

Abstract
Precursor messenger RNA (pre-mRNA) splicing is a critical step in the posttranscriptional regulation of gene expression, providing significant expansion of the functional proteome of eukaryotic organisms with limited gene numbers. Split eukaryotic genes contain intervening sequences or introns disrupting protein-coding exons, and intron removal occurs by repeated assembly of a large and highly dynamic ribonucleoprotein complex termed the spliceosome, which is composed of five small nuclear ribonucleoprotein particles, U1, U2, U4/U6, and U5. Biochemical studies over the past 10 years have allowed the isolation as well as compositional, functional, and structural analysis of splicing complexes at distinct stages along the spliceosome cycle. The average human gene contains eight exons and seven introns, producing an average of three or more alternatively spliced mRNA isoforms. Recent high-throughput sequencing studies indicate that 100% of human genes produce at least two alternative mRNA isoforms. Mechanisms of alternative splicing include RNA–protein interactions of splicing factors with regulatory sites termed silencers or enhancers, RNA–RNA base-pairing interactions, or chromatin-based effects that can change or determine splicing patterns. Disease-causing mutations can often occur in splice sites near intron borders or in exonic or intronic RNA regulatory silencer or enhancer elements, as well as in genes that encode splicing factors. Together, these studies provide mechanistic insights into how spliceosome assembly, dynamics, and catalysis occur; how alternative splicing is regulated and evolves; and how splicing can be disrupted by cis- and trans-acting mutations leading to disease states. These findings make the spliceosome an attractive new target for small-molecule, antisense, and genome-editing therapeutic interventions.
Keywords: intron, exon, pre-mRNA splicing, RNA-binding proteins, RNA structure, spliceosome, genomics, disease, splicing factors, enhancers, silencers
Introduction
One of the most unanticipated findings in molecular biology was the discovery that eukaryotic genes are discontinuous, with protein-coding segments or exons disrupted by noncoding segments or introns (1, 2). With advances in genome sequencing, it has become apparent that precursor messenger RNA (pre-mRNA) splicing can occur to a great extent that scales with organismal complexity (3, 4). Indeed, although the mouse and human genomes contain similar numbers of genes, alternative pre-mRNA splicing occurs in >95 to 100% of human genes, compared with ∼63% of mouse genes (Table 1) (5, 6). Thus, one function of alternative splicing is to significantly expand the form and function of the human proteome (7–9). Indeed, alternative splicing can serve many regulatory functions, from sex determination and diversity of neuronal wiring in the fruit fly to determination of the physiological function of membrane-bound receptors in the mammalian nervous system (10).
Table 1. Comparative genomics of splicing levels in several well-studied metazoansa.
| Humanb | Mouseb | Flyc | Wormc | |
|---|---|---|---|---|
| Genome size | 3,300 MB | 3,300 MB | 165 MB | 100 MB |
| Protein-coding genes | 22,180 | 22,740 | 13,937 | 20,541 |
| Multiexonic genes (percentage with 2+ isoforms) | 21,144 (88%) | 19,654 (63%) | 11,767 (45%) | 20,008 (25%) |
| Isoforms (average number per gene) | 215,170 (3.4) | 94,929 (2.4) | 29,173 (1.9) | 56,820 (1.2) |
| Average number of unique exons per gene (median) | 33 (26) | 22 (15) | 7.5 (4) | 8.6 (6) |
| Average number of unique introns per multiexonic gene (median) | 28 (21) | 19 (12) | 8.7 (5) | 7.2 (5) |
| Average exon length (median length) | 320 bp (145 bp) | 323 bp (141 bp) | 494 bp (272 bp) | 222 bp (157 bp) |
| Average intron length (median length) | 7,563 bp (1,964 bp) | 6,063 bp (1,693 bp) | 2,068 bp (642 bp) | 561 bp (354 bp) |
| Genes (all) | 63,677 | 39,179 | 15,682 | 46,726 |
| Isoforms (all) (average number per gene) | 215,170 (3.4) | 94,929 (2.4) | 29,173 (1.9) | 56,820 (1.2) |
On the basis of both initial (92, 93) and more recent deep (5, 6) RNA-sequencing (RNA-seq) data, 95% (92, 93) to 100% (5, 6) of human genes may encode two or more (2+) isoforms, and other vertebrates, especially primates, may be similar in that most of those genes also encode 2+ isoforms (5, 6). Relevant Drosophila RNA-seq data are from References 255 and 256, and relevant Caenorhabditis elegans RNA-seq data are from Reference 257.
The numbers are based on annotations from Ensembl (which does not use RNA-seq data for annotations). For current Ensembl versions of human and mouse gene/transcriptome annotations, see http://uswest.ensembl.org/Homo_sapiens/Info/Annotation and http://uswest.ensembl.org/Mus_musculus/Info/Annotation.
The Drosophila and C. elegans gene/transcriptome annotations were imported from FlyBASE and WormBASE, respectively; see http://uswest.ensembl.org/Drosophila_melanogaster/Info/Annotation and http://uswest.ensembl.org/Caenorhabditis_elegans/Info/Annotation.
Biochemical studies have demonstrated that the RNA cleavage and ligation reactions necessary for intron removal in protein-coding mRNAs (and long noncoding RNAs) occur in a large ribonucleoprotein (RNP) machine called the spliceosome (11, 12). The spliceosome functions in a complex and dynamic assembly, reaction, and disassembly cycle in which five small nuclear ribonucleoprotein (snRNP) complexes (U1, U2, U4/U6, and U5) recognize and assemble on each intron to ultimately form a catalytically active spliceosome (Figure 1). Over the past decade, remarkable progress has been made to isolate, purify, and characterize the protein composition and biochemical activities and to determine the structures of several of these distinct forms of the spliceosome as they proceed along the reaction pathway. The catalytic center of the spliceosome is also composed of RNA (13), so we can now definitively say that the spliceosome is a ribozyme, like the ribosome.
Figure 1.
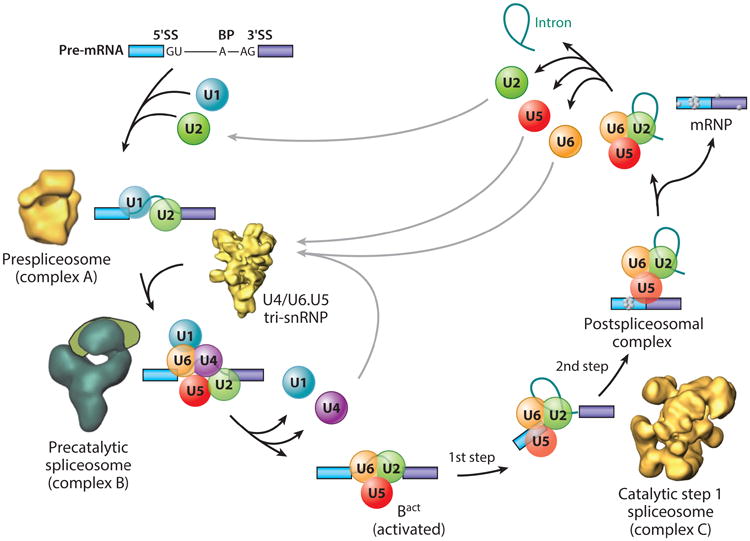
The spliceosome assembly and disassembly cycle, with known structures of individual complexes, as well as the cross-intron assembly and disassembly of the major (U1 and U2) spliceosome. Also depicted is the stepwise interaction of the spliceosomal small nuclear ribonucleoprotein (snRNP) particles (U1, U2, U4, U5, and U6) (colored circles) in the removal of an intron from a precursor messenger RNA (pre-mRNA) containing two exons (blue and purple); non-snRNP proteins are not shown. The spliceosomal complexes that can be resolved biochemically in mammalian splicing extracts are shown. The names of the complexes, as well as the first and second catalytic steps, are indicated. Also shown are the electron microscopy–derived structures of the purified prespliceosome (complex A) (56), the U4/U6.U5 tri-snRNP (249), the precatalytic spliceosome (complex B) (61, 250), and the catalytic step 1 spliceosome (complex C) (52, 60). Abbreviation: SS, splice site. Modified with permission from Reference 12.
Regarding alternative splicing, both single-gene and genome-wide methods have led to important insights into how alternative splicing patterns are set up and maintained in particular cell or tissue types (9, 14, 15). The role of cis-acting regulatory sequences and RNA-binding protein splicing factors that recognize and bind to these sites compose a common mechanism for setting up and maintaining alternative splicing patterns. These sites can be either intronic or exonic and can be positive (splicing enhancers) or negative (splicing silencers). In addition to RNA–protein recognition, RNA–RNA base pairing can specify site use, as is the case for the mutually exclusive exon 6 cluster in the Drosophila DSCAM gene (16). RNA–RNA base pairing can also occur in trans, exemplified by the small nucleolar RNA (snoRNA) HBII–5252B RNA, which regulates the serotonin receptor 2C transcript (17, 18). Finally, connections have been made between chromatin modifications (19–21), small RNA pathway components (Argonaute family members) (22–24), RNA polymerase II speed, and alternative splicing patterns (25–27).
Errors in alternative splicing can also lead to disease states (28, 29). Many cis-acting mutations in mapped human and mouse disease genes cause defects in pre-mRNA splicing, whether the mutations map at the intron–exon junction splice sites or at more remote sites, such as enhancers or silencers located in exons or introns (30–35). Moreover, for types of myeloid hematopoietic malignancies, especially myelodysplastic syndrome and chronic lymphocytic leukemia, mutations in 3′ splice-site recognition factors, such as U2AF and SF3b, are linked to disease in cancer patients (36–39). A variety of therapeutic strategies, such as small molecules (40, 41) and antisense oligonucleotides (42–44), as well as genome editing using CRISPR/Cas9 (45, 46), show promise for future intervention to ameliorate the diseasing-causing effects of human mutations on patterns of alternative splicing. Detailed biochemical knowledge of the spliceosome and how alternative patterns of splicing are set up and regulated will provide crucial information that can be used in these therapeutic endeavors.
Biochemistry of the Spliceosome
The spliceosome is a large and highly dynamic RNP machine. Biochemical purification and characterization of active splicing complexes have illuminated our understanding of the steps in the spliceosome cycle. They have also enabled the structural analysis of these staged complexes using electron microscopy methods (11, 12).
Spliceosome Purification, Assembly, Composition, and Structure
Spliceosome assembly needs to occur repeatedly every time an intron is removed from a pre-mRNA in a eukaryotic nucleus. Yeast and human spliceosomes have sedimentation values of 40 to 60S and masses of ∼4.8 MDa (11, 12). Many studies have described the stepwise assembly for the spliceosome, from E to A, to B, to Bact/B*, to C, to postspliceosomal complexes, and to the ultimate release of the intron lariat RNA, followed by snRNP recycling (Figure 1). The biochemistry of these ordered events has been intensively studied (11, 12). Among the most significant developments over the past decade relating to the biochemistry of the spliceosome have been the development and use of affinity purification, depletion, and reconstitution methods and other biochemical tricks to isolate and characterize spliceosomal complexes at distinct stages along the spliceosomal assembly pathway. The affinity purification methods have involved the use of “epitope-tagged” RNA substrates containing either a tobramycin RNA aptamer (47) or binding sites for the bacteriophage MS2 coat RNA-binding protein (48–50). These RNA substrates are first bound to immobilized tobramycin resin or a purified recombinant maltose-binding protein—phage MS2 coat fusion protein—and these RNAs are then incubated in splicing extracts from human, Drosophila (51), or yeast (52) cells. Following fractionation of the spliceosomal complexes by either gel filtration chromatography or velocity sedimentation in glycerol gradients, the RNA-protein complexes can be affinity-purified and analyzed by gel electrophoresis and/or mass spectrometry for protein and RNA composition and, in some cases, for catalytic activity. Collectively, these studies have provided an appreciation for the large, diverse, and dynamic protein composition of the spliceosome (>200 proteins in metazoans; ∼100 in yeast) (53) and also of how the protein composition of the spliceosome dynamically changes as the assembly and subsequent catalytic steps occur (11, 12).
Most interesting has been the ability through careful purification and analytical biochemistry to detect distinct proteins that either join or exit defined complexes at discrete places in the spliceosome cycle. In addition to the RNA–RNA interactions in the spliceosome, an extensive network of protein–protein interactions has been characterized (54). In some cases, biochemical depletion–reconstitution reactions have provided a biochemical assay to determine the function of specific proteins in, for instance, the transition from B to Bact/B*, where U1 and U4 snRNPs are ejected from the complex and ATP and GTP are hydrolyzed by the Brr2 and Snu114 proteins, respectively (55). As is the case for the ribosome, the use of ATP and/or GTP hydrolysis is used both to drive structural transitions and as a “proofreading” mechanism. It is clear that a number of DExD/H-box ATPases facilitate structural rearrangements in the spliceosome.
One of the underlying rationales for the purification of discrete splicing complexes is to determine the structure of these defined intermediates along the splicing pathway. Because of the dynamic nature of the spliceosome, structural biologists have largely turned to electron microscopy to assess the structures of a variety of spliceosomal complexes as well as isolated U snRNPs (Figure 1). As these studies have progressed, increasingly higher resolution structures have been determined for the A (56), B (57), Bact/B* (58), and C (59) complexes. Initial low-resolution (25-Å) studies on isolated C complexes (catalytically active, step I–blocked spliceosomes) produced a picture of three distinct structural domains with a diameter of ∼280 Å (60). Initial studies on the BΔU1 complex at ∼40-Å resolution revealed a stable triangular domain of ∼300-Å diameter linked via a flexible region to an upper domain (61). This upper head domain was found in various orientations with respect to the rest of the particle. Recall that the BΔU1 complex represents a precatalytic spliceosome and differs significantly in protein composition from the activated Bact/B* spliceosome (12). Biochemical comparisons of the human B complex (40S), Bact/B* complex (45S) (58), and C complex (40S) have indicated that the transition from B to Bact is accompanied by the loss of U1 and U4 snRNAs and of ∼35 proteins with the addition of 12 new proteins (51). The transition from Bact to C is accompanied by the loss of two proteins and the addition of nine new proteins (12).
Concomitant with these rearrangements and compositional changes are alterations in the electron microscopy images of these complexes (Figure 1). Interestingly, comparative biochemical analyses of human and Drosophila splicing complexes show remarkably similar protein compositions (51); yeast have similar complexes, but they contain fewer proteins (12, 62). Additional studies have provided pictures of the A complex or prespliceosome (containing U1 and U2 snRNPs) at a low resolution (∼40–50 Å), indicating a main globular body with smaller protruding elements (56). Higher-resolution structures will be possible with improvements in instrumentation and improved sample preparation using mild chemical fixation to limit sample heterogeneity.
Equally as impressive as the characterization of the structure and composition of spliceosomal complexes have been the purification, characterization, and electron microscopy structure determination of the spliceosomal snRNPs. We now have structures for U1 snRNP (∼240 kDa); U2 snRNP and the associated SF3b complex, which is the target for frequent mutations in myeloid cancers; and the U5, U4/U6, and U4/U6.U5 snRNPs (Figure 1) (63). Again, these structures have given us a glimpse into the organization of these RNA–protein complexes that compose the spliceosome, all of which have depended on careful and rigorous biochemical purification and characterization of these RNPs.
Although collectively important structural insights have been gained from these studies, electron microscopy does not yet routinely offer the resolution of X-ray crystallography. In a tour de force study, the human U1 snRNP was biochemically reconstituted from recombinant components (64). This structure beautifully illustrates the complex architecture and network of RNA–protein and protein–protein interactions required for this RNP complex. This reconstituted complex contained the seven common snRNP Sm proteins, the U1C protein, and a portion of the U1 70K protein, but it is lacking the U1 A protein. However, because of a previous X-ray structure of the U1 A protein bound to stem loop 2 of U1 snRNA (65), a complete structural model of U1 snRNP could be made (Figure 2). The U1 snRNA consists of four stem loops that form a four-helix junction with coaxially stacked helices. The structure also confirmed the common heptameric arrangement of the seven common snRNP Sm proteins bound to the U-rich Sm-binding site, found in all the spliceosomal snRNAs (Figure 2). In addition, the U1 70K protein contacts stem loop 1 of the U1 snRNA, with its N terminus contacting the Sm core and the U1 C protein. More recently, an X-ray structure of the native U1 snRNP was subjected to limited proteolysis at 4.4-Å resolution, showing details of the Sm protein–Sm RNA site interaction and multiple contacts of the U1 snRNP–specific 70K protein (66). In addition, X-ray crystallographic studies have revealed the core structure of U6 snRNP, containing most of the U6 snRNA and the four RNA recognition motif (RRM)-type RNA-binding domains of prp24 at 1.7 Å (67). Interestingly, RRMs 1, 2, and 4 of prp24 form an electropositive groove that binds double-stranded RNA and may play a role in the annealing of U6 and U4 snRNAs. Researchers have solved an X-ray structure of U4 snRNP that illuminates the complex interactions between the core U snRNP Sm proteins and their U-rich Sm RNA-binding site, common to all the U snRNPs (68). These structural studies have given us a way to better appreciate and understand the complex conformational transitions and RNA–protein and protein-protein interactions that occur during the splicing cycle in these large RNPs.
Figure 2.
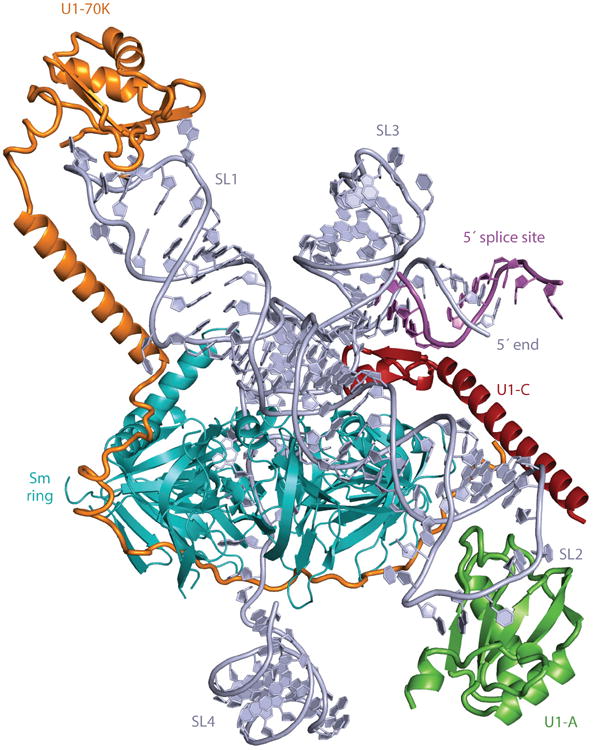
Overview of a model of the complete human U1 small nuclear ribonucleoprotein (snRNP) derived from X-ray crystal structures. Truncated stem loop 2 (SL2) was extended with an A-form RNA helix and, using the crystal structure of the U1A–RNA complex (64), was appended to the extended helix. The internal loop of SL2, consisting of four consecutive non-Watson-Crick base pairs, is in a position to interact with the Sm-B and Sm-D1 proteins. Closely matching images are found in the gallery of negatively stained images of U1 snRNP reported previously (251, 252).
Activities and Reconstitution of Splicing Complexes
Many of the spliceosomal complexes isolated to date were blocked at a particular step in the spliceosome cycle using various tricks. The purification and analysis of an active step I spliceosome provide an elegant example of the power of the affinity purification methods that have been developed for yeast spliceosomal complexes (62). The yeast spliceosome contains fewer proteins than does the mammalian spliceosome, but it has a conserved core design (52). The purification of human splicing complexes led to the isolation of an active salt-stable RNP core complex that was capable of being reactivated upon addition of a micrococcal nuclease-treated nuclear extract (69).
This biochemical complementation allowed the detection of important “second-step” splicing components. A more recent study reconstituted both steps of splicing with highly purified yeast spliceosomes by making use of a temperature-sensitive mutation in the Prp2 helicase to block splicing before the first catalytic step and then using recombinant Prp2, Spp2, and Cwc25 to complement the first step in splicing (62). This study showed a previously unknown role for Cwc25 in the first catalytic step of splicing and indicated that step 2 catalysis required Prp16, Slu7, Prp18, and Prp22. The data also suggested that Prp2 functions to remodel the spliceosome, destabilizing the SF3a and SF3b proteins. Purification of yeast spliceosomes also allowed a direct demonstration of the reversibility of the two catalytic steps of splicing (70). Although exceedingly challenging, these types of detailed biochemical analyses are necessary to understand the functional role and mechanism of action of individual proteins in the complex spliceosomal machine and where in the spliceosome cycle they function.
Single-Molecule Imaging of Splicing
The importance of functional assays for discrete biochemical steps in the splicing pathway and the ensemble averaging inherent in bulk biochemical assays led investigators to develop single–RNA molecule assays to detect both spliceosome assembly and catalysis (71). These systems used a powerful combination of yeast genetic engineering for fluorescent protein tagging; chemical biology to attach bright fluorescent dyes to RNA and protein molecules; and a type of total internal reflection multiwavelength fluorescence microscopy, termed multiwavelength colocalization single-molecule spectroscopy (CoSMoS) (71). In these assays, fluorescently labeled pre-mRNAs are attached via a biotin moiety to the surface of a polyethylene glycol–coated coverslip or microscope slide. Yeast splicing extract, generated from genetically engineered yeast strains carrying fusion proteins of spliceosome proteins that are fluorescently labeled using highly specific chemical modification reactions, is then flowed into the reaction chamber containing the immobilized pre-mRNA, which can be visualized in the CoSMoS microscope (72, 73). Binding of spliceosome components to the pre-mRNA can be detected by the colocalization of two fluorochrome-labeled macromolecules of distinct wavelengths. Time-course experiments can be done using video recording to follow both spliceosome assembly and intron removal. Through these methods, several new insights have emerged. First, many of the initial pre-mRNA-binding events by U1 and U2 snRNPs do not yield productive spliceosomes (73). Second, using a variety of introns, a study showed that both U1-first and U2-first binding events could give rise to active spliceosomes (74). This finding has implications for the assembly of spliceosomes across large introns and for alternative splicing events that use intron or exon definitions. Finally, using fluorescence resonance energy transfer experiments, a study followed the intron ends in real time and found that they came together only when catalytically active spliceosomes were formed (75). These types of experiments also provided insights into the RNA conformational dynamics that occur during intron assembly and removal during the splicing cycle (76). Looking at individual RNA molecules has given us an even better appreciation of the dynamics of spliceosome assembly and intron removal.
RNA–RNA Base Pairing and RNA and Protein Structures in the Active Site of the Spliceosome
One of the reasons that the spliceosome contains many DEAD/H-box RNA-dependent ATP-ases/helicases is that alterations in RNA–RNA base pairing need to occur at multiple points along the spliceosome assembly and catalysis pathway (12). For instance, as spliceosome assembly precedes the 5′ splice site, the U1 snRNA base pairing that occurs in the initial E complex is disrupted and replaced by a U6 snRNA interaction at the 5′ splice site after engagement of the U4/U6.U5 tri-snRNP complex (12). Most dramatically, the initial joining of the tri-snRNP complex to form the B complex contains U4 and U6 snRNAs that are base paired. Upon catalytic activation of the spliceosome, this U4–U6 base pairing is disrupted and U6 snRNA forms an internal stem loop that creates a critical catalytic metal-binding platform (Figure 3) and a base-paired complex with U2 snRNA. This U2–U6 base-paired complex forms the active site of the spliceosome, where the catalytic transesterification reactions of intron excision and exon joining occur (Figure 3). This structure bears remarkable similarity to the domain V region of self-splicing group II introns (13, 77, 78), which also use a lariat 2′–5′ mechanism for group II intron removal. On the basis of the similarity between the U2–U6 snRNA base pairing and the group II domain V structure and mechanism, it was speculated that the spliceosome used RNA-mediated catalysis, much like the ribosome.
Figure 3.

Chemistry of precursor messenger RNA (pre-mRNA) splicing and U2/U6 model showing sites that are sensitive to sulfur substitutions and rescued by thiophilic metal. (a) Reaction scheme (top) and transition state diagrams (bottom) for the two steps of nuclear pre-mRNA splicing. (b) Two-metal model for the RNA catalytic core of the spliceosome. For branching, R1 represents the 29 hydroxyl of the branch adenosine, R2 represents the intron, and R3 represents the pro-Sp oxygen. For exon ligation, R1 represents the 39 oxygen leaving group, R2 represents the pro-Sp oxygen, and R3 represents the 39 exon. (c) Model of group II intron domain V during hydrolysis [PDB 4FAQ (77)]. Throughout, the reactive oxygens are colored red, the pre-mRNA scissile phosphate is depicted in a transition state, and interactions between specific ligands and the reactive oxygens mediated by M1 and M2 are shown as light purple dashed lines. Modified with permission from Reference 13.
A critical protein factor, the U5 snRNP protein prp8, is close to the active center of the spliceosome (79, 80). Genetic experiments suggested an intimate involvement of the protein at the heart of the spliceosome (79). Moreover, structural analysis of the prp8 protein revealed two interesting domains: one similar to the RNase H/RuvC superfamily of nucleases and the other similar to the reverse transcriptase (RT) enzyme superfamily (80–84). These findings suggested that the catalytic core of the spliceosome may be an RNP enzyme, much like telomerase with both RNA and protein components, but that the RNase H or RT domains of prp8 may use acidic amino acid residues to coordinate catalytic metal ions, like the TERT subunit of telomerase (84).
Previous genetic studies of prp8 showed that it plays a critical role in both the first and second steps of splicing (79) and in the transition of the active site from the first to second catalytic steps. Photochemical cross-linking data also indicated that the prp8 protein was intimately located within the heart of the spliceosome. Genetic and more recent structural studies indicate that prp8 plays a role, along with prp16 and U6 snRNA, in alternative U2 snRNA and prp8 protein conformations, thereby modulating the first and second catalytic steps of splicing (85). Thus, both biochemical and genetic data make prp8 a good candidate for a protein component of the spliceosome that lies at its catalytic center. The realization that prp8 has both RNase H and RT domains strengthened the idea that pre-mRNA splicing evolved from the mechanistically related, but protein-free, self-splicing group II intron RNA moieties.
The Spliceosome Is a Ribozyme
Several lines of evidence have suggested that the catalytic center of the spliceosome is composed of RNA. In addition to the structural similarity between the U2 and U6 snRNA base pairing, small segments of synthetic, purified, protein-free U2 and U6 snRNAs could function to catalytically generate a phosphotriester bond by using the branched adenosine residue at the branch point as the nucleophile (86, 87). The locations of catalytic metal ions in self-cleaving and self-splicing ribozymes can often be determined by an experiment called metal ion rescue. Using chemical synthesis, this approach involves the substitution of oxygen for sulfur atoms at various locations surrounding the putative catalytic RNA residues. Normally, as indicated by X-ray structures of a number of ribozymes, oxygen atoms serve to coordinate active-site metal ions, normally magnesium, for catalysis (77). However, sulfur interacts poorly with oxygen, and these sulfur substitutions are typically inactive in the presence of magnesium. Often these sulfur-substituted ribozymes become active in the presence of more thiophilic metal ions, such as manganese or cadmium. Early sulfur substitutions in the spliceosome active site suggested that RNA was the catalytic entity (88), but the structures and domains of the prp8 protein brought these findings into question (80, 84). Earlier biochemical studies on yeast U6 snRNP showed that functional snRNP could be reconstituted with in vitro–synthesized U6 snRNA (89).
Using this reconstitution assay, researchers tested a vast array of sulfur substitutions in the yeast U6 snRNA for splicing activity in yeast splicing extracts in the presence of different metal ions (13). These studies showed that sulfur substitutions at critical positions in divalent metal ions in the U2–U6 snRNA complex were inactive in magnesium but could be reactivated for both splicing steps in the presence of manganese or cadmium. Interestingly, the U6 catalytic metal ligands correspond to positions observed to localize catalytic metal ions in the structures of group II intron RNAs. Also, double-sulfur substitutions in U6 snRNA and the substrate pre-mRNA have provided evidence that these U6-bound metal ions serve a catalytic role by interacting directly with scissile phosphates, rather than simply functioning structurally. These studies, along with a mutational analysis of putative metal-coordinating amino acid residues in prp8 that had no effect on the activity of the spliceosome (13), indicate that, similar to the group II introns, the spliceosome active site is a ribozyme that catalyzes both steps in pre-mRNA splicing (Figure 3a).
Alternative Splicing: Prevalence, Tissue Specificity, and Disease Connections
Splice Sites
The major class of introns in metazoans is composed of the U2 type and contains loosely defined consensus sequences for the 5′ splice site, the intron branch point, and the 3′ splice site (Figure 4). During initial intron recognition, U1 snRNA base-pairs with the 5′ splice site and U2 snRNA base-pairs with the intron branch-point sequence (12). By contrast, the splice-site sequences in the budding yeast Saccharomyces cerevisiae are very highly conserved, and this conservation is correlated with the fact that the vast majority of yeast introns are constitutively spliced, with only a few examples of alternative splicing (12). However, in metazoans, these degenerate consensus splice sites may be a key feature that allows the generation of diverse alternative splicing patterns and may also lead to a requirement for additional protein factors to stabilize or target specific splice sites in a given tissue or cell type. Nonetheless, these consensus splice sites can be targets for mutations that affect pre-mRNA splicing patterns and can lead to disease states (29–35). In fact, early human genetic studies indicated that many of the thalassemia mutations in the β-globin gene, which are common in human populations, affect splice sites and give rise to aberrant splicing patterns (90, 91). More recent studies indicate that a large fraction of human and mouse disease gene mutations affect the splicing process (30–35). Finally, many so-called silent mutations can affect pre-mRNA splicing and other RNA processing reactions (31, 35).
Figure 4.
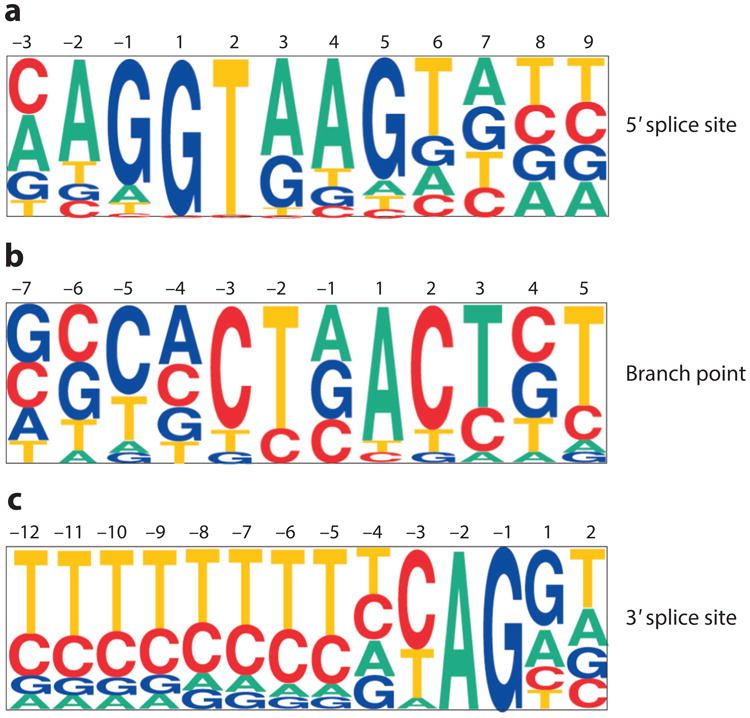
Pictograms of the major U2-dependent intron class consensus splice-site signals. Approximately 20,000 5′ and 3′ splice sites from annotated GenBank files were extracted and aligned as described elsewhere (253, 254). In these pictograms, the size of a letter corresponds to the frequency with which that base is present at each position in a compilation of splice sites. (a) Major class 5′ splice-site consensus sequence. The position labeled 1 is the first nucleotide of the intron, and the position labeled –1 is the last nucleotide of the upstream exon. (b) Major class branch-site consensus. A small database of experimentally confirmed branch sites (166) was used to generate this pictogram. The position labeled 1 is the branch-site residue. (c) Major class 3′ splice-site consensus. The position labeled –1 is the last nucleotide of the intron, and the position labeled 1 is the first nucleotide of the downstream exon. Modified with permission from Reference 254.
Prevalence of Alterative Splicing and Correlation with Organismal Complexity
An interesting outcome of the sequencing of the human and other model organism genomes was the realization that humans do not have many more genes than other commonly studied model organisms, such as mice, fruit flies, or worms (Table 1) (see http://www.ensembl.org/index.html). This observation raises the question of how humans can be so much more morphologically and behaviorally complex than these other metazoans. One possibility is that the role and extent of alternative pre-mRNA splicing increase with increasing organismal complexity. Consistent with this idea, characterization of expressed complementary DNA (cDNA) clone sequence tags indicates an increase in the prevalence and extent of alternative splicing that correlates with organismal complexity (Table 1) (3, 4). The current Ensembl annotations [which do not take into account recent RNA-sequencing (RNA-seq) data] indicate that, for multiexon protein-coding genes, Caenorhabditis elegans has 25% that undergo alternative splicing, Drosophila has 45%, mice have 63%, and humans have 88%. This general trend is consistent with a role for alternative splicing in organismal complexity. In fact, humans have the largest average number of mRNA isoforms per gene (Table 1). The most current estimates, based on RNA-seq data, indicate that >95–100% of human genes generate at least two alternative pre-mRNA isoforms (with an average of seven mRNA isoforms per gene) (Table 1) (92, 93). Moreover, an analysis of expression of the human transcriptome (based on References 5 and 6) indicates that alternative splicing may be a key aspect related to the phenotypic complexity of Homo sapiens. Thus, alternative pre-mRNA splicing plays key roles in gene expression and in the diversification of both the transcriptome and the encoded proteome, with humans having the largest extent of alternative splicing.
Disease Mutations, Cancer, and Neurodegenerative Diseases
It has long been known that disease mutations can affect splicing by altering either the splice sites or exonic or intronic sequence regulatory motifs, termed silencers or enhancers (see the section titled Silencers and Enhancers, below). Among the first examples of human disease mutations affecting splicing were the β-globin thalassemia mutations (discussed above) (90, 91) and mutations in the SMN-2 gene, which give rise to spinal muscular atrophy (94, 95). The splicing factor hnRNPA1, which binds to a regulatory site in the SMN-2 transcript, plays a key role in regulating the splicing of SMN-2 pre-mRNA as well as the splicing of the pyruvate kinase pre-mRNA in cancer (96–99). More extensive surveys of silent and missense mutations in a variety of disease genes have linked non-splice-site point mutations in exonic or intronic splicing silencers and enhancers to defects in RNA processing (30–35). Studies showing that RNA regulatory elements deep within an intron can control splicing of an exon that is kilobases away indicate that exome-sequencing strategies to identify base changes associated with disease may be missing important mutations (100). Such links between silent mutations have been found in DNA damage and repair factors, such as ATM, BRCA1, and MLH1, which have direct roles in cancer pathways (31, 99, 101).
Also disease causing are the cis-acting mutations in a prion-like domain in the C-terminal region (the glycine-rich domain) of hnRNPA1 that are linked to the degenerative muscle disease amyotrophic lateral sclerosis (102). Prion-like domains are rich in asparagine, glutamine, tyrosine, and glycine residues and are found in hnRNPA2B1, hnRNPA1, TDP-43, FUS, EWSR1, and TAF15 (103). Mutations in the prion-like domains of the RNA-binding proteins TDP-43 and FUS are also linked to amyotrophic lateral sclerosis. These low-complexity sequences are common in heterogeneous nuclear ribonucleoprotein particles (hnRNPs), some of which can form fibrils, and can interact with the C-terminal domain (CTD) of RNA polymerase II (104).
One of the most exciting findings in this area in the past few years comes from the Cancer Genome Project, in which genomic DNA from a variety of human tumors was sequenced and analyzed. A surprising finding was that there are recurring somatic mutations in genes encoding 3′ splice-site recognition protein components and serine–arginine repeat (SR) splicing factors, namely U2AF1 (U2AF35), SRSF2 (SC35), SF3B1 (SF3B155 or SAP155), and ZRSR2 (URP) (29, 36, 38, 105-107). Functional assays showed that overexpression of mutant versions of these factors could alter splicing patterns and that splicing patterns were also altered in patient samples, indicating that 3′ splice-site use patterns were affected (108). Thus, somatic mutations in genes encoding well-studied splicing factors are correlated with at least two types of cancer, indicating that aberrant splicing patterns are directly linked to the disease phenotype (39, 99).
Previous genome-wide studies have shown that in different tumors there are altered patterns of splicing. However, global patterns of spliced pre-mRNA isoforms cannot pinpoint the causal change responsible for a cancer phenotype. Likewise, cis-acting mutations found in 3′ splice-site factors led to the discovery of many alterations in 3′ splice-site use, but the challenge now is to find one or several “causal” spliced pre-mRNAs that lead to a cancer cell. Studies of overexpression of the splicing factor SRSF1 (ASF/SF2) showed that it acts as an oncogene, leading to tumors in mice (109). Several causal target genes in the mTORc1 pathway that linked to the cancer phenotype have been identified (110). Thus, overexpression of splicing factors can also lead to cancer.
The Spliceosome as a Target for Small-Molecule and Nucleic Acid Therapeutics
Cancer genomics has identified cis-acting mutations in several 3′ splice-site factors (29, 36, 38, 105–108). Interestingly, chemical genetics and chemical biology studies have identified small-molecule splicing inhibitors, such as spliceostatin (111, 112), that also target the 3′ splice-site factor SF3b (see the section titled Prevalence of Alterative Splicing and Correlation with Organismal Complexity, above). These compounds have previously been used as anticancer agents because they cause cell cycle arrest. Moreover, large-scale compound screens have led to the discovery of additional organic compounds that inhibit splicing at different stages (113, 114). Taken together with the cancer-causing mutations in these factors, such studies give good leads on small-molecule therapeutic applications for compounds that act on the spliceosome (40, 41).
In addition to small-molecule therapeutics, antisense oligonucleotides have been used extensively to alter and control splicing patterns in vivo (42, 43). Most dramatically, a method called TSUNAMI has been used to correct a spinal muscular atrophy–like syndrome in a mouse model by altering SMN-2 pre-mRNA splicing patterns (44, 115–117). Thus, the use of antisense oligonucleotides may also be a viable therapeutic alternative to small-molecule therapy.
Most recently, very efficient genome editing using the CRISPR/Cas9 system, performed via hydrodynamic mouse tail vein injection, allowed correction of a single-point mutation in a mouse liver model causing a splicing defect and normal liver development and function (46). This proof-of-principle experiment used a mouse model with a point mutation in the FAH (fumarylacetoacetate hydrolase) gene leading to tyrosinemia and liver disease. Researchers showed that this gene could be edited to the wild-type allele using Cas9 in whole animals, thereby rescuing the body-weight-loss phenotype associated with the disease mutation. Thus, the future of using genome editing to correct the many disease-causing defects in pre-mRNA splicing is bright (45).
Alternative Splicing: Mechanisms
RNA–Protein Interactions: Heterogeneous Nuclear Ribonucleoproteins and Serine–Arginine Repeat Proteins
It has long been known that extrinsic, nonspliceosomal RNA-binding proteins play a role in splice-site selection and activity (14, 118–120). Generally, these proteins can be divided into three classes: the classical/canonical hnRNPs (121); SR proteins (122–125); and tissue-specific RNA-binding proteins, such as nova (126), neuronal PTB/hnRNPI (126), the Rbfox family (100, 127–129), and the muscleblind/CELF family (130, 131). In some cases, hnRNP proteins act as splicing repressors, and SR proteins act as splicing activators. The tissue-specific RNA-binding protein splicing factors, such as nova or Rbfox, can act as either activators or repressors (126, 129). A recent study showed that SR proteins can both cooperate and compete in splicing regulation (132). SR proteins and their binding to RNA have been studied extensively, using both in vitro binding and selection assays. These proteins can also recognize short RNA sequence motifs, which can function as splicing “enhancers” when bound to exons (124, 125), but they can repress splicing when bound to introns. hnRNP proteins also possess sequence-specific RNA-binding activity, and these motifs often can function in a variety of assays as splicing “silencers” (121). However, in some cases, hnRNP proteins, such as hnRNPL, can activate splicing. Thus, splicing factors, often depending on the position in a pre-mRNA to which they bind, can act as activators or repressors.
One well-studied family of RNA-binding proteins of the hnRNP class consists of hnRNPA1, A2, and A3 (121, 133). There are also four homologs of the hnRNPA/B proteins in Drosophila. Characterization of hnRNPA1 and A2 in mammals (134) and hrp48, 40, 38, and 36 in Drosophila (135) indicates that in some cases the family members can function redundantly or with overlapping specificity in the regulation of pre-mRNA splicing (134–136). In vitro RNA binding (137), binding site selection experiments (SELEX) (135, 138), and genome-wide approaches (134, 135) indicated that these proteins have specific affinities for a variety of RNA sequences and regulate overlapping, yet distinct, populations of transcripts. Human hnRNPA1 is a well-studied splicing repressor that interacts with silencer elements (96, 97, 139–142). Mechanistically, hnRNPA1 has different modes of action, including (a) binding to exonic or intronic splicing silencer elements to repress exon inclusion by steric action (31), as is also the case for hrp48 and the Drosophila P element exonic splicing silencer (see below); (b) binding of hnRNPA1 to a higher-affinity binding site that promotes cooperative binding and “spreading” hnRNPA1 proteins to adjacent lower-affinity binding sites (140); and (c) interaction hnRNPA1 proteins bound to intronic silencer elements on both sides of an alternative exon, resulting in loop formation and exclusion of the exon (143). In some cases, hnRNPA1 can act as a splicing activator (133, 136, 144, 145).
hnRNPL (and the related hnRNPLL) can bind to both exonic and intronic RNA sites and acts as an enhancer or repressor of exon inclusion (146). hnRNPL has been best studied as a splicing repressor of the CD45 gene (147) and acts in conjunction with hnRNPA1 to induce extended U1 snRNP–pre-mRNA interactions (148). hnRNPL can also interfere with 3′ splice-site recognition by U2AF65 (149). In vivo binding studies on hnRNPL, using individual-nucleotide-resolution UV cross-linking and immunoprecipitation (iCLIP), also indicate that hnRNPL exhibits a binding preference for C/A motifs that correlates with the in vitro binding SELEX consensus sequence (150). Genome-wide mapping revealed that hnRNPL preferably binds to introns and the 3′ untranslated region (UTR) (150). hnRNPL may function as a repressor when bound to intronic regions upstream of alternative exons and as an enhancer when bound to downstream introns (150).
Silencers and Enhancers
Studies of splicing regulation using in vitro biochemical assays led to the discovery of cis-acting elements that promote (enhancers) or inhibit (silencers) splicing activity from nearby splice sites. These regulatory elements can be located either in exons or in introns (Figure 5). One of the first exonic splicing silencers defined was found in the Drosophila transposable P element pre-mRNA, whose activity blocks splicing of the P element transposase pre-mRNA in somatic cells (151, 152). This silencer binds U1 snRNP to a pseudo-5′ splice site, the hnRNP proteins PSI and hrp48, and other RNA-binding proteins (151–154). Both exonic and intronic splicing silencers (abbreviated ESS and ISS, respectively), regulatory motifs that bind the splicing repressor protein hnRNPA1, have been identified and characterized; they regulate splicing of the HIV pre-mRNA (139, 155, 156). In addition, exonic and intronic splicing enhancers have been defined, typically as binding sites for the SR protein class of splicing activators (Figure 5) (122, 124, 125).
Figure 5.

Positive and negative control of precursor messenger RNA (pre-mRNA) splicing by cis-acting intronic and exonic silencers and enhancers. Diagram of a segment of a typical metazoan pre-mRNA with exon and surrounding introns indicated. Intronic and exonic splicing enhancers (ISE, red box; ESE, purple box) and intronic and exonic splicing silencers (ISS, orange box; ESS, brown box) are indicated. Serine–arginine repeat (SR) proteins generally act to promote splicing from nearby splice sites by interacting with splicing enhancers. Heterogeneous nuclear ribonucleoprotein particle (hnRNP) proteins generally act to inhibit splicing from nearby splice sites by interacting with splicing silencers.
In addition to individual gene studies, a number of clever selection strategies, carried out either in vitro or in vivo, have identified both splicing enhancers (157–159) and silencers (160–162). A biochemical study of in vitro-selected silencers identified hnRNPA1, which appeared to affect U1 snRNP binding across a nearby exon to effect silencing (162). More recent biochemical studies incorporated an extensive in vivo screen for exonic splicing silencers coupled with the use of RNA affinity purification and mass spectrometry to identify proteins that bound to the comprehensive collection of splicing silencer motifs defined bioinformatically from the in vivo selections (163,164). These large-scale studies further point to the critical role of hnRNP proteins in splicing silencer activity.
Interactions between Small Nuclear Ribonucleoproteins and Splicing Factors
U1 snRNP can bind in vitro to both normal and cryptic 5′ splice sites in the β-globin pre-mRNA (165, 166). More recent studies have shown that U1 snRNP plays a role in the suppression of premature cleavage and polyadenylation (PCPA) by binding to non-5′ splice-site sequences (see the section titled U1 Small Nuclear Ribonucleoprotein and Premature Cleavage and Polyadenylation, below) (167,168). A general and important principle of splicing regulation that has emerged is that snRNP binding to specific sites may be enhanced by interactions early in spliceosome assembly. In addition, intron–exon definition is governed by non-snRNP splicing factors, namely hnRNP and SR proteins (118). For example, the splicing repressor protein PSI has an auxiliary domain, C terminal to the four KH-type RNA-binding domains, that interacts directly with the U1 snRNP 70K protein and promotes U1 snRNP binding to the P element exonic splicing silencer (169). Both hnRNPA1 and hnRNPLL can promote U1 snRNP binding to specific pre-mRNAs during splicing silencing (148). SRSF1 (ASF/SF2) can promote proximal 5′ splice-site use by increasing U1 snRNP–pre-mRNA binding (170). The splicing activator protein, TIA-1, also binds U-rich intronic regulatory elements adjacent to 5′ splice sites and interacts directly with the U1 snRNP C protein to promote 5′ splice-site use (171). Interestingly, in vitro selection (SELEX) assays showed that the U1 C protein possesses a site-specific RNA-binding activity, which may facilitate U1 snRNP binding to particular sites in the transcriptome (172). Finally, a recent study showed that the splicing repressor PTB/hnRNPI interacts directly with U1 snRNA in the intact snRNP complex to mediate splicing repression of the c-src N1 exon (173). Similarly, recognition of 3′ splice sites by U2 snRNP requires the RNA-binding proteins SF1/BBP and the heterodimeric U2 snRNP auxiliary factor (U2AFLS/U2AFSS) (9). In many cases, these interactions result in cooperative assembly of proteins, snRNPs, and the pre-mRNA substrate, as was shown for the doublesex splicing enhancer in vitro (174) and for the C. elegans splicing factors ASD-2 and SUP-12 (175). Thus, one key step to the initial stages of intron recognition and spliceosome assembly is positioning snRNPs correctly on a given pre-mRNA through cooperative interactions with nonspliceosomal RNA-binding factors.
RNA–RNA Base Pairing
A role for RNA–RNA secondary structures in controlling alternative splicing has long been suggested (176), but recent experiments have illuminated concrete examples of how RNA–RNA recognition can act to dictate splice-site choice (177). In addition, recent studies using chemical probes to study RNA structures in vivo, coupled with high-throughput cDNA sequencing, highlight that regions of the transcriptome can be highly structured (178). It is also likely that there are transcriptome RNA–RNA dynamics that must occur, as nascent pre-mRNAs synthesized by RNA polymerase II are folded cotranscriptionally, spliced, polyadenylated, and bound by nuclear RNA-binding proteins in preparation for export of mature mRNAs to the cytoplasm. High-density RNA structure mapping of nuclear RNAs promises to illuminate the dynamics of pre-mRNA structure in vivo (178).
The best example of cis-acting RNA–RNA base pairing controlling alternative splicing comes from the Drosophila DSCAM gene (179). Here, four clusters of alternative exons, used in a mutually exclusive manner, can combinatorially generate >36,000 distinct spliced mature mRNA isoforms, approximately three times more than the number of genes in the fruit fly genome (179). DSCAM is an immunoglobulin superfamily member that plays a role in neuronal connectivity in development. The exon 6 cluster contains 48 alternative exons that are used in a mutually exclusive manner (16). A conserved “docking” site is complementary to a conserved “selector” site upstream from each of the 48 alternative exons (16, 179). There is both phylogenetic and experimental evidence that RNA–RNA base pairing between the docking and selector sites dictates which of 48 alternative exons in the exon 6 cluster are used to make the mature DSCAM mRNA (180). RNA interference (RNAi) and RNA-binding assays also identified the hnRNP protein, hrp36, as a repressor that functions to enforce the mutual exclusivity by allowing only one exon from the exon 6 cluster to be spliced into the mature DSCAM mRNA (Figure 6) (16). A new study using reporter transgenes in flies showed that the patterns of DSCAM splicing in the Drosophila nervous system are probabilistic (181).
Figure 6.
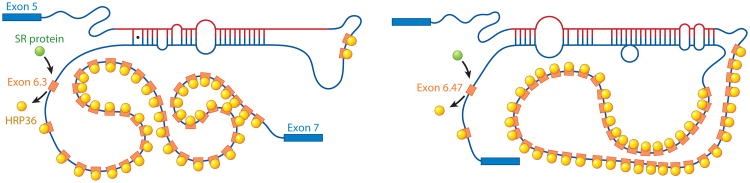
RNA–RNA and RNA–protein interactions that regulate mutually exclusive splicing of the Drosophila DSCAM exon 6 exon cluster. A model for the mechanism by which the heterogeneous nuclear ribonucleoprotein (hnRNP) hrp36 prevents the inclusion of multiple DSCAM exon 6 variants. hrp36 (yellow circles) binds to all the exon 6 variants (orange) and represses their inclusion. When the conserved cis-acting RNA selector sequence upstream of a specific exon interacts by RNA–RNA base pairing with the conserved exon 6 cis-acting RNA docking site located upstream of the exon 6 cluster of 48 exons, it results in the derepression of hrp36 on the exon immediately downstream, but not for the other 47 exon 6 variants. In this way, only a single exon 6 variant is included. hrp36 competes with serine–arginine repeat (SR) proteins (green circles) for binding to the exon 6 variants. In the absence of hrp36, these activators can bind to all the exon 6 variants and function to enhance their splicing to other exon 6 exons. Figure modeled on data from Reference 16.
The DSCAM gene remains the best example of cis-acting RNA secondary structures controlling splice-site choice. In addition, recent studies have indicated that splicing of DSCAM exons 4 and 9, as well as the 14-3-3xi pre-mRNA (182) and splicing targets of the Rbfox proteins (100), use RNA–RNA base pairing to control splicing patterns.
What about the possibility of trans-acting RNA–RNA base pairing affecting alternative splicing patterns? There has been one report of a small RNA controlling alternative splicing. The snoRNA HBII-52 appears to regulate alternative splicing of the serotonin receptor 2C pre-mRNA (17, 18). Here, an 18-nucleotide complementary RNA region between the snoRNA and a splicing silencer element in the serotonin receptor pre-RNA leads to alternative exon usage. Given the highly flexible nature of microRNA–target mRNA base pairing and the finding of Argonaute family members in the nucleus (see the section titled Connections Between Alternative Splicing and Small RNA Pathways, below), other examples of trans-acting small RNAs controlling alternative splicing may be found.
Chromatin
Several examples of links between chromatin and splicing have been discovered (20, 26, 183, 184). First, the speed of RNA polymerase II appears to correlate with splicing patterns (see the section titled Cotranscriptional Splicing: Connections to Chromatin, below) (25, 185), which apparently can be influenced by histone modifications. Second, there are “adapters” that link specific histone modifications or “marks” to splicing factors. Finally, many protein–protein interactions between chromatin-binding/remodeling proteins and splicing factors or spliceosome components have been observed. The first example of a direct, functional interaction of a chromatin-binding protein and a spliceosome component was the interaction between CHD1 and U2 snRNP (186). CHD1 contains two tandem chromodomains and binds tightly to covalently modified histone H3(H3K4me3) (186). Functional in vivo and in vitro assays established a link between CHD1 and splicing. Another example of a chromatin-splicing connection is the binding of the splicing repressor protein PTB/hnRNPI to the histone-binding adapter protein MRG15 (187). MRG15 binds to the modified histone H3K36me3 mark and may serve as an adapter to target PTB/hnRNPI. Perturbations of MRG15 alter the splicing of the FGFR2 gene exon IIIb, as well as other exons. In addition to acting to recruit splicing factors, histone modifications may alter chromatin states and change RNA polymerase II transcription rates (25, 26). For instance, repressive histone methylation marks, such as H3K9me3 and H3K27me3, can recruit heterochromatin protein 1 and slow RNA polymerase II, leading to changes in splicing patterns (188). Thus, discrete histone modifications on chromatin may be used to target splicing factors to specific genes or exons/introns to control splice-site use or to change the rate of RNA polymerase II transcription through a chromosomal locus, resulting in splicing pattern changes.
Alternative Splicing: Insights from Genomics
Access to genome and cDNA sequences, synthetic oligonucleotide microarrays, and more recently, high-throughput cDNA sequencing has led to broad genome-wide assessment of alternative splicing patterns. These technologies have also revealed how RNA-binding proteins and intrinsic spliceosomal components act to control hundreds to thousands of alternative splicing events in metazoan tissues.
Genome-Wide Studies of Alternative Splicing
Comparative genome sequencing has led to the realization that complex multicellular eukaryotes do not scale in complexity with gene number: Humans, mice, worms, and fruit flies each have 15,000–25,000 genes. One hint of the complexity of splicing came initially from expressed sequence tag sequencing, which suggested that higher levels of alternative splicing scaled with organismal complexity. This idea made some sense in light of the fact that newly sequenced metazoan genomes had approximately 15,000–25,000 genes. One of the early breakthrough technologies was the use of inkjet long DNA oligonucleotide synthesis methods to generate splice junction microarrays, coupled with bioinformatics of annotated splicing events, to demonstrate wide variations of alternative splicing in different human tissues (119, 189, 190). This technology, coupled with RNAi in Drosophila cells, gave the first glimpse into how many splicing events hnRNP or SR protein splicing factors could regulate (191). This method was subsequently overtaken by high-throughput sequencing, which demonstrated that >95%–100% of human genes can generate at least two alternatively spliced isoforms (92, 93). More recent and extensive sequencing and comparisons among several vertebrate species indicate that essentially all multiexon genes can undergo alternative splicing (5, 6). However, our ability to sequence more deeply and detect novel alternatively spliced mRNA isoforms does not necessarily mean that these low-abundance species are functional at the biological level and may represent low-level errors made by the spliceosome. Nonetheless, these recent sequencing and comparative genomics studies indicate the great variety and extent of alternative splicing that multiexon genes can undergo in different metazoan tissues. Many of these alternative splicing events are conserved across multiple species and in a tissue-specific manner (5, 6).
Genome-Wide RNA–Protein Interaction Maps, RNA Structure Maps, and Alternative Splicing Patterns
Along with advances in genome-wide technologies to detect alternative splicing patterns, different methods have been developed to detect the binding of RNA-binding proteins and splicing factors to regions of cellular transcripts in vivo. Initially, immunopurifications with affinity-purified antibodies were used in conjunction with microarrays to detect transcripts that could be enriched using antibodies to specific splicing factors (for examples, see References 192 and 193). Several cautionary notes have appeared regarding the use of native, low-salt conditions to retrieve RNA-binding proteins, which could, in principle, exchange between different RNAs during cell lysis and various biochemical manipulations (194, 195). As an alternative to native immunoprecipitation, a method called CLIP (UV cross-linking and immunoprecipitation) (196) that uses in vivo cross-linking of RNA to bound proteins with shortwave UV light (254 nm), much the same as Choi & Dreyfuss (197) had done initially to identify in vivo RNA-bound hnRNP proteins. Improvements in high-throughput sequencing (HTS) coupled with the CLIP method led to HTS-CLIP (198) or CLIP-seq (128,199,200) that resulted in a large increase in the ability to recover and map protein-bound RNA tags to the transcriptome. In addition, two variations on the original CLIP method, iCLIP (201) and photoactivatable ribonucleoside-enhanced cross-linking and immunoprecipitation (PAR-CLIP) (202), have been developed to improve the efficiency of recovery and sequencing of protein-bound RNA-binding site tags. To generate a covalent photochemical adduct, the UV cross-linking methods described above rely on a specific juxtaposition of the RNA nucleotide bases and appropriately positioned amino acid side chains in the protein of interest. Thus, because the UV cross-linking efficiency of protein to RNA in cells is rather low (1–2%), it is likely that the CLIP methods do not retrieve all the protein-bound sites for a given protein in the transcriptome. In addition, the covalent UV-induced protein–RNA cross-links likely trap transient (and possibly nonspecific) RNA-binding sites, as has been recently shown for DNA-binding transcription factors in mouse nuclei (203). Owing to these limitations, several alternative methods, termed RIPiT (using formaldehyde cross-linking and high-throughput sequencing) (204), PIP-seq (a ribonuclease-mediated protein–RNA footprinting method in conjunction with high-throughput sequencing) (205), and RNA Bind-n-Seq (using purified protein and high-throughput sequencing to generate a series of RNA-binding motifs, with associated affinities) (206), have recently been described. Finally, using all available data, a comprehensive compendium of RNA-binding motifs for many RNA-binding proteins has been assembled (207). Taken together, these methods allow protein-binding sites in the complex metazoan transcriptome to be identified. Finally, several recent studies have used cell-permeable chemical probes, either 2′-acylation SHAPE reagents (208) or dimethyl sulfate (178, 209, 210), to probe RNA structures in living cells. These new methods will also shed light on protein binding site regions and the role of RNA structure in alternative splicing in the future.
The application of HTS-CLIP/CLIP-seq methods to study a large number of RNA-binding proteins has provided a transcriptome-wide view of the locations of binding regions for these proteins. These “RNA maps” have provided many insights into how and where different RNA-binding proteins function in alternative splicing as well as many other processes (15, 211, 212). It is likely that a single RNA-binding protein has many different roles to play in the cell. One of the best-studied neuronal splicing factors, nova-1, is a great example of the predictive power of a genome-wide RNA map to show whether the protein would act as a splicing activator or repressor in different pre-mRNAs depending on where the protein is bound (Figure 7) (198). Interestingly, this “nova RNA map” and the RNA-binding specificity of nova protein are conserved between mammals and Drosophila (213). Other tissue-specific splicing factors, such as muscleblind (130, 131, 214) and Rbfox(100, 127–129), also show this position-dependent activator/repressor activity. One of the great powers of genomics is to illuminate patterns in genome-wide data in a statistically significant way, and the “nova RNA map” is a great example of this type of data analysis (Figure 7). Moreover, this type of data can be combined with mathematical modeling to provide new insights in to the “splicing code”and splicing “networks” to reveal new biology (129, 215, 216). In the future, RNA maps will be integrated with splicing profiling data, transcriptome structure probing, and human genetic variation to provide insights into how mutations can affect the splicing process in Homo sapiens.
Figure 7.
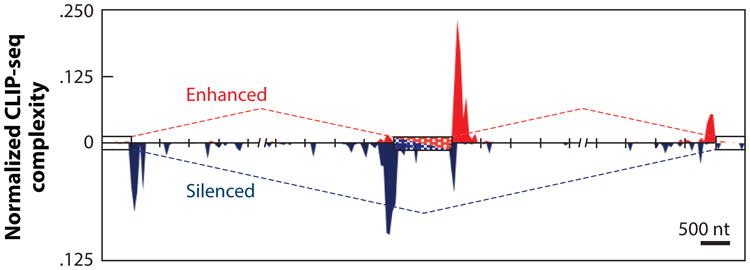
The RNA map of splicing regulation by the neuronal nova protein. A nova RNA splicing map for cassette exons generated by integrating the high-throughput sequencing cross-linking and immunoprecipitation (HTS-CLIP)/CLIP-sequencing (CLIP-seq) experimental identification of nova-binding sites and splice junction microarray data. The red dashed line (top) indicates enhanced exon inclusion by nova, and red peaks indicate the density and location of HTS-CLIP/CLIP-seq tags. The blue dashed line (bottom) indicates enhanced exon skipping by nova, and blue peaks indicate the density and location of HTS-CLIP/CLIP-seq tags. Modified with permission from Reference 198.
Cotranscriptional Splicing: Connections to Chromatin
A variety of data indicate that splicing can occur during transcription of protein-coding and non-coding RNA genes by RNA polymerase II (217). The spliceosomal snRNPs are recruited to active genes during transcription (218–220). Classic biochemical fractionation and RNA metabolic labeling studies indicated that hnRNA existed as polyadenylated species and that its size was reduced prior to arrival in the cytoplasm (221). More recent studies have used biochemical fractionation of chromatin and high-throughput sequencing in several mammalian, Drosophila, and yeast systems to demonstrate that splicing can occur cotranscriptionally on nascent transcripts (218–220, 222–230). However, given the average rate of RNA polymerase II elongation in vivo, ∼3–4 kb/min (221), it is not surprising that, for long genes with large introns, splicing may not occur before the gene is finished transcribing and cleavage and polyadenylation of the transcripts may occur prior to completion of intron splicing, a process that takes minutes to complete. Indeed, some spliced and polyadenylated transcripts stay associated with chromatin at the gene locus after processing but prior to nuclear export to the cytoplasm (231). However, at least some transcripts appear to be “posttranscriptionally” spliced (221), as evidenced both by genome-wide data (223) and by the fact that an antibody to the active, phosphorylated form of SF3b detected spliceosomes in the nucleoplasm away from chromatin (232).
A series of studies have suggested a connection between the speed of RNA polymerase II and alternative splicing patterns (25, 27, 185). One such study used a “slow” α-amanitin-resistant mutant of the largest subunit of the RNA polymerase II and found distinct splicing patterns depending on whether the gene was transcribed by the wild-type or “slow” form of RNA polymerase II (185). Another study showed that transfection of small interfering RNAs targeted to the region near the fibronectin EDI exon could cause a switch in the splicing pattern that correlated with a change in histone modifications (increased dimethylation at K9 and trimethylation at K27 of histone H3) at the target site, leading to the recruitment of the heterochromatin-associated protein HP1-α, which is thought to slow RNA polymerase II transcription (188). Thus, the histone modifications on chromatin may cause changes in alternative splicing patterns by affecting the RNA polymerase II transcription rate.
Connections to Other Rna Processing Reactions
There is an abundance of evidence that pre-mRNA splicing is coordinated with transcription and other RNA processing reactions (233, 234). A key factor in this coordination is the CTD of the largest subunit of RNA polymerase II (234, 235). Phosphorylation of this CTD region is used to coordinate different stages in the transcription cycle, and the CTD interacts with a variety of proteins, including splicing factors. Biochemical studies have indicated that RNA polymerase II has direct effects on pre-mRNA splicing (236) and polyadenylation (237). These studies reconstituted these RNA processing reactions in vitro and showed that addition of purified phosphorylated RNA polymerase II or the phosphorylated recombinant, purified CTD stimulated both of these RNA processing reactions. In a coupled in vitro RNA polymerase II transcription-splicing system, SR splicing factors were required for the coupling of transcription with splicing (238). Connections between pre-RNA splicing and polyadenylation were also shown in vivo, when a polyadenylation enhancer was found to bind U1 snRNP and the SR splicing factor SRp20 (SRSF3) (239).
U1 Small Nuclear Ribonucleoprotein and Premature Cleavage and Polyadenylation
One of the most exciting findings in the past few years was the connection between U1 snRNP and suppression of pre-mRNA PCPA, a process called telescripting (167, 168). U1 snRNP is an integral component of the splicing machinery and is the most abundant spliceosomal snRNP (∼1 million copies per cell). Dreyfuss and colleagues (168) used antisense morpholino oligonucleotides in conjunction with genome-wide approaches to examine the effect of U1 snRNP on the transcriptome. They found that blocking the 5′ end of U1 by transfection of antisense morpholino oligonucleotides into human cells led to a dramatic increase in prematurely terminated, polyadenylated gene transcripts terminating within 1 kb of the transcription start sites. In addition to normal 5′ splice sites, U1 snRNP also binds to many specific sites in the pre-mRNA. This may be reminiscent of the binding of U1 snRNP to a pseudo-5′ splice site in the Drosophila P element exonic splicing silencer (i.e., U1 binding to nonfunctional splice site–like sequences) (169). This telescripting process also occurred in mouse and Drosophila cells (167). Interestingly, a recent study indicates a role for U1 snRNP and polyadenylation signals in termination of the short promoter-associated antisense transcripts (paRNAs) and promoter directionality (240). Thus, these studies revealed a new and unexpected function for U1 snRNP in suppression of PCPA and “transcriptome surveillance.”
Connections Between Alternative Splicing and Small Rna Pathways
A very interesting connection has been made recently between components of the small RNA pathways, namely the Argonaute family members and alternative splicing patterns. Several results suggested that small RNA pathways could impinge on alternative splicing. Specifically, it appears that a trans-acting snoRNA, HBII-52, can regulate alternative splicing patterns (17, 18). There is also highly mismatched base pairing between microRNAs and their target mRNAs (22). Data from HTS-CLIP/CLIP-seq/iCLIP experiments with human (241), mouse (242), C. elegans (243), and Drosophila (244) Argonaute family members also showed that the proteins bound both introns and exons as well as canonical 3′ UTRs. Thus, small RNA pathways may play a role in controlling alternative splicing (23, 24).
Because of the well-characterized slicer function of the Argonaute family of proteins in posttranscriptional gene silencing occurs in the cytoplasm, there was a question about whether Argonaute proteins may be present at appreciable levels in the nucleus. However, experiments from Drosophila showed that Argonaute-2 (Ago-2) played a role in the nucleus by interacting with insulator-binding proteins on chromatin (245) and Ago-2-bound small RNAs from the heat-shock gene promoter regions (246), both of which are nuclear functions. More recently, Ago-2 was found to associate with small antisense promoter-associated transcripts in mouse embryonic stem cells that are homozygous mutant for Dicer (247). Importantly, epitope-tagged human Ago-2 associates with chromatin and copurifies from chromatin with splicing factors (248). In Drosophila, cell fractionation experiments and chromatin immunoprecipitation sequencing (ChIP-seq) data showed that Ago-2 was both in the nucleus and associated with chromatin (244). In human cells, RNAi knockdown of Ago-2 affects alternative splicing by using exon microarrays and specifically modulates splicing of the CD44 gene (248). In Drosophila, Ago-2, but not other components of the somatic RNAi machinery (Ago-1, Dicer-1, or Dicer-2), plays a role in alternative splicing, as indicated by RNAi to Ago-2, Ago-2 mutants, and both splice junction microarrays and RNA-seq (244). Interestingly, a catalytic mutant of Ago-2 is still able to control splicing, whereas a deletion of the Ago-2 gene is not (244). iCLIP/CLIP-seq data from Drosophila have shown that Ago-2 binds in vivo to a glycine-rich RNA-binding motif present in the splicing target transcripts (244), similar to that found in mouse embryonic stem cells that are homozygous mutant for Dicer (242), thus suggesting that Ago-2 possesses intrinsic RNA-binding activity without being programmed by small RNAs. In Drosophila, ChIP-seq and RNAi experiments revealed a second nuclear function for Ago-2: Ago-2 binds to chromatin at the promoters of genes whose transcription is derepressed upon Ago-2 RNAi depletion (244). Interestingly, these promoter sites also colocalize with Polycomb group repressor proteins, which may provide a mechanism for Ago-2-mediated transcriptional repression (244). Thus, Argo-2 plays an unexpected role in several nuclear processes, including the regulation of alternative splicing.
Conclusions
Alternative pre-mRNA splicing is a critical component of the regulation of gene expression pathways in metazoans. Combining alternative exons in different patterns dramatically expands the proteomic of metazoan genomes and contributes significantly to the identity, development, and diversity of cells, tissue, and organs. Biochemical and structural studies have illuminated the composition, structure, and dynamics to the spliceosome, a ribozyme with an evolutionary history related to that of the self-splicing group II introns. New breakthroughs in cryo–electron microscopy and image collection will soon lead to improved high-resolution structures of the spliceosome.
Most importantly, mutations at the intron–exon borders in splice sites or in intronic or exonic splicing regulatory RNA sequences, often “silent” mutations, affect the splicing process, leading to disease in humans. The Cancer Genome Project has revealed mutations in gene-encoding splicing factors that are linked to specific cancer types. Moreover, small-molecule compounds, antisense oligonucleotides, and genome-editing approaches hold promise for correcting the effects of these mutations common in human genetic variation and disease. Breakthroughs in high-throughput sequencing technologies promise to identify and link many more human mutations to disease. Finally, high-throughput sequencing coupled with in vivo probes of RNA structure promises to reveal global aspects of RNA folding throughout the transcriptome and show how this organization may be disrupted by human disease-causing mutations.
Acknowledgments
We thank Marco Blanchette, Bob Darnell, Brent Graveley, Reinhard Lührmann, Kiyoshi Nagai, Joe Piccirilli, and Jon Staley for the figures. We thank Chris Burge, Jason Merkin, Courtney French, and Bob Waterston for help with Table 1. We thank Doug Black, Chris Burge, and Brent Graveley for discussions. We apologize to those whose work could not be cited as primary literature, but due to the breadth of topics discussed and to constraints on the number of references, we have often had to refer to some peoples' work by referencing related review articles. Many important contributions have been made in this area in the past 12 years, since the last Annual Review of Biochemistry review on alternative splicing by Doug Black appeared. We especially thank Doug Black, Reinhard Lührmann, Kiyoshi Nagai, Jon Staley, Cindy Will, and Rio lab members for critical review of the manuscript. Work in our lab on pre-mRNA splicing is supported by National Institutes of Health (NIH) grants (R01 GM094890 and R01 GM097352) and the NIH Center for RNA Systems Biology (P50 GM102706; J. Cate, principal investigator).
Footnotes
Note Added In Proof: We want to highlight the fact that a new 3.3-Å structural model of U1 snRNP has just been published by the Nagai lab (258).
Disclosure Statement: The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Literature Cited
- 1.Sharp PA. Split genes and RNA splicing. Cell. 1994;77:805–15. doi: 10.1016/0092-8674(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 2.Sharp PA. The discovery of split genes and RNA splicing. Trends Biochem Sci. 2005;30:279–81. doi: 10.1016/j.tibs.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 3.Kim E, Magen A, Ast G. Different levels of alternative splicing among eukaryotes. Nucleic Acids Res. 2007;35:125–31. doi: 10.1093/nar/gkl924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim H, Klein R, Majewski J, Ott J. Estimating rates of alternative splicing in mammals and invertebrates. Nat Genet. 2004;36:915–17. doi: 10.1038/ng0904-915. [DOI] [PubMed] [Google Scholar]
- 5.Merkin J, Russell C, Chen P, Burge CB. Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science. 2012;338:1593–99. doi: 10.1126/science.1228186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, et al. The evolutionary landscape of alternative splicing in vertebrate species. Science. 2012;338:1587–93. doi: 10.1126/science.1230612. [DOI] [PubMed] [Google Scholar]
- 7.Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–63. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Irimia M, Blencowe BJ. Alternative splicing: decoding an expansive regulatory layer. Curr Opin Cell Biol. 2012;24:323–32. doi: 10.1016/j.ceb.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 9.Braunschweig U, Gueroussov S, Plocik AM, Graveley BR, Blencowe BJ. Dynamic integration of splicing within gene regulatory pathways. Cell. 2013;152:1252–69. doi: 10.1016/j.cell.2013.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelemen O, Convertini P, Zhang Z, Wen Y, Shen M, et al. Function of alternative splicing. Gene. 2013;514:1–30. doi: 10.1016/j.gene.2012.07.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–18. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 12.Will CL, Lührmann R. Spliceosome structure and function. Cold Spring Harb Perspect Biol. 2011;3:a003707. doi: 10.1101/cshperspect.a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fica SM, Tuttle N, Novak T, Li NS, Lu J, et al. RNA catalyses nuclear pre-mRNA splicing. Nature. 2013;503:229–34. doi: 10.1038/nature12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009;10:741–54. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Licatalosi DD, Darnell RB. RNA processing and its regulation: global insights into biological networks. Nat Rev Genet. 2010;11:75–87. doi: 10.1038/nrg2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olson S, Blanchette M, Park J, Savva Y, Yeo GW, et al. A regulator of Dscam mutually exclusive splicing fidelity. Nat Struct Mol Biol. 2007;14:1134–40. doi: 10.1038/nsmb1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kishore S, Stamm S. Regulation of alternative splicing by snoRNAs. Cold Spring Harb Symp Quant Biol. 2006;71:329–34. doi: 10.1101/sqb.2006.71.024. [DOI] [PubMed] [Google Scholar]
- 18.Kishore S, Stamm S. The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science. 2006;311:230–32. doi: 10.1126/science.1118265. [DOI] [PubMed] [Google Scholar]
- 19.Allemand E, Batsche E, Muchardt C. Splicing, transcription, and chromatin: a menage a trois. Curr Opin Genet Dev. 2008;18:145–51. doi: 10.1016/j.gde.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 20.Luco RF, Alló M, Schor IE, Kornblihtt AR, Misteli T. Epigenetics in alternative pre-mRNA splicing. Cell. 2011;144:16–26. doi: 10.1016/j.cell.2010.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou HL, Luo G, Wise JA, Lou H. Regulation of alternative splicing by local histone modifications: potential roles for RNA-guided mechanisms. Nucleic Acids Res. 2014;42:701–13. doi: 10.1093/nar/gkt875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meister G. Argonaute proteins: functional insights and emerging roles. Nat Rev Genet. 2013;14:447–59. doi: 10.1038/nrg3462. [DOI] [PubMed] [Google Scholar]
- 23.Huang V, Li LC. Demystifying the nuclear function of Argonaute proteins. RNA Biol. 2014;11:18–24. doi: 10.4161/rna.27604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cecere G, Grishok A. A nuclear perspective on RNAi pathways in metazoans. Biochim Biophys Acta. 2014;1839:223–33. doi: 10.1016/j.bbagrm.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dujardin G, Lafaille C, Petrillo E, Buggiano V, Gómez Acuña LI, et al. Transcriptional elongation and alternative splicing. Biochim Biophys Acta. 2013;1829:134–40. doi: 10.1016/j.bbagrm.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 26.Gómez Acuña LI, Fiszbein A, Alló M, Schor IE, Kornblihtt AR. Connections between chromatin signatures and splicing. Wiley Interdiscip Rev RNA. 2013;4:77–91. doi: 10.1002/wrna.1142. [DOI] [PubMed] [Google Scholar]
- 27.Dujardin G, Lafaille C, de la Mata M, Marasco LE, Muñoz MJ, et al. How slow RNA polymerase II elongation favors alternative exon skipping. Mol Cell. 2014;54:683–90. doi: 10.1016/j.molcel.2014.03.044. [DOI] [PubMed] [Google Scholar]
- 28.Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–93. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Padgett RA. New connections between splicing and human disease. Trends Genet. 2012;28:147–54. doi: 10.1016/j.tig.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet. 1992;90:41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- 31.Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3:285–98. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 32.Pagani F, Baralle FE. Genomic variants in exons and introns: identifying the splicing spoilers. Nat Rev Genet. 2004;5:389–96. doi: 10.1038/nrg1327. [DOI] [PubMed] [Google Scholar]
- 33.Krawczak M, Thomas NS, Hundreiser B, Mort M, Wittig M, et al. Single base-pair substitutions in exon–intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Hum Mutat. 2007;28:150–58. doi: 10.1002/humu.20400. [DOI] [PubMed] [Google Scholar]
- 34.ElSharawy AGA, Hundreiser B, Brosch M, Wittig M, Huse K, et al. Systematic evaluation of the effect of common SNPs on pre-mRNA splicing. Hum Mutat. 2009;30:625–32. doi: 10.1002/humu.20906. [DOI] [PubMed] [Google Scholar]
- 35.Supek F, Miñana B, Valcárcel J, Gabaldón T, Lehner B. Synonymous mutations frequently act as driver mutations in human cancers. Cell. 2014;156:1324–35. doi: 10.1016/j.cell.2014.01.051. [DOI] [PubMed] [Google Scholar]
- 36.Ogawa S. Splicing factor mutations in myelodysplasia. Int J Hematol. 2012;96:438–42. doi: 10.1007/s12185-012-1182-y. [DOI] [PubMed] [Google Scholar]
- 37.Maciejewski JP, Padgett RA. Defects in spliceosomal machinery: a new pathway of leukaemogenesis. Br J Haematol. 2012;158:165–73. doi: 10.1111/j.1365-2141.2012.09158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119:3203–10. doi: 10.1182/blood-2011-12-399774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Auboeuf D, Carno-Fonseca M, Valcárcel J, Biamonti G. Alternative splicing and cancer. J Nucleic Acids. 2012;2012:363809. doi: 10.1155/2012/363809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tazi J, Durand S, Jeanteur P. The spliceosome: a novel multi-faceted target for therapy. Trends Biochem Sci. 2005;30:469–78. doi: 10.1016/j.tibs.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 41.Bonnal S, Vigevani L, Valcárcel J. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov. 2012;11:847–59. doi: 10.1038/nrd3823. [DOI] [PubMed] [Google Scholar]
- 42.Cartegni L, Krainer AR. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat Struct Biol. 2003;10:120–25. doi: 10.1038/nsb887. [DOI] [PubMed] [Google Scholar]
- 43.Hua Y, Vickers TA, Okunola HL, Bennett F, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834–48. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sahashi K, Hua Y, Ling KK, Hung G, Rigo F, et al. TSUNAMI: an antisense method to phenocopy splicing-associated diseases in animals. Genes Dev. 2012;26:1874–84. doi: 10.1101/gad.197418.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods. 2013;10:957–63. doi: 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol. 2014;32:551–53. doi: 10.1038/nbt.2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hartmuth K, Vornlocher HP, Lührmann R. Tobramycin affinity tag purification of spliceosomes. Methods Mol Biol. 2004;257:47–64. doi: 10.1385/1-59259-750-5:047. [DOI] [PubMed] [Google Scholar]
- 48.Zhou Z, Licklider LJ, Gygi SP, Reed R. Comprehensive proteomic analysis of the human spliceosome. Nature. 2002;419:182–85. doi: 10.1038/nature01031. [DOI] [PubMed] [Google Scholar]
- 49.Zhou Z, Sim J, Griffith J, Reed R. Purification and electron microscopic visualization of functional human spliceosomes. PNAS. 2002;99:12203–7. doi: 10.1073/pnas.182427099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jurica MS, Moore MJ. Capturing splicing complexes to study structure and mechanism. Methods. 2002;28:336–45. doi: 10.1016/s1046-2023(02)00240-2. [DOI] [PubMed] [Google Scholar]
- 51.Herold N, Will CL, Wolf E, Kastner B, Urlaub H, Lührmann R. Conservation of the protein composition and electron microscopy structure of Drosophila melanogaster and human spliceosomal complexes. Mol Cell Biol. 2009;29:281–301. doi: 10.1128/MCB.01415-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fabrizio P, Dannenberg J, Dube P, Kastner P, Stark H, et al. The evolutionarily conserved core design of the catalytic activation step of the yeast spliceosome. Mol Cell. 2009;36:593–608. doi: 10.1016/j.molcel.2009.09.040. [DOI] [PubMed] [Google Scholar]
- 53.Jurica MS, Moore MJ. Pre-mRNA splicing: awash in a sea of proteins. Mol Cell. 2003;12:5–14. doi: 10.1016/s1097-2765(03)00270-3. [DOI] [PubMed] [Google Scholar]
- 54.Hegele A, Kamburov A, Grossman A, Sourlis C, Wowro S, et al. Dynamic protein–protein interaction wiring of the human spliceosome. Mol Cell. 2012;45:567–80. doi: 10.1016/j.molcel.2011.12.034. [DOI] [PubMed] [Google Scholar]
- 55.Häcker I, Sander B, Golas MM, Karagöz E, Kastner B, et al. Localization of Prp8, Brr2, Snu114 and U4/U6 proteins in the yeast tri-snRNP by electron microscopy. Nat Struct Mol Biol. 2008;15:1206–12. doi: 10.1038/nsmb.1506. [DOI] [PubMed] [Google Scholar]
- 56.Behzadnia N, Golas MM, Harmuth K, Sander B, Deckert J, et al. Composition and three-dimensional EM structure of double affinity-purified, human prespliceosomal A complexes. EMBO J. 2007;26:1737–48. doi: 10.1038/sj.emboj.7601631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deckert J, Harmuth K, Boehringer D, Behzadnia N, Will CL, et al. Protein composition and electron microscopy structure of affinity-purified human spliceosomal B complexes isolated under physiological conditions. Mol Cell Biol. 2006;26:5528–43. doi: 10.1128/MCB.00582-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bessonov S, Anokhina M, Krasauskas A, Golas MM, Sander B, et al. Characterization of purified human Bact spliceosomal complexes reveals compositional and morphological changes during spliceo-some activation and first step catalysis. RNA. 2010;16:2384–403. doi: 10.1261/rna.2456210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Golas MM, Sander B, Bessonov S, Grote M, Wolf E, et al. 3D cryo-EM structure of an active step I spliceosome and localization of its catalytic core. Mol Cell. 2010;40:927–38. doi: 10.1016/j.molcel.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 60.Jurica MS, Sousa D, Moore MJ, Girgorieff N. Three-dimensional structure of C complex spliceosomes by electron microscopy. Nat Struct Mol Biol. 2004;11:265–69. doi: 10.1038/nsmb728. [DOI] [PubMed] [Google Scholar]
- 61.Boehringer D, Makarov EM, Sander B, Markaova OV, Kastner B, et al. Three-dimensional structure of a pre-catalytic human spliceosomal complex B. Nat Struct Mol Biol. 2004;11:463–68. doi: 10.1038/nsmb761. [DOI] [PubMed] [Google Scholar]
- 62.Warkocki Z, Odenwälder P, Schmitzov´ J, Platzmann F, Stark H, et al. Reconstitution of both steps of Saccharomyces cerevisiae splicing with purified spliceosomal components. Nat Struct Mol Biol. 2009;16:1237–43. doi: 10.1038/nsmb.1729. [DOI] [PubMed] [Google Scholar]
- 63.Stark H, Lührmann R. Cryon–electron microscopy of spliceosomal components. Annu Rev Biophys Biomol Struct. 2006;35:435–57. doi: 10.1146/annurev.biophys.35.040405.101953. [DOI] [PubMed] [Google Scholar]
- 64.Pomeranz Krummel DA, Oubridge C, Leung AKW, Li J, Nagai K. Crystal structure of human spliceosomal U1 snRNP at 5.5 Å resolution. Nature. 2009;458:475–80. doi: 10.1038/nature07851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oubridge C, Ito N, Evans PR, Teo CH, Nagai K. Crystal structure at 1.92 Å resolution of the RNA-binding domain of the U1A spliceosomal protein complexed with an RNA hairpin. Nature. 1994;372:432–38. doi: 10.1038/372432a0. [DOI] [PubMed] [Google Scholar]
- 66.Weber G, Trowitzsch S, Kastner B, Lührmann R, Wahl MC. Functional organization of the Sm core in the crystal structure of human U1 snRNP. EMBO J. 2010;29:4172–84. doi: 10.1038/emboj.2010.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Montemayor EJ, Curran EC, Liao HH, Andrews KL, Treba CN, et al. Core structure of the U6 small nuclear ribonucleoprotein at 1.7-Å resolution. Nat Struct Mol Biol. 2014;21:544–51. doi: 10.1038/nsmb.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leung AK, Nagai K, Li J. Structure of the spliceosomal U4 snRNP core domain and its implication for snRNP biogenesis. Nature. 2011;473:536–39. doi: 10.1038/nature09956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bessonov S, Anokhina M, Will CL, Urlaub H, Lührmann R. Isolation of an active step I spliceosome and composition of its RNP core. Nature. 2008;452:846–50. doi: 10.1038/nature06842. [DOI] [PubMed] [Google Scholar]
- 70.Tseng CK, Cheng SC. Both catalytic steps of nuclear pre-mRNA splicing are reversible. Science. 2008;320:1782–84. doi: 10.1126/science.1158993. [DOI] [PubMed] [Google Scholar]
- 71.Hoskins AA, Gelles J, Moore MJ. New insights into the spliceosome by single molecule fluorescence microscopy. Curr Opin Chem Biol. 2011;15:864–70. doi: 10.1016/j.cbpa.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Crawford DJ, Hoskins AA, Friedman LJ, Gelles J, Moore MJ. Visualizing the splicing of single pre-mRNA molecules in whole cell extract. RNA. 2008;14:170–79. doi: 10.1261/rna.794808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hoskins AA, Friedman LJ, Gallagher SS, Crawford DJ, Anderson EG, et al. Ordered and dynamic assembly of single spliceosomes. Science. 2011;331:1289–95. doi: 10.1126/science.1198830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shcherbakova I, Hoskins AA, Friedman LJ, Serebrov V, Correa IR, Jr, et al. Alternative spliceosome assembly pathways revealed by single-molecule fluorescence microscopy. Cell Rep. 2013;5:151–65. doi: 10.1016/j.celrep.2013.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Crawford DJ, Hoskins AA, Friedman LJ, Gelles J, Moore MJ. Single-molecule colocalization FRET evidence that spliceosome activation precedes stable approach of 5′ splice site and branch site. PNAS. 2013;110:6783–88. doi: 10.1073/pnas.1219305110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Abelson J, Blanco M, Ditzler MA, Fuller F, Aravamudhan P, et al. Conformational dynamics of single pre-mRNA molecules during in vitro splicing. Nat Struct Mol Biol. 2010;17:504–12. doi: 10.1038/nsmb.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marcia M, Pyle AM. Visualizing group II intron catalysis through the stages of splicing. Cell. 2012;151:497–507. doi: 10.1016/j.cell.2012.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fica SM, Mefford MA, Piccirilli JA, Staley JP. Evidence for a group II intron–like catalytic triplex in the spliceosome. Nat Struct Mol Biol. 2014;21:464–71. doi: 10.1038/nsmb.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smith DJ, Query CC, Konarska MM. “Nought may endure but mutability”: spliceosome dynamics and the regulation of splicing. Mol Cell. 2008;30:657–66. doi: 10.1016/j.molcel.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Newman AJ, Nagai K. Structural studies of the spliceosome: blind men and an elephant. Curr Opin Struct Biol. 2010;20:82–89. doi: 10.1016/j.sbi.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 81.Pena V, Rozov A, Fabrizio P, Lührmann R, Wahl MC. Structure and function of an RNase H domain at the heart of the spliceosome. EMBO J. 2008;27:2929–40. doi: 10.1038/emboj.2008.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schellenberg MJ, Ritchie DB, MacMillan AM. Pre-mRNA splicing: a complex picture in higher definition. Trends Biochem Sci. 2008;33:243–46. doi: 10.1016/j.tibs.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 83.Galej WP, Oubridge C, Newman AJ, Nagai K. Crystal structure of Prp8 reveals active site cavity of the spliceosome. Nature. 2013;493:638–43. doi: 10.1038/nature11843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Galej WP, Nguyen THD, Newman AJ, Nagai K. Structural studies of the spliceosome: zooming into the heart of the machine. Curr Opin Struct Biol. 2014;25:57–66. doi: 10.1016/j.sbi.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schellenberg MJ, Wu T, Ritchie DB, Fica S, Staley JP, et al. A conformational switch in PRP8 mediates metal ion coordination that promotes pre-mRNA exon ligation. Nat Struct Mol Biol. 2013;20:728–34. doi: 10.1038/nsmb.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Valadkhan S, Manley JL. Splicing-related catalysis by protein-free snRNAs. Nature. 2001;413:701–7. doi: 10.1038/35099500. [DOI] [PubMed] [Google Scholar]
- 87.Valadkhan S, Manley JL. Characterization of the catalytic activity of U2 and U6 snRNAs. RNA. 2003;9:892–904. doi: 10.1261/rna.5440303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sontheimer EJ, Sun S, Piccirilli JA. Metal ion catalysis during splicing of premessenger RNA. Nature. 1997;388:801–5. doi: 10.1038/42068. [DOI] [PubMed] [Google Scholar]
- 89.Fabrizio P, McPheeters DS, Abelson J. In vitro assembly of yeast U6 snRNP: a functional assay. Genes Dev. 1989;3:2137–50. doi: 10.1101/gad.3.12b.2137. [DOI] [PubMed] [Google Scholar]
- 90.Treisman R, Orkin SH, Maniatis T. Specific transcription and RNA splicing defects in five cloned β-thalassaemia genes. Nature. 1983;302:591–96. doi: 10.1038/302591a0. [DOI] [PubMed] [Google Scholar]
- 91.Treisman R, Proudfoot NJ, Shander M, Maniatis T. A single-base change at a splice site in a β0-thalassemic gene causes abnormal RNA splicing. Cell. 1982;29:903–11. doi: 10.1016/0092-8674(82)90452-4. [DOI] [PubMed] [Google Scholar]
- 92.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–15. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 93.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–76. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet. 2003;34:460–63. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- 95.Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002;30:377–84. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- 96.David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364–68. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kashima T, Rao N, David CJ, Manley JL. hnRNP A1 functions with specificity in repression of SMN2 exon 7 splicing. Hum Mol Genet. 2007;16:3149–59. doi: 10.1093/hmg/ddm276. [DOI] [PubMed] [Google Scholar]
- 98.Wang Z, Chatterjee D, Jeon HY, Akerman M, Vander Heiden MG, et al. Exon-centric regulation of pyruvate kinase M alternative splicing via mutually exclusive exons. J Mol Cell Biol. 2012;4:79–87. doi: 10.1093/jmcb/mjr030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 2010;24:2343–64. doi: 10.1101/gad.1973010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lovci MT, Ghanem D, Marr H, Arnold J, Gee S, et al. Rbfox proteins regulate alternative mRNA splicing through evolutionarily conserved RNA bridges. Nat Struct Mol Biol. 2013;20:1434–42. doi: 10.1038/nsmb.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang J, Manley JL. Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov. 2013;3:1228–37. doi: 10.1158/2159-8290.CD-13-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–73. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012;1462:61–80. doi: 10.1016/j.brainres.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kwon I, Kato M, Xiang S, Wu L, Theodoropoulos P, et al. Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell. 2013;155:1049–60. doi: 10.1016/j.cell.2013.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hahn CN, Scott HS. Spliceosome mutations in hematopoietic malignancies. Nat Genet. 2012;44:9–10. doi: 10.1038/ng.1045. [DOI] [PubMed] [Google Scholar]
- 106.Przychodzen B, Jerez A, Guinta K, Sekeres MA, Padgett R, et al. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood. 2013;122:999–1006. doi: 10.1182/blood-2013-01-480970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–20. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brooks AN, Choi PS, de Waal L, Sharifnia T, Imielinksi M, et al. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLOS ONE. 2014;9:e87361. doi: 10.1371/journal.pone.0087361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol. 2007;14:185–93. doi: 10.1038/nsmb1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Karni R, Hippo Y, Lowe SW, Krainer AR. The splicing-factor oncoprotein SF2/ASF activates mTORC1. PNAS. 2008;105:15323–27. doi: 10.1073/pnas.0801376105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol. 2007;3:576–83. doi: 10.1038/nchembio.2007.18. [DOI] [PubMed] [Google Scholar]
- 112.Roybal GA, Jurica MS. Spliceostatin A inhibits spliceosome assembly subsequent to prespliceosome formation. Nucleic Acids Res. 2010;38:6664–72. doi: 10.1093/nar/gkq494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Effenberger KA, Perriman RJ, Bray WM, Lokey RS, Ares M, Jr, Jurica MS. A high-throughput splicing assay identifies new classes of inhibitors of human and yeast spliceosomes. J Biomol Screen. 2013;18:1110–20. doi: 10.1177/1087057113493117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ilagan JO, Jurica MS. Isolation and accumulation of spliceosomal assembly intermediates. Methods Mol Biol. 2014;1126:179–92. doi: 10.1007/978-1-62703-980-2_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Passini MA, Bu J, Richards AM, Kinnecom C, Sardi SP, et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. 2011;3:72ra18. doi: 10.1126/scitranslmed.3001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hua Y, Sahashi K, Rigo F, Hung G, Horev G, et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478:123–26. doi: 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rigo F, Hua Y, Krainer AR, Bennett CF. Antisense-based therapy for the treatment of spinal muscular atrophy. J Cell Biol. 2012;199:21–25. doi: 10.1083/jcb.201207087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 119.Blencowe BJ. Alternative splicing: new insights from global analyses. Cell. 2006;126:37–47. doi: 10.1016/j.cell.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 120.Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–13. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Martinez-Contreras R, Cloutier P, Shkreta L, Fisette JF, Revil T, Chabot B. hnRNP proteins and splicing control. Adv Exp Med Biol. 2007;623:123–47. doi: 10.1007/978-0-387-77374-2_8. [DOI] [PubMed] [Google Scholar]
- 122.Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem J. 2009;417:15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- 123.Manley JL, Krainer AR. A rational nomenclature for serine/arginine-rich protein splicing factors (SR proteins) Genes Dev. 2010;24:1073–74. doi: 10.1101/gad.1934910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Änkö ML. Regulation of gene expression programmes by serine–arginine rich splicing factors. Semin Cell Dev Biol. 2014;32:11–21. doi: 10.1016/j.semcdb.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 125.Zhou Z, Fu XD. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma. 2013;122:191–207. doi: 10.1007/s00412-013-0407-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Darnell RB. RNA protein interaction in neurons. Annu Rev Neurosci. 2013;36:243–70. doi: 10.1146/annurev-neuro-062912-114322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang C, Zhang Z, Castle J, Sun S, Johnson J, et al. Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev. 2008;22:2550–63. doi: 10.1101/gad.1703108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yeo GW, Coufal NG, Liang TY, Peng GE, Fu XD, Gage FH. An RNA code for the FOX2 splicing regulator revealed by mapping RNA–protein interactions in stem cells. Nat Struct Mol Biol. 2009;16:130–37. doi: 10.1038/nsmb.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Weyn-Vanhentenryck SM, Mele A, Yan Q, Sun S, Farny N, et al. HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep. 2014;6:1139–52. doi: 10.1016/j.celrep.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang ET, Cody NAL, Jog S, Biancolella M, Wang TT, et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell. 2012;150:710–24. doi: 10.1016/j.cell.2012.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Han H, Irimia M, Ross PJ, Sung HK, Alipanahi B, et al. MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature. 2013;498:241–45. doi: 10.1038/nature12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pandit S, Zhou Y, Shiue L, Coutinho-Mansfield G, Li H, et al. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol Cell. 2013;50:223–35. doi: 10.1016/j.molcel.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jean-Philippe J, Paz S, Caputi M. hnRNP A1: the Swiss army knife of gene expression. Int J Mol Sci. 2013;14:18999–9024. doi: 10.3390/ijms140918999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Huelga SC, Vu AQ, Arnold JD, Liang TY, Liu PP, et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 2012;1:167–78. doi: 10.1016/j.celrep.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Blanchette M, Green RE, MacArthur S, Brooks AN, Brenner SE, et al. Genome-wide analysis of alternative pre-mRNA splicing and RNA-binding specificities of the Drosophila hnRNP A/B family members. Mol Cell. 2009;33:438–49. doi: 10.1016/j.molcel.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Venables JP, Koh CS, Froehlich U, Couture S, Inkle L, et al. Multiple and specific mRNA processing targets for the major human hnRNP proteins. Mol Cell Biol. 2008;28:6033–43. doi: 10.1128/MCB.00726-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Abdul-Manan N, O'Malley SM, Williams KR. Origins of binding specificity of the A1 heterogeneous nuclear ribonucleoprotein. Biochemistry. 1996;35:3545–54. doi: 10.1021/bi952298p. [DOI] [PubMed] [Google Scholar]
- 138.Burd CG, Dreyfuss G. RNA binding specificity of hnRNP A1: significance of hnRNP A1 high-affinity binding sites in pre-mRNA splicing. EMBO J. 1994;13:1197–204. doi: 10.1002/j.1460-2075.1994.tb06369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Caputi M, Mayeda A, Krainer AR, Zahler AM. hnRNP A/B proteins are required for inhibition of HIV-1 pre-mRNA splicing. EMBO J. 1999;18:4060–67. doi: 10.1093/emboj/18.14.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhu J, Mayeda A, Krainer AR. Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Mol Cell. 2001;8:1351–61. doi: 10.1016/s1097-2765(01)00409-9. [DOI] [PubMed] [Google Scholar]
- 141.Okunola HL, Krainer AR. Cooperative-binding and splicing-repressive properties of hnRNP A1. Mol Cell Biol. 2009;29:5620–31. doi: 10.1128/MCB.01678-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. PNAS. 2010;107:1894–99. doi: 10.1073/pnas.0914845107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Blanchette M, Chabot B. Modulation of exon skipping by high-affinity hnRNP A1–binding sites and by intron elements that repress splice site utilization. EMBO J. 1999;18:1939–52. doi: 10.1093/emboj/18.7.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Martinez-Contreras R, Fisette JF, Nasim FH, Madden R, Cordeau M, Chabot B. Intronic binding sites for hnRNP A/B and hnRNP F/H proteins stimulate pre-mRNA splicing. PLOS Biol. 2006;4:e21. doi: 10.1371/journal.pbio.0040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Oh HK, Lee E, Jang HN, Lee J, Moon H, et al. hnRNP A1 contacts exon 5 to promote exon 6 inclusion of apoptotic Fas gene. Apoptosis. 2013;18:825–35. doi: 10.1007/s10495-013-0824-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Motta-Mena LB, Heyd F, Lynch KW. Context-dependent regulatory mechanism of the splicing factor hnRNP L. Mol Cell. 2010;37:223–34. doi: 10.1016/j.molcel.2009.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.House AE, Lynch KW. An exonic splicing silencer represses spliceosome assembly after ATP-dependent exon recognition. Nat Struct Mol Biol. 2006;13:937–44. doi: 10.1038/nsmb1149. [DOI] [PubMed] [Google Scholar]
- 148.Chiou NT, Shankarling G, Lynch KW. hnRNP L and hnRNP A1 induce extended U1 snRNA interactions with an exon to repress spliceosome assembly. Mol Cell. 2013;49:972–82. doi: 10.1016/j.molcel.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Heiner M, Hui J, Schreiner S, Hung LH, Bindereif A. HnRNPL-mediated regulation of mammalian alternative splicing by interference with splice site recognition. RNA Biol. 2010;7:56–64. doi: 10.4161/rna.7.1.10402. [DOI] [PubMed] [Google Scholar]
- 150.Rossbach O, Hung LH, Khrameeva E, Schreiner S, König J, et al. Crosslinking-immunoprecipitation (iCLIP) analysis reveals global regulatory roles of hnRNP L. RNA Biol. 2014;11:146–55. doi: 10.4161/rna.27991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Siebel CW, Fresco LD, Rio DC. The mechanism of somatic inhibition of Drosophila P-element pre-mRNA splicing: multiprotein complexes at an exon pseudo-5′ splice site control U1 snRNP binding. Genes Dev. 1992;6:1386–401. doi: 10.1101/gad.6.8.1386. [DOI] [PubMed] [Google Scholar]
- 152.Siebel CW, Rio DC. Regulated splicing of the Drosophila P transposable element third intron in vitro: somatic repression. Science. 1990;248:1200–8. doi: 10.1126/science.2161558. [DOI] [PubMed] [Google Scholar]
- 153.Siebel CW, Kanaar R, Rio DC. Regulation of tissue-specific P-element pre-mRNA splicing requires the RNA-binding protein PSI. Genes Dev. 1994;8:1713–25. doi: 10.1101/gad.8.14.1713. [DOI] [PubMed] [Google Scholar]
- 154.Siebel CW, Admon A, Rio DC. Soma-specific expression and cloning of PSI, a negative regulator of P element pre-mRNA splicing. Genes Dev. 1995;9:269–83. doi: 10.1101/gad.9.3.269. [DOI] [PubMed] [Google Scholar]
- 155.Tange TO, Damgaard CK, Guth S, Valc´rcel J, Kjems J. The hnRNP A1 protein regulates HIV-1 tat splicing via a novel intron silencer element. EMBO J. 2001;20:5748–58. doi: 10.1093/emboj/20.20.5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Damgaard CK, Tange TO, Kjems J. hnRNP A1 controls HIV-1 mRNA splicing through cooperative binding to intron and exon splicing silencers in the context of a conserved secondary structure. RNA. 2002;8:1401–15. doi: 10.1017/s1355838202023075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Schaal TD, Maniatis T. Selection and characterization of pre-mRNA splicing enhancers: identification of novel SR protein–specific enhancer sequences. Mol Cell Biol. 1999;19:1705–19. doi: 10.1128/mcb.19.3.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Liu HX, Chew SL, Cartengi L, Zhang MQ, Krainer AR. Exonic splicing enhancer motif recognized by human SC35 under splicing conditions. Mol Cell Biol. 2000;20:1063–71. doi: 10.1128/mcb.20.3.1063-1071.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Fairbrother WG, Yeh RF, Sharp PA, Burge CB. Predictive identification of exonic splicing enhancers in human genes. Science. 2002;297:1007–13. doi: 10.1126/science.1073774. [DOI] [PubMed] [Google Scholar]
- 160.Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB. Systematic identification and analysis of exonic splicing silencers. Cell. 2004;119:831–45. doi: 10.1016/j.cell.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 161.Wang Z, Xiao X, Van Nostrand E, Burge CB. General and specific functions of exonic splicing silencers in splicing control. Mol Cell. 2006;23:61–70. doi: 10.1016/j.molcel.2006.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yu Y, Maroney PA, Denker JA, Zhang XHF, Dybkov O, et al. Dynamic regulation of alternative splicing by silencers that modulate 5′ splice site competition. Cell. 2008;135:1224–36. doi: 10.1016/j.cell.2008.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wang Y, Xiao X, Zhang J, Choudhury R, Robertson A, et al. A complex network of factors with overlapping affinities represses splicing through intronic elements. Nat Struct Mol Biol. 2013;20:36–45. doi: 10.1038/nsmb.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang Y, Wang Z. Systematical identification of splicing regulatory cis-elements and cognate trans-factors. Methods. 2014;65:350–58. doi: 10.1016/j.ymeth.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chabot B, Steitz JA. Recognition of mutant and cryptic 5′ splice sites by the U1 small nuclear ribonucleoprotein in vitro. Mol Cell Biol. 1987;7:698–707. doi: 10.1128/mcb.7.2.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nelson KK, Green MR. Splice site selection and ribonucleoprotein complex assembly during in vitro pre-mRNA splicing. Genes Dev. 1988;2:319–29. doi: 10.1101/gad.2.3.319. [DOI] [PubMed] [Google Scholar]
- 167.Berg MG, Singh LN, Younis I, Liu Q, Pinto AM, et al. U1 snRNP determines mRNA length and regulates isoform expression. Cell. 2012;150:53–64. doi: 10.1016/j.cell.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kaida D, Berg MG, Younis I, Kasim M, Singh LN, et al. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature. 2010;468:664–68. doi: 10.1038/nature09479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Labourier E, Adams MD, Rio DC. Modulation of P-element pre-mRNA splicing by a direct interaction between PSI and U1 snRNP 70K protein. Mol Cell. 2001;8:363–73. doi: 10.1016/s1097-2765(01)00311-2. [DOI] [PubMed] [Google Scholar]
- 170.Kohtz JD, Jamison SF, Will CL, Zuo P, Lührmann R, et al. Protein–protein interactions and 5′-splice-site recognition in mammalian mRNA precursors. Nature. 1994;368:119–24. doi: 10.1038/368119a0. [DOI] [PubMed] [Google Scholar]
- 171.Förch P, Puig O, Martínez C, Séraphin B, Valc´rcel J. The splicing regulator TIA-1 interacts with U1-C to promote U1 snRNP recruitment to 5′ splice sites. EMBO J. 2002;21:6882–92. doi: 10.1093/emboj/cdf668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Du H, Rosbash M. The U1 snRNP protein U1C recognizes the 5′ splice site in the absence of base pairing. Nature. 2002;419:86–90. doi: 10.1038/nature00947. [DOI] [PubMed] [Google Scholar]
- 173.Sharma S, Maris C, Allain FHT, Black DL. U1 snRNA directly interacts with polypyrimidine tract–binding protein during splicing repression. Mol Cell. 2011;41:579–88. doi: 10.1016/j.molcel.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lynch KW, Maniatis T. Assembly of specific SR protein complexes on distinct regulatory elements of the Drosophila doublesex splicing enhancer. Genes Dev. 1996;10:2089–101. doi: 10.1101/gad.10.16.2089. [DOI] [PubMed] [Google Scholar]
- 175.Ohno G, Ono K, Togo M, Watanabe Y, Ono S, et al. Muscle-specific splicing factors ASD-2 and SUP-12 cooperatively switch alternative pre-mRNA processing patterns of the ADF/cofilin gene in Caenorhabditis elegans. PLOS Genet. 2012;8:e1002991. doi: 10.1371/journal.pgen.1002991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Buratti E, Baralle FE. Influence of RNA secondary structure on the pre-mRNA splicing process. Mol Cell Biol. 2004;24:10505–14. doi: 10.1128/MCB.24.24.10505-10514.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.McManus CJ, Graveley BR. RNA structure and the mechanisms of alternative splicing. Curr Opin Genet Dev. 2011;21:373–79. doi: 10.1016/j.gde.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature. 2014;505:701–5. doi: 10.1038/nature12894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Graveley BR. Mutually exclusive splicing of the insect Dscam pre-mRNA directed by competing intronic RNA secondary structures. Cell. 2005;123:65–73. doi: 10.1016/j.cell.2005.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.May GE, Olson S, McManus CJ, Graveley BR. Competing RNA secondary structures are required for mutually exclusive splicing of the Dscam exon 6 cluster. RNA. 2011;17:222–29. doi: 10.1261/rna.2521311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Miura SK, Martins A, Zhang KX, Graveley BR, Zipursky SL. Probabilistic splicing of Dscam1 establishes identity at the level of single neurons. Cell. 2013;155:1166–77. doi: 10.1016/j.cell.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yang Y, Zhan L, Zhang W, Sun F, Wang W, et al. RNA secondary structure in mutually exclusive splicing. Nat Struct Mol Biol. 2011;18:159–68. doi: 10.1038/nsmb.1959. [DOI] [PubMed] [Google Scholar]
- 183.de Almeida SF, Carmo-Fonseca M. Design principles of interconnections between chromatin and pre-mRNA splicing. Trends Biochem Sci. 2012;37:248–53. doi: 10.1016/j.tibs.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 184.Iannone C, Valc´rcel J. Chromatin's thread to alternative splicing regulation. Chromosoma. 2013;122:465–74. doi: 10.1007/s00412-013-0425-x. [DOI] [PubMed] [Google Scholar]
- 185.de la Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, et al. A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell. 2003;12:525–32. doi: 10.1016/j.molcel.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 186.Sims RJ, 3rd, Millhouse S, Chen CF, Lewis BA, Erdjument-Bromage H, et al. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol Cell. 2007;28:665–76. doi: 10.1016/j.molcel.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010;327:996–1000. doi: 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Alló M, Buggiano V, Fededa JP, Petrillo E, Schor I, et al. Control of alternative splicing through siRNA-mediated transcriptional gene silencing. Nat Struct Mol Biol. 2009;16:717–24. doi: 10.1038/nsmb.1620. [DOI] [PubMed] [Google Scholar]
- 189.Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, et al. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003;302:2141–44. doi: 10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- 190.Pan Q, Shai O, Misquitta C, Zhang W, Saltzman AL, et al. Revealing global regulatory features of mammalian alternative splicing using a quantitative microarray platform. Mol Cell. 2004;16:929–41. doi: 10.1016/j.molcel.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 191.Blanchette M, Green RE, Brenner SE, Rio DC. Global analysis of positive and negative pre-mRNA splicing regulators in Drosophila. Genes Dev. 2005;19:1306–14. doi: 10.1101/gad.1314205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–87. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- 193.Labourier E, Blanchette M, Feiger JW, Adams MD, Rio DC. The KH-type RNA-binding protein PSI is required for Drosophila viability, male fertility, and cellular mRNA processing. Genes Dev. 2002;16:72–84. doi: 10.1101/gad.948602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Mili S, Steitz JA. Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA. 2004;10:1692–94. doi: 10.1261/rna.7151404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Pawlicki JM, Steitz JA. Nuclear networking fashions pre-messenger RNA and primary microRNA transcripts for function. Trends Cell Biol. 2010;20:52–61. doi: 10.1016/j.tcb.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–15. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- 197.Choi YD, Dreyfuss G. Monoclonal antibody characterization of the C proteins of heterogeneous nuclear ribonucleoprotein complexes in vertebrate cells. J Cell Biol. 1984;99:1997–204. doi: 10.1083/jcb.99.6.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–69. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Sanford JR, Wang X, Mort M, Vanduyn N, Cooper DN, et al. Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res. 2009;19:381–94. doi: 10.1101/gr.082503.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Xue Y, Zhou Y, Wu T, Zhu T, Ji X, et al. Genome-wide analysis of PTB–RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol Cell. 2009;36:996–1006. doi: 10.1016/j.molcel.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Huppertz I, Attig J, D'Ambrogio A, Easton LE, Sibley CR, et al. iCLIP: protein-RNA interactions at nucleotide resolution. Methods. 2014;65:274–87. doi: 10.1016/j.ymeth.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser L, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–41. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Chen J, Zhang Z, Li L, Chen BC, Revyakin A, et al. Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell. 2014;156:1274–85. doi: 10.1016/j.cell.2014.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Singh G, Ricci EP, Moore MJ. RIPiT-seq: a high-throughput approach for footprinting RNA:protein complexes. Methods. 2014;65:320–32. doi: 10.1016/j.ymeth.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Silverman IM, Li F, Alexander A, Goff L, Trapnell C, et al. RNase-mediated protein footprint sequencing reveals protein-binding sites throughout the human transcriptome. Genome Biol. 2014;15:R3. doi: 10.1186/gb-2014-15-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lambert N, Robertson A, Jangi M, McGeary S, Sharp PA, Burge CB. RNA Bind-n-Seq: quantitative assessment of the sequence and structural binding specificity of RNA binding proteins. Mol Cell. 2014;54:887–900. doi: 10.1016/j.molcel.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–77. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Spitale RC, Crisalli P, Flynn RA, Torre EA, Kool ET, Chang HY. RNA SHAPE analysis in living cells. Nat Chem Biol. 2013;9:18–20. doi: 10.1038/nchembio.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ding Y, Tang Y, Kwok CK, Zhang Y, Bevilacqua PC, Assmann SM. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature. 2014;505:696–700. doi: 10.1038/nature12756. [DOI] [PubMed] [Google Scholar]
- 210.Talkish J, May G, Lin Y, Woolford JL, Jr, McManus CJ. Mod-seq: high-throughput sequencing for chemical probing of RNA structure. RNA. 2014;20:713–20. doi: 10.1261/rna.042218.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Witten JT, Ule J. Understanding splicing regulation through RNA splicing maps. Trends Genet. 2011;27:89–97. doi: 10.1016/j.tig.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.König J, Zarnack K, Luscombe NM, Ule J. Protein-RNA interactions: new genomic technologies and perspectives. Nat Rev Genet. 2011;13:77–83. doi: 10.1038/nrg3141. [DOI] [PubMed] [Google Scholar]
- 213.Brooks AN, Yang L, Duff MO, Hansen KD, Park JW, et al. Conservation of an RNA regulatory map between Drosophila and mammals. Genome Res. 2011;21:193–202. doi: 10.1101/gr.108662.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Charizanis K, Lee KY, Batra R, Goodwin M, Zhang C, et al. Muscleblind-like 2–mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron. 2012;75:437–50. doi: 10.1016/j.neuron.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Barash Y, Calarco JA, Gao W, Pan Q, Wang X, et al. Deciphering the splicing code. Nature. 2010;465:53–59. doi: 10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- 216.Zhang C, Frias MA, Mele A, Ruggiu M, Eom T, et al. Integrative modeling defines the nova splicing-regulatory network and its combinatorial controls. Science. 2010;329:439–43. doi: 10.1126/science.1191150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Perales R, Bentley D. “Cotranscriptionality”: the transcription elongation complex as a nexus for nuclear transactions. Mol Cell. 2009;36:178–91. doi: 10.1016/j.molcel.2009.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Lacadie SA, Rosbash M. Cotranscriptional spliceosome assembly dynamics and the role of U1 snRNA:5′ss base pairing in yeast. Mol Cell. 2005;19:65–75. doi: 10.1016/j.molcel.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 219.Görnemann J, Kotovic KM, Hujer K, Neugebauer KM. Cotranscriptional spliceosome assembly occurs in a stepwise fashion and requires the cap binding complex. Mol Cell. 2005;19:53–63. doi: 10.1016/j.molcel.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 220.Listerman I, Sapra AK, Neugebauer KM. Cotranscriptional coupling of splicing factor recruitment and precursor messenger RNA splicing in mammalian cells. Nat Struct Mol Biol. 2006;13:815–22. doi: 10.1038/nsmb1135. [DOI] [PubMed] [Google Scholar]
- 221.Darnell JE., Jr Reflections on the history of pre-mRNA processing and highlights of current knowledge: a unified picture. RNA. 2013;19:443–60. doi: 10.1261/rna.038596.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Rodriguez J, Menet JS, Rosbash M. Nascent-seq indicates widespread cotranscriptional RNA editing in Drosophila. Mol Cell. 2012;47:27–37. doi: 10.1016/j.molcel.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Khodor YL, Menet JS, Tolan M, Rosbash M. Cotranscriptional splicing efficiency differs dramatically between Drosophila and mouse. RNA. 2012;18:2174–86. doi: 10.1261/rna.034090.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Khodor YL, Rodriguez J, Abruzzi KC, Tang CHA, Marr MT, II, Rosbash M. Nascent-seq indicates widespread cotranscriptional pre-mRNA splicing in Drosophila. Genes Dev. 2011;25:2502–12. doi: 10.1101/gad.178962.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Lacadie SA, Tardiff DF, Kadener S, Rosbash M. In vivo commitment to yeast cotranscriptional splicing is sensitive to transcription elongation mutants. Genes Dev. 2006;20:2055–66. doi: 10.1101/gad.1434706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Carrillo Oesterreich F, Preibisch S, Neugebauer KM. Global analysis of nascent RNA reveals transcriptional pausing in terminal exons. Mol Cell. 2010;40:571–81. doi: 10.1016/j.molcel.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 227.Pandya-Jones A, Black DL. Co-transcriptional splicing of constitutive and alternative exons. RNA. 2009;15:1896–908. doi: 10.1261/rna.1714509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Pandya-Jones A, Bhatt DM, Lin CH, Tong AJ, Smale ST, Black DL. Splicing kinetics and transcript release from the chromatin compartment limit the rate of lipid A–induced gene expression. RNA. 2013;19:811–27. doi: 10.1261/rna.039081.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Bhatt DM, Pandya-Jones A, Tong AJ, Barozzi I, Lissner MM, et al. Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions. Cell. 2012;150:279–90. doi: 10.1016/j.cell.2012.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Brugiolo M, Herzel L, Neugebauer KM. Counting on co-transcriptional splicing. F1000Prime Rep. 2013;5:9. doi: 10.12703/P5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Brody Y, Neufeld N, Bieberstein N, Causse SZ, Böhnlein EM, et al. The in vivo kinetics of RNA polymerase II elongation during co-transcriptional splicing. PLOS Biol. 2011;9:e1000573. doi: 10.1371/journal.pbio.1000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Girard C, Will CL, Peng J, Makarov EM, Kastner B, et al. posttranscriptional spliceosomes are retained in nuclear speckles until splicing completion. Nat Commun. 2012;3:994. doi: 10.1038/ncomms1998. [DOI] [PubMed] [Google Scholar]
- 233.Hirose Y, Manley JL. RNA polymerase II and the integration of nuclear events. Genes Dev. 2000;14:1415–29. [PubMed] [Google Scholar]
- 234.Hsin JP, Manley JL. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 2012;26:2119–37. doi: 10.1101/gad.200303.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.David CJ, Manley JL. The RNA polymerase C-terminal domain: a new role in spliceosome assembly. Transcription. 2011;2:221–25. doi: 10.4161/trns.2.5.17272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Hirose Y, Tacke R, Manley JL. Phosphorylated RNA polymerase II stimulates pre-mRNA splicing. Genes Dev. 1999;13:1234–39. doi: 10.1101/gad.13.10.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Hirose Y, Manley JL. RNA polymerase II is an essential mRNA polyadenylation factor. Nature. 1998;395:93–96. doi: 10.1038/25786. [DOI] [PubMed] [Google Scholar]
- 238.Das R, Yu J, Zhang Z, Gygi MP, Krainer AR, et al. SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol Cell. 2007;26:867–81. doi: 10.1016/j.molcel.2007.05.036. [DOI] [PubMed] [Google Scholar]
- 239.Lou H, Gagel RF, Berget SM. An intron enhancer recognized by splicing factors activates polyadenylation. Genes Dev. 1996;10:208–19. doi: 10.1101/gad.10.2.208. [DOI] [PubMed] [Google Scholar]
- 240.Almada AE, Wu X, Kriz AJ, Burge CB, Sharp PA. Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature. 2013;499:360–63. doi: 10.1038/nature12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA–mRNA interaction maps. Nature. 2009;460:479–86. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Leung AK, Young AG, Bhutkar A, Zheng GX, Bosson AD, et al. Genome-wide identification of Ago2 binding sites from mouse embryonic stem cells with and without mature microRNAs. Nat Struct Mol Biol. 2011;18:237–44. doi: 10.1038/nsmb.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Zisoulis DG, Lovci MT, Wilbert ML, Hutt KR, Liang TY, et al. Comprehensive discovery of endogenous Argonaute binding sites in Caenorhabditis elegans. Nat Struct Mol Biol. 2010;17:173–79. doi: 10.1038/nsmb.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Taliaferro JM, Aspden JL, Bradley T, Marwha D, Blanchette M, Rio DC. Two new and distinct roles for Drosophila Argonaute-2 in the nucleus: alternative pre-mRNA splicing and transcriptional repression. Genes Dev. 2013;27:378–89. doi: 10.1101/gad.210708.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Moshkovich N, Nisha P, Boyle PJ, Thompson BA, Dale RK, Lei EP. RNAi-independent role for Argonaute2 in CTCF/CP190 chromatin insulator function. Genes Dev. 2011;25:1686–701. doi: 10.1101/gad.16651211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Cernilogar FM, Onorati MC, Kothe GO, Burroughs AM, Parsi KM, et al. Chromatin-associated RNA interference components contribute to transcriptional regulation in Drosophila. Nature. 2011;480:391–95. doi: 10.1038/nature10492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Zamudio JR, Kelly TJ, Sharp PA. Argonaute-bound small RNAs from promoter-proximal RNA polymerase II. Cell. 2014;156:920–34. doi: 10.1016/j.cell.2014.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Ameyar-Zazoua M, Rachez C, Souidi M, Robin P, Fritsch L, et al. Argonaute proteins couple chromatin silencing to alternative splicing. Nat Struct Mol Biol. 2012;19:998–1004. doi: 10.1038/nsmb.2373. [DOI] [PubMed] [Google Scholar]
- 249.Sander B, Golas MM, Makarov EM, Brahms H, Kastner B, et al. Organization of core spliceosomal components U5 snRNA loop I and U4/U6 di-snRNP within U4/U6.U5 tri-snRNP as revealed by electron cryomicroscopy. Mol Cell. 2006;24:267–78. doi: 10.1016/j.molcel.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 250.Wolf E, Kastner B, Deckert J, Merz C, Stark H, Lührmann R. Exon, intron and splice site locations in the spliceosomal B complex. EMBO J. 2009;28:2283–92. doi: 10.1038/emboj.2009.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Kastner B, Lührmann R. Electron microscopy of U1 small nuclear ribonucleoprotein particles: shape of the particle and position of the 5′ RNA terminus. EMBO J. 1989;8:277–86. doi: 10.1002/j.1460-2075.1989.tb03374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Kastner B, Kornstädt U, Bach M, Lührmann R. Structure of the small nuclear RNP particle U1: identification of the two structural protuberances with RNP-antigens A and 70K. J Cell Biol. 1992;116:839–49. doi: 10.1083/jcb.116.4.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Burge CB, Tuschl T, Sharp PA. Splicing of Precurors to mRNAs by the Spliceosome. Cold Spring Harbor, NY: Cold Spring Harbor Lab. Press; 1999. pp. 525–60. [Google Scholar]
- 254.Padgett RA, Burge CB. Encyclopedia of Life Sciences. New York: Wiley; 2005. Splice sites; pp. 1–7. [Google Scholar]
- 255.Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, et al. The developmental transcriptome of Drosophila melanogaster. Nature. 2011;471:473–79. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Brown JB, Boley N, Eisman R, May GE, Soiber MH, et al. Diversity and dynamics of the Drosophila transcriptome. Nature. 2014;512:393–99. doi: 10.1038/nature12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Ramani AK, Calarco JA, Pan Q, Mavandadi S, Wang Y, et al. Genome-wide analysis of alternative splicing in Caenorhabditis elegans. Genome Res. 2011;21:342–48. doi: 10.1101/gr.114645.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Kondo Y, Oubridge C, van Roon AM, Nagai K. Crystal structure of human U1 snRNP, a small nuclear ribonucleoprotein particle, reveals the mechanism of 5′ splice site recognition. eLife. 2015;4:e04986. doi: 10.7554/eLife.04986. [DOI] [PMC free article] [PubMed] [Google Scholar]