between the two helices of the same monomer resembles the same distance predicted from X-ray fiber diffraction (9.5 Å). The region near the turn can expand to a maximum distance of 20 Å while previous X-ray fiber diffraction data suggest 16 Å (see Figure S12). In the dimer, the parallel growing motif leads to protofibrils with a diameter equal to the length of the helical seam of the monomer; approximately 30–32 Å, in agreement with the diameter of the fibrils measured from previous and current EM imaging.20 Overall, the intra- and intermolecular distances are qualitatively consistent with the reflections obtained from X-ray fiber diffraction used to construct the previous model. The major difference lies in the arrangement of the dimer. The model proposed by Lazar et al. had the two helix-turn-helix monomers stacked on top of each other in an anti-parallel manner, rather than the parallel manner shown here. A revised model of the peptide fibrils showing the parallel arrangement is shown in Figure 9. The previous X-ray data cannot distinguish between parallel and anti-parallel arrangements.
Figure 9.
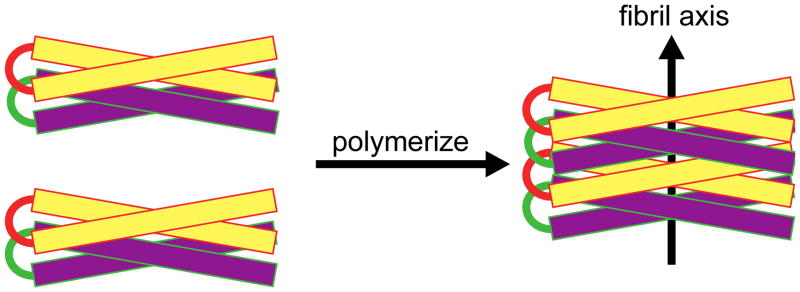
Model of peptide fibrils. The α-helical axes are perpendicular to the long axis of the fibril. Helix-turn-helix dimers are shown stacking in a parallel manner to grow the individual fibril. For clarity, two colors (yellow and purple) are used to show the helix-turn-helix peptides stacking.
CONCLUSIONS
Some conclusions can be drawn from the comparison between modeling distance measurements, IM-MS data, and the previous X-ray fiber diffraction data.20
The formation of the helix-turn-helix monomer correlates with non-β fibrillization of the wt and D26N mutant peptides.
The turn region is essential for aggregation. The 18A monomer adopts an α-helix, but the dimer is globular, preventing fibrillization from occurring.
In the process of dimerization, a helix-turn-helix with a “short turn” (as seen in the aMD simulations of the monomers) undergoes some conformational transitions to elongate the turn and separate the two helices inside the monomer (to a maximum distance of 20 Å). This occurs without disrupting the hydrophobic surface needed to stabilize the native state, allowing another helix-turn-helix monomer to make contact. This transition can occur due to the presence of the E21-R24 salt bridge. The E21Q mutation significantly weakens this salt bridge and destabilizes the helix-turn-helix.
From the basis of dimer stability and comparison between models and experimental IM-MS dimer cross sections, the parallel stacking of the two monomers initiates the fiber formation process instead of the elongation through side-by-side interactions in other directions (Figure 8).
According to the IM-MS and aMD data, the wt and D26N peptides favor the native helix-turn-helix N state monomers, whereas the E21Q peptide prefers the partially folded P state. The E21Q mutant cannot aggregate into fibrils unless it is heated to a higher temperature (343K) to overcome the barrier between the P and N states.
The E21-R24 salt-bridge is very important for helix-turn-helix formation, whereas the K19-D26 salt bridge appears to be as a negative factor. Consequently, the wt peptide and the D26N mutant can aggregate into fibrils. The mutation on the turn (E21Q) has a more profound impact towards the aggregation propensities of the peptides than the mutation on the helical sides (D26N).
The current work illustrates that the helix-turn-helix motif is essential in the fibrillogenesis of apo A-I mimetic peptides. Because the helix-turn-helix motif is observed in the C-terminal region of native apo A-I, and α-helical fibrils have been observed in some C-terminal apo A-I mutants, we predict that the C-terminal helix-turn-helix motif in apo A-I is essential for α-helical fibrillogenesis.
Supplementary Material
Acknowledgments
We thank Yimei Chen in the Electron Microscopy Facility at the University of Chicago for help collecting EM images and Stanley Anderson for help with PSA calculations.
Funding sources
We gratefully acknowledge support from the National Science Foundation grants CHE-1301032 (M.T.B) and MCB-1158577 (J.-E.S), the National Institutes of Health grant 1RO1AG047116-01 (M.T.B), the David and Lucile Packard Foundation (J.-E.S), the Westmont Provost’s Office for summer research funding (A.B, D.C, T.V, H.T, N.D and B.T) and the Allan Nishimura Summer Research Fund (K.L.C). This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number OCI-1053575. The authors acknowledge the Texas Advanced Computing Center (TACC) at The University of Texas at Austin for providing HPC resources through the XSEDE grant number TG-MCA05S027 (J.-E.S.). We acknowledge support from the Center for Scientific Computing at the CNSI and MRL: an NSF MRSEC (DMR-1121053) and NSF CNS-0960316.
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information. aMD simulation protocol, intrinsic tryptophan fluorescence, additional Figures showing the theoretical structures of the wt obtained from aMD and in-vacuo T-REMD simulations, additional ATDs, models of the dimer and a plot of RMSD fluctuation as a function of time obtained from explicit solvent simulations of the dimers. A movie showing the folding of the wt peptide into a helix-turn-helix structure. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hardy J, Selkoe DJ. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 2.Ittner LM, Gotz J. Amyloid-Beta and Tau--a Toxic Pas De Deux in Alzheimer’s Disease. Nat Rev Neurosci. 2011;12:65–72. doi: 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- 3.Maries E, Dass B, Collier TJ, Kordower JH, Steece-Collier K. The Role of Alpha-Synuclein in Parkinson’s Disease: Insights from Animal Models. Nature Reviews Neuroscience. 2003;4:727–738. doi: 10.1038/nrn1199. [DOI] [PubMed] [Google Scholar]
- 4.de la Paz ML, Goldie K, Zurdo J, Lacroix E, Dobson CM, Hoenger A, Serrano L. De Novo Designed Peptide-Based Amyloid Fibrils. Proc Natl Acad Sci USA. 2002;99:16052–16057. doi: 10.1073/pnas.252340199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Do TD, Economou NJ, LaPointe NE, Kincannon WM, Bleiholder C, Feinstein SC, Teplow DB, Buratto SK, Bowers MT. Factors That Drive Peptide Assembly and Fibril Formation: Experimental and Theoretical Analysis of Sup35 Nnqqny Mutants. J Phys Chem B. 2013;117:8436–8446. doi: 10.1021/jp4046287. [DOI] [PubMed] [Google Scholar]
- 6.von Bergen M, Barghorn S, Biernat J, Mandelkow EM, Mandelkow E. Tau Aggregation Is Driven by a Transition from Random Coil to Beta Sheet Structure. Bba-Mol Basis Dis. 2005;1739:158–166. doi: 10.1016/j.bbadis.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 7.Celej MS, Sarroukh R, Goormaghtigh E, Fidelio GD, Ruysschaert JM, Raussens V. Toxic Prefibrillar Alpha-Synuclein Amyloid Oligomers Adopt a Distinctive Antiparallel Beta-Sheet Structure. Biochem J. 2012;443:719–726. doi: 10.1042/BJ20111924. [DOI] [PubMed] [Google Scholar]
- 8.Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, Elliott JI, Nostrand WEV, Smith SO. Structural Conversion of Neurotoxic Amyloid-B1–42 Oligomers to Fibrils. Nat Struct Mol Biol. 2010;17:561–567. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldschmidt L, Teng PK, Riek R, Eisenberg D. Identifying the Amylome, Proteins Capable of Forming Amyloid-Like Fibrils. Proc Natl Acad Sci USA. 2010;107:3487–3492. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colletiera J-P, Laganowskya A, Landaua M, Zhaoa M, Soriagaa AB, Goldschmidta L, Flotb D, Cascioa D, Sawayaa MR, Eisenberga D. Molecular Basis for Amyloid-B Polymorphism. Proc Natl Acad Sci USA. 2011:16938–16943. doi: 10.1073/pnas.1112600108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eisenberg D, Jucker M. The Amyloid State of Proteins in Human Diseases. Cell. 2012;148:1188–1203. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang LL, Che MX, Zhao J, Zhou CJ, Xie MY, Li HY, He JH, Hu HY. Structural Transformation of the Amyloidogenic Core Region of Tdp-43 Protein Initiates Its Aggregation and Cytoplasmic Inclusion. J Biol Chem. 2013;288:19614–19624. doi: 10.1074/jbc.M113.463828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sivanandam VN, Jayaraman M, Hoop CL, Kodali R, Wetzel R, van der Wel PCA. The Aggregation-Enhancing Huntingtin N-Terminus Is Helical in Amyloid Fibrils. J Am Chem Soc. 2011;133:4558–4566. doi: 10.1021/ja110715f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sadqi M, Hernandez F, Pan UM, Perez M, Schaeberle MD, Avila J, Munoz V. Alpha-Helix Structure in Alzheimer’s Disease Aggregates of Tau-Protein. Biochemistry. 2002;41:7150–7155. doi: 10.1021/bi025777e. [DOI] [PubMed] [Google Scholar]
- 15.Pannuzzo M, Raudino A, Milardi D, La Rosa C, Karttunen M. Alpha-Helical Structures Drive Early Stages of Self-Assembly of Amyloidogenic Amyloid Polypeptide Aggregate Formation in Membranes. Sci Rep. 2013;3:2781. doi: 10.1038/srep02781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoshino T, Mahmood MI, Mori K, Matsuzaki K. Binding and Aggregation Mechanism of Amyloid Beta-Peptides onto the Gm1 Ganglioside-Containing Lipid Membrane. J Phys Chem B. 2013;117:8085–8094. doi: 10.1021/jp4029062. [DOI] [PubMed] [Google Scholar]
- 17.Georgieva ER, Xiao S, Borbat PP, Freed JH, Eliezer D. Tau Binds to Lipid Membrane Surfaces Via Short Amphipathic Helices Located in Its Microtubule-Binding Repeats. Biophys J. 2014;107:1441–1452. doi: 10.1016/j.bpj.2014.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pertinhez TA, Bouchard M, Tomlinson EJ, Wain R, Ferguson SJ, Dobson CM, Smith LJ. Amyloid Fibril Formation by a Helical Cytochrome. FEBS Lett. 2001;495:184–186. doi: 10.1016/s0014-5793(01)02384-5. [DOI] [PubMed] [Google Scholar]
- 19.Fezoui Y, Hartley DM, Walsh DM, Selkoe DJ, Osterhout JJ, Teplow DB. A De Novo Designed Helix-Turn-Helix Peptide Forms Nontoxic Amyloid Fibrils. Nat Struct Biol. 2000;7:1095–1099. doi: 10.1038/81937. [DOI] [PubMed] [Google Scholar]
- 20.Lazar KL, Miller-Auer H, Getz GS, Orgel JPRO, Meredith SC. Helix-Turn-Helix Peptides That Form Alpha-Helical Fibrils: Turn Sequences Drive Fibril Structure. Biochemistry. 2005;44:12681–12689. doi: 10.1021/bi0509705. [DOI] [PubMed] [Google Scholar]
- 21.Brouillette CG, Anantharamaiah GM, Engler JA, Borhani DW. Structural Models of Human Apolipoprotein a-I: A Critical Analysis and Review. BbA-Mol Cell Biol L. 2001;1531:4–46. doi: 10.1016/s1388-1981(01)00081-6. [DOI] [PubMed] [Google Scholar]
- 22.Hatters DM, Zhong N, Rutenber E, Weisgraber KH. Amino-Terminal Domain Stability Mediates Apolipoprotein E Aggregation into Neurotoxic Fibrils. J Mol Biol. 2006;361:932–944. doi: 10.1016/j.jmb.2006.06.080. [DOI] [PubMed] [Google Scholar]
- 23.Kirkitadze MD, Condron MM, Teplow DB. Identification and Characterization of Key Kinetic Intermediates in Amyloid Beta-Protein Fibrillogenesis. J Mol Biol. 2001;312:1103–1119. doi: 10.1006/jmbi.2001.4970. [DOI] [PubMed] [Google Scholar]
- 24.Mangione P, Sunde M, Giorgetti S, Stoppini M, Esposito G, Gianelli L, Obici L, Asti L, Andreola A, Viglino P, Merlini G, Bellotti V. Amyloid Fibrils Derived from the Apolipoprotein A1 Leu174ser Variant Contain Elements of Ordered Helical Structure. Protein Sci. 2001;10:187–199. doi: 10.1110/ps.29201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petrlova J, Duong T, Cochran MC, Axelsson A, Morgelin M, Roberts LM, Lagerstedt JO. The Fibrillogenic L178h Variant of Apolipoprotein a-I Forms Helical Fibrils. J Lipid Res. 2012;53:390–398. doi: 10.1194/jlr.M020883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anantharamaiah GM, Jones JI, Brouillette CG, Schmidt CF, Chung BH, Hughes TA, Bhown AS, Segrest JP. Studies of Synthetic Peptide Analogs of the Amphipathic Helix - Structure of Complexes with Dimyristoyl Phosphatidylcholine. J Biol Chem. 1985;260:248–255. [PubMed] [Google Scholar]
- 27.Morriset JD, David JSK, Pownall HJ, Gotto AM. Interaction of an Apolipoprotein (Apolp-Alanine) with Phosphatidylcholine. Biochemistry. 1973;12:1290–1299. doi: 10.1021/bi00731a008. [DOI] [PubMed] [Google Scholar]
- 28.Kemper PR, Dupuis NF, Bowers MT. A New, Higher Resolution, Ion Mobility Mass Spectrometer. Int J Mass Spectrom. 2009;287:46–57. [Google Scholar]
- 29.Wyttenbach T, Kemper PR, Bowers MT. Design of a New Electrospray Ion Mobility Mass Spectrometer. Int J Mass Spectrom. 2001;212:13–23. [Google Scholar]
- 30.Mason EA. Transport Properties of Ions in Gases. 99. John Wiley & Sons; 1988. [Google Scholar]
- 31.Bushnell JE, Kemper PR, Bazan GC, Bowers MT. The Determination of Cis-Trans Conformations in Tetrahedral P-Phenylene Vinylene Oligomers. J Phys Chem A. 2004;108:7730–7735. [Google Scholar]
- 32.Lindorff-Larsen K, Piana S, Dror RO, Shaw DE. How Fast-Folding Proteins Fold. Science. 2011;334:517–520. doi: 10.1126/science.1208351. [DOI] [PubMed] [Google Scholar]
- 33.Levitt M, Warshel A. Computer-Simulation of Protein Folding. Nature. 1975;253:694–698. doi: 10.1038/253694a0. [DOI] [PubMed] [Google Scholar]
- 34.Zwier MC, Chong LT. Reaching Biological Timescales with All-Atom Molecular Dynamics Simulations. Curr Opin Pharmacol. 2010;10:745–752. doi: 10.1016/j.coph.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 35.Weber JK, Pande VS. Protein Folding Is Mechanistically Robust. Biophys J. 2012;102:859–867. doi: 10.1016/j.bpj.2012.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Faradjian AK, Elber R. Computing Time Scales from Reaction Coordinates by Milestoning. J Chem Phys. 2004;120:10880–10889. doi: 10.1063/1.1738640. [DOI] [PubMed] [Google Scholar]
- 37.Hamelberg D, Mongan J, McCammon JA. Accelerated Molecular Dynamics: A Promising and Efficient Simulation Method for Biomolecules. J Chem Phys. 2004;120:11919–11929. doi: 10.1063/1.1755656. [DOI] [PubMed] [Google Scholar]
- 38.Voter AF. Hyperdynamics: Accelerated Molecular Dynamics of Infrequent Events. Phys Rev Lett. 1997;78:3908–3911. [Google Scholar]
- 39.Pierce LCT, Salomon-Ferrer R, de Oliveira CAF, McCammon JA, Walker RC. Routine Access to Millisecond Time Scale Events with Accelerated Molecular Dynamics. J Chem Theory Comput. 2012;8:2997–3002. doi: 10.1021/ct300284c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sugita Y, Okamoto Y. Replica-Exchange Molecular Dynamics Method for Protein Folding. Chem Phys Lett. 1999;314:141–151. [Google Scholar]
- 41.Morriss-Andrews A, Shea JE. Computational Studies of Protein Aggregation: Methods and Applications. Ann Rev Phys Chem. 2015;66:643–666. doi: 10.1146/annurev-physchem-040513-103738. [DOI] [PubMed] [Google Scholar]
- 42.Mesleh MF, Hunter JM, Shvartsburg AA, Schatz GC, Jarrold MF. Structural Information from Ion Mobility Measurements: Effects of the Long Range Potential. J Phys Chem A. 1996;100:16082–16086. [Google Scholar]
- 43.Shvartsburg AA, Jarrold MF. An Exact Hard-Spheres Scattering Model for the Mobilities of Polyatomic Ions. Chem Phys Lett. 1996;261:86–91. [Google Scholar]
- 44.Bleiholder C, Wyttenbach T, Bowers MT. A Novel Projection Approximation Algorithm for the Fast and Accurate Computation of Molecular Collision Cross Sections (I). Method. Int J Mass Spectrom. 2011;308:1–10. [Google Scholar]
- 45.Bleiholder C, Contreras S, Do TD, Bowers MT. A Novel Projection Approximation Algorithm for the Fast and Accurate Computation of Molecular Collision Cross Sections (Ii). Parameterization and Application to Biomolecules. Int J Mass Spectrom. 2013;345–347:89–96. [Google Scholar]
- 46.Anderson SE, Bleiholder C, Brocker ER, Stang PJ, Bowers MT. A Novel Projection Approximation Algorithm for the Fast and Accurate Computation of Molecular Collision Cross Sections (Iii): Application to Supramolecular Coordination-Driven Assemblies with Complex Shapes. Int J Mass Spectrom. 2012;330:78–84. [Google Scholar]
- 47.Bleiholder C, Contreras S, Bowers MT. A Novel Projection Approximation Algorithm for the Fast and Accurate Computation of Molecular Collision Cross Sections (Iv). Application to Polypeptides. Int J Mass Spectrom. 2013;354:275–280. [Google Scholar]
- 48.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman P. A Point-Charge Force field for Molecular Mechanics Simulations of Proteins Based on Condensed-Phase Quantum Mechanical Calculations. J Comp Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 49.Lei H, Wu C, Wang ZX, Zhou Y, Duan Y. Folding Processes of the B Domain of Protein a to the Native State Observed in All-Atom Ab Initio Folding Simulations. J Chem Phys. 2008;128:235105. doi: 10.1063/1.2937135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lei H, Duan Y. Two-Stage Folding of Hp-35 from Ab Initio Simulations. J Mol Biol. 2007;370:196–206. doi: 10.1016/j.jmb.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lei H, Wu C, Liu H, Duan Y. Folding Free-Energy Landscape of Villin Headpiece Subdomain from Molecular Dynamics Simulations. Proc Natl Acad Sci USA. 2007;104:4925–4930. doi: 10.1073/pnas.0608432104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Case DA, Darden TA, Cheatham TE, I, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Wang B, Hayik S, Roitberg A, Seabra G, Kolossváry I, Wong KF, Paesani F, Vanicek J, Liu J, Wu XS, Brozell R, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M-J, Cui G, Roe DR, Mathews DH, Seetin MG, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. Amber. Vol. 12. University of California; San Francisco: 2012. [Google Scholar]
- 53.Mishra VK, Anantharamaiah GM, Segrest JP, Palgunachari MN, Chaddha M, Sham SW, Krishna NR. Association of a Model Class a (Apolipoprotein) Amphipathic Alpha Helical Peptide with Lipid: High Resolution Nmr Studies of Peptide. Lipid Discoidal Complexes. J Biol Chem. 2006;281:6511–6519. doi: 10.1074/jbc.M511475200. [DOI] [PubMed] [Google Scholar]
- 54.Dupuis NF, Wu C, Shea JE, Bowers MT. Human Islet Amyloid Polypeptide Monomers Form Ordered B-Hairpins: A Possible Amyloidogenic Conformation. J Am Chem Soc. 2009;131:18283–18292. doi: 10.1021/ja903814q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Daura X, Gademann K, Jaun B, Seebach D, Gunsteren WFv, Mark AE. Peptide Folding: When Simulation Meets Experiment. Angew Chem Int Ed. 1999;38:236–240. [Google Scholar]
- 56.Abraham MJ, Gready JE. Optimization of Parameters for Molecular Dynamics Simulation Using Smooth Particle-Mesh Ewald in Gromacs 4.5. J Comput Chem. 2011;32:2031–2040. doi: 10.1002/jcc.21773. [DOI] [PubMed] [Google Scholar]
- 57.Hess B, Kutzner C, Spoel Dvd, Lindahl E. Gromacs 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput. 2008;4:435–437. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 58.Spoel DVD, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. Gromacs: Fast, Flexible, and Free. J Comp Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 59.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 60.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. Lincs: A Linear Constraint Solver for Molecular Simulations. J Comput Chem. 1997;18:1463–1472. [Google Scholar]
- 61.Miyamoto S, Kollman PA. Settle - an Analytical Version of the Shake and Rattle Algorithm for Rigid Water Models. J Comput Chem. 1992;13:952–962. [Google Scholar]
- 62.Darden T, York D, Pedersen L. Particle Mesh Ewald: An N·Log(N) Method for Ewald Sums in Large Systems. J Chem Phys. 1993;98:10089–10093. [Google Scholar]
- 63.Braga C, Travis KP. A Configurational Temperature Nose-Hoover Thermostat. J Chem Phys. 2005;123:134101. doi: 10.1063/1.2013227. [DOI] [PubMed] [Google Scholar]
- 64.Kim DE, Chivian D, Baker D. Protein Structure Prediction and Analysis Using the Robetta Server. Nucleic Acids Res. 2004;32:W526–W531. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chivian D, Kim DE, Malmstrom L, Bradley P, Robertson T, Murphy P, Strauss CEM, Bonneau R, Rohl CA, Baker D. Automated Prediction of Casp-5 Structures Using the Robetta Server. Proteins-Structure Function and Bioinformatics. 2003;53:524–533. doi: 10.1002/prot.10529. [DOI] [PubMed] [Google Scholar]
- 66.Mishra VK, Palgunachari MN. Interaction of Model Class A1, Class A2, and Class Y Amphipathic Helical Peptides with Membranes. Biochemistry. 1996;35:11210–11220. doi: 10.1021/bi960760f. [DOI] [PubMed] [Google Scholar]
- 67.Dusa A, Kaylor J, Edridge S, Bodner N, Hong DP, Fink AL. Characterization of Oligomers During Alpha-Synuclein Aggregation Using Intrinsic Tryptophan Fluorescence. Biochemistry. 2006;45:2752–2760. doi: 10.1021/bi051426z. [DOI] [PubMed] [Google Scholar]
- 68.Hristova K, Wimley WC, Mishra VK, Anantharamiah GM, Segrest JP, White SH. An Amphipathic Alpha-Helix at a Membrane Interface: A Structural Study Using a Novel X-Ray Diffraction Method. J Mol Biol. 1999;290:99–117. doi: 10.1006/jmbi.1999.2840. [DOI] [PubMed] [Google Scholar]
- 69.West MW, Wang W, Patterson J, Mancias JD, Beasley JR, Hecht MH. De Novo Amyloid Proteins from Designed Combinatorial Libraries. Proc Natl Acad Sci USA. 1999;96:11211–11216. doi: 10.1073/pnas.96.20.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.