

Summary
Akt is frequently activated in human cancers. However, it is unknown whether systemic inhibition of a single Akt isoform, could regress cancer progression in cancers that are not driven by Akt activation. We systemically deleted Akt1, after tumor onset, in p53−/− mice, which develop tumors independently of Akt activation. Systemic Akt1 deletion regresses thymic lymphoma in p53−/− mice emulating p53 restoration. Furthermore, pharmacological inhibition of Akt selectively kills thymic lymphoma cells and not primary thymocytes. Mechanistically, Akt1 inhibition in p53−/− thymic lymphoma inhibits Skp2 expression, and induces FasL, which is the primary cause of cell death. Skp2 exerts resistance to cell death by antagonizing the induction of FasL and reducing FAS expression, which is linked to cyclin D1 expression. The results established a new paradigm whereby systemic Akt1 inhibition is sufficient to regress tumors that are not driven by Akt activation, and a new mechanism of cell survival by Skp2.
Graphical Abstract
Introduction
The serine/threonine kinase Akt is perhaps the most frequently activated oncoprotein in human cancers (reviewed in (Bhaskar and Hay, 2007)). The pathways leading to its activation as well as Akt itself are being targeted for cancer therapy. However, there are three Akt isoforms (Akt1-3) encoded by three separate genes. These isoforms are about 85% identical to each other, and it is impossible to distinguish among them with respect to substrate specificity in in-vitro kinase assay (Walker et al., 1998). Nevertheless, it is possible that the different isoforms are functionally different in vivo. It is unknown, however, which isoform should be preferentially targeted and for which type of cancer, without eliciting adverse physiological consequences. It is also not known if Akt could be targeted for therapy of cancers that are not driven directly or indirectly by Akt activation.
Previously we and others showed that germ line deletion of Akt1 exerts resistance to Pten deletion induced tumorigenesis (Chen et al., 2006), skin carcinogenesis (Skeen et al., 2006), mammary and salivary gland tumorigenesis (Ju et al., 2007; Maroulakou et al., 2007; Skeen et al., 2006), and lung carcinogenesis (Hollander et al., 2011). The germ line deletion of Akt1 in the mouse does not have adverse physiological consequences, and Akt1−/− mice live significantly longer than control WT mice (Chen et al., 2006). By contrast germ line deletion of Akt2 induces insulin resistance and does not inhibit tumor development in mouse models of breast cancer and in Pten+/− mice (Maroulakou et al., 2007; Xu et al., 2012). However, studies using germ line deletions of Akt isoforms cannot determine if Akt is required for tumor progression and do not emulate drug therapy. To address whether systemic Akt1 deletion after tumor onset could inhibit tumor progression even in tumors, which are not driven by Akt activation, we examined the ability of systemic Akt1 deletion to inhibit tumor progression in p53−/− mice. We found that the systemic deletion of Akt1 was sufficient to regress or halt the progression of thymic lymphoma in p53−/− mice similar to what observed after p53 restoration in these mice (Ventura et al., 2007). The deletion of Akt1 in p53−/− thymic lymphoma is sufficient to inhibit cell cycle progression and induces apoptosis, which could be recapitulated by a pharmacological inhibitor of Akt. The cytotoxic effect of Akt inhibition on p53−/− thymic lymphoma is selective as it is not cytotoxic to normal thymocytes. However, hyperactivation of Akt in p53−/− thymic lymphoma exerts resistance to pharmacological inhibitor of Akt. The induction of FasL expression by the inhibition of Akt and activation of FOXO appears to be the predominant cause of cell death. Interestingly ectopic expression of Skp2 in the p53−/− thymic lymphoma cells also exerts resistance to Akt inhibition. Skp2 exerts resistance to cell death, at least in part by, the inhibition of FasL and Fas expression. It was recently reported that D-cyclins promote survival of hematopoietic cells through inhibition of Fas and FasL expression in E2F1 dependent manner (Choi et al., 2014). Consistently we found that the ectopic expression of Skp2 also elevates Cyclin D1 expression and reduces p27 expression, which are known to activate E2F1, and thus could explain the mechanism by which Skp2 inhibits Fas and FasL expression. Taken together these results established a new paradigm whereby Akt1 inhibition is therapeutic even for cancers that are not driven by Akt activation. These results suggest that Akt inhibitors could be used effectively to treat cancers that are driven by the loss of p53 such as in Li-Fraumeni syndrome (LFS) patients. Although only about 20% LFS patients have a compete loss of p53, most of the p53 mutations in LFS are occurring in the DNA binding domain and act in a dominant manner to inhibit Wt p53 (Malkin, 2011). The results also suggest that high Skp2 expression could predict resistance to Akt inhibition and that the inhibition of both Akt and Skp2 might be considered for a long-term therapeutic effect.
Results
Systemic deletion of Akt1 in p53−/− mice increases lifespan and halts the progression of thymic lymphoma in these mice
To determine the ability of systemic Akt1 ablation to regress or attenuate tumor progression, we have generated Akt1f/f mice (Fig. S1A, B) and crossed these mice with R26CreERT2 mice, in which CreERT2 was inserted in the ubiquitously expressed ROSA26 locus (Ventura et al., 2007), to generate Akt1f/f/R26CreERT2 mice (Fig. 1A and Fig. S1C). CreERT2 is an inducible Cre recombinase fused in frame to a mutated ligand-binding domain of estrogen receptor, which responds only to hydroxytamoxifen (4-OHT) and not to estrogen. To determine if systemic Akt1 inhibition could regress or inhibit progression of tumors that are not driven by lesions that activate Akt, we crossed Akt1f/f/R26CreERT2 mice with p53−/− mice to generate p53−/−Akt1f/f/R26CreERT2 mice (Fig. 1A). Injection of tamoxifen to the mice 6 weeks after birth successfully deletes Akt1 in all tissues tested (Fig. 1B and Fig. S1C). The p53−/− mice develop thymic lymphoma with an early onset, and eventually develop also sarcoma (Ventura et al., 2007). The mice die of these tumors within 6 months after birth, and restoration of p53 regresses the lymphoma with increased apoptosis, and inhibits the growth of sarcoma, indicating that these tumors are highly dependent on p53 deletion (Ventura et al., 2007). We have generated cohorts of p53−/−Akt1f/f/R26CreERT2 and p53−/−R26CreERT2 mice. The mice were injected with tamoxifen for 7 consecutive days, six weeks after birth when they start developing thymic lymphoma. As shown in Fig. 1C, the systemic deletion of Akt1 substantially increased the median lifespan of p53−/− mice from 16 weeks to 29 weeks. We did not see a significant change in the lifespan of control p53−/−R26CreERT2 mice (not shown).
Figure 1. Systemic deletion of Akt1 in p53−/− mice after tumor onset increased lifespan.

A. Schematic illustration showing crossing of mice to generate p53−/−Akt1f/f/R26CreERT2 mice. B. Immunoblot showing Akt1 deletion in several mouse tissues after injection of tamoxifen (Tam) to 6-week-old p53−/−Akt1f/f/R26CreERT2 mice. C. Kaplan-Meier plot showing life span of p53−/− or p53−/−Akt1f/f/R26CreERT2 mice after tamoxifen injection at 6 weeks of age (n=10).
To further substantiate our findings and demonstrate that systemic Akt1 ablation is sufficient to regress or halt thymic lymphoma progression in p53−/− mice, we monitored in vivo tumor progression by magnetic resonance imaging (MRI) prior to and after treatment with tamoxifen. Cohorts of p53−/−Akt1f/f/R26CreERT2 and p53−/−R26CreERT2 mice were monitored to detect in vivo lymphoma as previously described (Ventura et al., 2007). When tumors detected by MRI reached the volume of approximately 200mm3, mice were treated with tamoxifen, and tumor growth was followed as previously described (Ventura et al., 2007). As shown in Fig. 2A,B tumor size was significantly reduced 12–16 days after the administration of tamoxifen into p53−/−Akt1f/f/R26CreERT2 mice, while at the same time in untreated mice tumors grew approximately by about 5-fold (Fig. 2A). Injection of tamoxifen into control p53−/−R26CreERT2 mice did not reduce tumor growth (Fig. 2B). In general the deletion of Akt1 by tamoxifen treatment either halts or regresses tumor growth while at the same time in control mice tumor grew by 2–6 fold (Fig. 2C). Moreover, analysis of sections from the tumor mass that was left after Akt1 deletion revealed a massive apoptosis as indicated by cleaved caspase 3 staining and a relatively few Ki67 positive cells (Fig. 2D). These results indicate that systemic deletion of Akt1 after tumor onset in p53−/− mice, which emulate drug therapy, is sufficient to regress p53−/− thymic lymphoma. Moreover, the effect of Akt1 deletion on thymic lymphoma in p53−/− mice is similar to what observed after restoration of p53 in these mice (Ventura et al., 2007).
Figure 2. Systemic Akt1 deletion in p53−/− mice halts or regresses tumor growth in vivo.

A,B. MRI images (left) and calculated tumor volume (right) in response to tamoxifen of p53−/−Akt1f/f/R26CreERT2 (A and B) or p53−/−R26CreERT2 mice (B). ***p<0.001 Tam vs. vehicle; ‡‡‡ p<0.0001, ‡‡ p<0.005, ‡ p<0.01 vs. day 0. C. Summary of individual tumor responses to systemic Akt1 deletion 16 days after tamoxifen treatment or vehicle alone of p53−/−Akt1f/f/R26CreERT2 mice. D. Representative, tumor sections from 2 tumors after regression as determined by MRI sowing cleaved caspase 3 (CC3) and Ki67 staining. Data are presented as an average of 3 measurements ± SEM.
The deletion of Akt1 in p53−/− thymic lymphoma cells induces cell death and inhibits cell proliferation
To further establish the role of Akt1 in p53−/− thymic lymphoma, cell lines were isolated from thymic lymphoma tumors in p53−/−R26CreERT2 and p53−/−Akt1f/f/R26CreERT2 mice and were exposed to (4-OHT) in vitro (Fig. 3A). The deletion of Akt1 in these lymphoma cell lines profoundly inhibited the growth of the cells (Fig. 3B). This effect is due to both induction of apoptosis and inhibition of proliferation as measured by BrdU incorporation (Fig. 3C). Thus, Akt1 deletion is sufficient to induce both cell death and cell cycle arrest in p53−/− thymic lymphoma. In general Akt1 deletion by itself has only cytostatic effect and can only render cells more susceptible to cell death by the presence of other apoptotic stimuli (Chen et al., 2001). Notably, the p53−/− thymocytes do not display activation of Akt when compared to control thymocytes, and Akt activity did not significantly change after re-expressing p53 in the p53−/− thymic lymphoma cells (Fig. S2). Thus, clearly Akt is not hyperactivated in thymic lymphoma driven by p53 deletion. Ectopic expression of a myristoylated Akt1 (mAkt), a constitutively active form of Akt, exerts resistance to the deletion of Akt1 by 4-OHT (Fig. S2 C,D). Finally, we injected either p53−/−/R26CreERT2 or p53−/−Akt1f/f/R26CreERT2 lymphoma cells into nude mice and followed tumor growth (Fig. 4A). When tumor size reached 60mm3 in volume the mice were treated with tamoxifen. While tumor growth of p53−/−/R26CreERT2 lymphoma cells was not affected by tamoxifen treatment, tumor growth of p53−/−Akt1f/f/R26CreERT2 lymphoma cells in the treated mice was halted when compared to tumor growth of untreated mice Fig. 4B, C). When, tumor section samples were analyzed for cell proliferation or cell death as measured by BrdU incorporation and cleaved caspase 3 respectively, a profound reduction in proliferation and increased apoptosis were observed (Fig. 4D). Thus, the in vivo results recapitulate the results obtained ex-vivo.
Figure 3. Ex-vivo deletion of Akt1 in thymic lymphoma cells derived from p53−/−Akt1f/f/R26CreERT2 mice inhibits proliferation and induces cell death.

A. Strategy of isolating thymic lymphoma cell lines (top), and immunoblot showing Akt1 deletion after exposure to 4-OHT (bottom). B. Proliferation of two independent isolated cell lines in response to 4-OHT. Cells were exposed to 4-OHT for 3 days and cell number was determined in the indicated time points. Data are presented as an average of three independent experiments in triplicates ± SEM. ***p<0.001 **p<0.01 *p<0.05 vs. 4-OHT 300nM & 1μM. C. BrdU (Bromodeoxyuridine) incorporation as measured by anti-BrdU staining and cell death as measured by PI (Propidium iodide) staining using flow cytometry. Cells were exposed to 4-OHT for 3 days and were subjected to BrdU incorporation and PI staining at the indicated time points). Data are presented as an average of three independent experiments in triplicates ± SEM. * p < 0.05 ns- not significant.
Figure 4. The deletion of Akt1 inhibits the growth of xenographic p53−/− thymic lymphoma tumors by inhibiting proliferation and inducing apoptosis.

A. Flow chart depicting the strategy to determine tumor response in xenograft assays. Cells isolated from the thymic lymphoma were cultured and inoculated into nude mice. When tumor size reached 60mm3 mice were treated with tamoxifen and tumor growth was determined. B. Tumor growth curves of p53−/−R26CreERT2 or p53−/−Akt1f/f/R26CreERT2 cells in response to tamoxifen. Data are presented as an average of ten tumors in five mice per each cell line and per each treatment ± SEM (n=9). **p<0.001 vs. Tam. C. Representative tumors from vehicle or tamoxifen treated mice (left) and immunoblot showing the extent by which Akt1 deletion affected total serine 473 phosphorylation of Akt in various tumors (right). D. Representative tumor sections from vehicle or tamoxifen treated mice showing cleaved caspase 3(CC3) and BrdU incorporation (left panels). Quantification of CC3 or BrdU positive cells in randomly selected 6 fields from 3 different tumors (right panels). Data are presented as an average ± SEM. **p< 0.001.
We then wanted to find out whether pharmacological inhibition of Akt1 could recapitulate Akt1 deletion. Since there are no specific inhibitors of Akt1, we employed MK2206, which is an allosteric inhibitor of Akt that is commonly used in clinical trials. MK2206 inhibits Akt1, Akt2, and Akt3 with IC50 of 8nM, 12nM, and 65nM respectively (Hirai et al., 2010). We first exposed the thymic lymphoma cells to MK2206 and compared its effect to the effect induced by Akt1 deletion. We found a similar effect of MK2206 on the proliferation of the cells (Fig. 5A) as well on BrdU incorporation (Fig. 5B) and apoptosis (Fig. 5C) to that observed after the induction of Akt1 deletion. Likewise in vivo tumor growth of these cells was halted after treatment with MK2206 (Fig. 5D) similar to what was observed after systemic deletion of Akt1. Thus, the pan-Akt inhibitor could recapitulate the conditional deletion of Akt1 to inhibit the progression of cancer that is not driven by oncogenic signaling that activates Akt. Importantly, the effect of MK2206 on p53−/− thymic lymphoma is selective because we found that the cytotoxic effect on primary normal thymocytes is minimal in comparison to the lymphoma cells (Fig. 5E). Taken together these results showed that Akt activity is also required for tumors that are not driven by signaling pathways that activate Akt, and that Akt ablation is therapeutic for thymic lymphoma induced by p53 deletion.
Figure 5. MK2206 treatment recapitulates the response to Akt1 deletion and selectively kills lymphoma cells in vitro.

A. Proliferation of p53−/−R26CreERT2 or p53−/− Akt1f/f/R26CreERT2 lymphoma cells in response to two doses of MK2206. Data are presented as an average of three independent experiments ± SEM. ***p<0.001 **p<0.01 vs. DMSO. B, C. BrdU incorporation (B) and cell death (C) in response to MK2206. Data are presented as an average of three independent experiments in triplicates ± SEM ***p<0.001 **p<0.01 *p<0.01 vs. DMSO in each group. D. Xenographic tumor growth of p53−/−Akt1f/f/R26CreERT2 lymphoma cells in response to MK2206. When tumor size reached 60mm3, mice were treated with either capistol or MK2206. Data are presented as an average of ten tumors in five mice per capistol or MK2206 treatment ± SEM (n=9) ***p<0.001. E. Assessment of apoptosis in response to 1μM MK2206 of normal thymocytes isolated from 3 individual mice or of p53−/− thymic lymphoma cells. Data are presented as an average of three independent experiments in triplicates ± SEM. ***p<0.001 vs. DMSO and thymic lymphoma vs. normal primary thymocytes. F. Fold change in apoptosis in response to MK2206 in the colorectal HCT116, SW48, and DLD-1 and their isogenic p53−/− cell lines. Cells were exposed to either the indicated concentrations of MK2206 or DMSO, and apoptosis was quantified 48h later. Data presented as an average of two independent experiments in triplicates ± SEM. ***p<0.001 p53−/− vs. WT.
Interestingly, we previously showed that germ line deletion of Akt1 sensitized primary thymocytes to killing by gamma-irradiation (Chen et al., 2001). Thus, it is possible that the deletion of p53, which increases DNA damage, similarly sensitizes thymocytes to killing by Akt inhibition. Consistently, we found that human cancer cells lacking p53 are profoundly more sensitive to cell death after inhibition of Akt then cancer cells proficient for p53. We employed well-established 3 pairs of colorectal cancer cell lines in which p53 is either WT or was somatically deleted ((Sur et al., 2009), Fig. S3). As shown in Fig. 5F, the p53-deficient cells are highly sensitive to cell death after exposure to MK2206 when compared to the p53-proficient cells. These results suggest that the hypersensitivity to Akt inhibition exerted by p53 deficiency could be extended to different types of cancer.
The mechanisms by which the deletion of Akt1 inhibits proliferation and induces cell death in p53−/− thymic lymphoma
We previously showed that mTORC1 is the most critical downstream effector of Akt, required for cell proliferation and tumorigenesis (Skeen et al., 2006). Our results showed that upon deletion of Akt1 by 4-OHT, there was a decrease in FOXO phosphorylation (Fig. 6A) and a marked reduction in mTORC1 activity as measured by the phosphorylation of S6 and 4EBP1 (Fig. 6B). As we previously showed, mTORC1 activity and its downstream effector, the initiation factor of mRNA translation, eIF4E, are required for the Skp2 mRNA translation, and therefore for its expression at the protein level (Nogueira et al., 2012). Indeed, we found a significant decrease in Skp2 expression following the reduced mTORC1 activity (Fig. 6B). Skp2 is a F-box protein that mediates the degradation of p27 and other cell cycle inhibitory proteins (Nakayama et al., 2004; Pagano, 2004). Similarly, Akt inhibition by MK2206 inhibited FOXO phosphorylation and mTORC1 activity and elevated p27 expression (Fig. 6C). Thus, the reduced mTORC1 activity in combination with the induced FOXO activity could explain the high level of p27 as well as the inhibition of proliferation following Akt inhibition in p53−/− thymic lymphoma. We also found a decrease in the anti-apoptotic protein Mcl-1 after Akt inhibition (Fig. 6C). This is consistent with the high dependency of Mcl-1 mRNA translation on mTORC1 activity and eIF4E (Wendel et al., 2007), as well as the increase of MCL-1 protein stability by Akt through the inhibition of GSK3 (Maurer et al., 2006). In addition, the mRNAs for the FOXO-targets pro-apoptotic proteins Bim and FasL (Fig. 6D, Fig. 7C), as well as FasL protein, which its expression as determined by FACS analysis, overlaps with cells undergoing cell death (Fig. 6E and Fig. S4), were elevated after Akt inhibition. Thus, the reduced Mcl-1 expression together with the higher expression of Bim and FasL could explain the induction of cell death after Akt inhibition. Surprisingly, however, we found that ectopic expression of Skp2 in p53−/− thymic lymphoma exerted resistance to cell death induced by Akt inhibition almost to the same extent as by mAkt (Fig. 7A). Analysis of protein extracts derived from these cells before and after treatment showed that Skp2 protein expression declined in control treated but only modestly in mAkt treated cells, and the elevation of p27 was observed in control treated cells but not in Skp2 or mAkt treated cells (Fig. 7B). Unlike in mAkt treated cells, mTORC1 activity, as measured by S6K1 phosphorylation and 4EBP1 mobility shift, as well as FOXO phosphorylation are still inhibited to a similar extent in control and Skp2 expressing cells. MCL-1 protein levels were reduced to a similar extent in control and Skp2 expressing cells (Fig. 7B). Nonetheless, cleaved caspase 3 levels were profoundly reduced in Skp2 expression cells, consistent with the resistance of these cells to cell death induced by Akt inhibition. Thus, Skp2 could partially recapitulate the resistance to cell death exerted by activated Akt in p53−/− thymic lymphoma cells and overrides mTORC1 inhibition and FOXO activation. Therefore, the resistance to cell death induced by Skp2 overexpression seems to be independent of mTORC1 activity, FOXO phosphorylation or Mcl-1 expression. These results were unexpected but although we did not observe a significant change in the induction of Bim mRNA in Skp2 expressing cells, we found that Fas ligand (FasL) expression was significantly reduced (Fig. 7C). This could explain in part the resistance of p53−/− thymic lymphoma cells overexpressing Skp2 to Akt inhibition. To verify the role of FasL in apoptosis, we monitored caspase 8 cleavage and found that it is increased in the control treated cells but is inhibited in Skp2 overexpressing cells and mAkt expressing cells (Fig. 7D), to a similar extent as observed with caspase 3 cleavage (Fig. 7B). Furthermore, inhibition of caspase 8 activity or FAS silencing inhibited cell death induced by Akt inhibition. We employed the caspase 8 inhibitor Z-LETD-FMK and showed that it markedly inhibited cell death induced by Akt inhibition (Fig. 7E), without affecting the induction of FAS or FasL (Fig. S5). Silencing FAS using siRNA (Fig. S6A) inhibited the induction of FAS, and inhibited apoptosis induced by Akt inhibition (Fig. 7F). All together these results strongly indicate that the predominant initiator of apoptosis, induced by Akt inhibition in p53−/− thymic lymphoma, is FasL. Consistently, although we found that Bim mRNA levels are highly induced by the inhibition of Akt, we did not find a significant change in the major expressed isoform BIM-EL protein, and only small changes in the alternatively spliced isoform BIM-L (Fig. S6B). Only in cells expressing activated Akt the protein levels of the three BIM isoforms were decreased.
Figure 6. Activation of FOXO and inhibition of mTORC1 and Skp2 expression in response to Akt1 deletion or Akt inhibition by MK2206.

A. FOXO phosphorylation at day 4 (D4) or day 5 (D5) after deletion of Akt1 by 4OHT in p53−/−Akt1f/f/R26CreERT2 lymphoma cells. B. Phosphorylation of, 4EBP1, S6, and the expression of Skp2 at day 4 or day 5 after deletion of Akt1 by 4OHT in p53−/−Akt1f/f/R26CreERT2 lymphoma cells. C. Immunoblot showing FOXO phosphorylation, mTORC1 activity as measured by 4EBP1 mobility shift and S6 phosphorylation, p27 and MCL-1 protein levels, and CC3 after treatment of p53−/−Akt1f/f/R26CreERT2 lymphoma cells with MK2206 at the indicated time points. D. Fas ligand (FasL) and Bim mRNA levels as measured by quantitative RT-PCR after treatment of p53−/−Akt1f/f/R26CreERT2 lymphoma cells with MK2206 for 18h. Data are presented as an average of three independent RNA preparations ± SEM. **p<0.001 *p<0.05. E. FACS analysis of FasL expression in cells treated with DMSO or 1μM MK-2206 for 18h. Data are presented as an average of 3 independent experiments in triplicates ± SEM. ** p<0.005.
Figure 7. Ectopic expression of Skp2 or mAkt in p53−/− thymic lymphoma cells exerts resistance to MK2206.

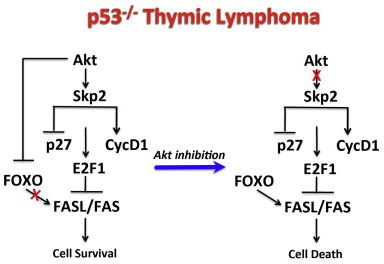
A. Induction of cell death of control GFP expressing cells, Skp2 or mAkt expressing cells after treatment with MK2206 for 18h. Data are presented as an average of three independent experiments ± SEM. B. Immunoblot showing the phosphorylation of Skp2 expression, FOXO3 phosphorylation, p27 expression, mTORC1 activity as measured by S6 phosphorylation and 4EBP1 mobility shift, Mcl-1 and cleaved caspase 3 (CC3) expression in GFP, Skp2 or mAkt expressing cells following treatment with MK2206 for 18h. C. Expression of FasL and Bim as measured by RT-PCR in GFP, Skp2 or mAkt expressing cells following treatment with MK2206 for 18 hours. Data are presented as an average of three independent RNA preparations ± SEM. **p<0.01 *p<0.05 vs. GFP; ‡‡ p<0.01 ‡p<0.05 vs. DMSO in each group. D. Immunoblot showing the level of cleaved caspase 8 in GFP, Skp2 or mAkt expressing cells following treatment with 1μM MK2206 for 18h.E. Induction of cell death of p53−/− thymic lymphoma cells after treatment with 1μM MK2206 for 18h in presence or absence of cell permeable caspase-8 inhibitor (Z-LETD-FMK). Data are presented as an average of 3 independent experiments in triplicates ± SEM. *** p<0.0001 and ** p<0.005 compared to DMSO. ‡‡‡ p<0.0001 compared to MK2206. F. Cells were treated with DMSO or MK2206 for 18h after knockdown of Fas using a pool of 4 siRNAs. Following treatment, cell death was assessed by flow (PI staining). Experiments were done twice in triplicate. Statistical analysis: ***p<0.0001 compared to control siRNA. ‡‡‡ p<0.0001 compared to DMSO. G. Immunoblot showing Cyclin D1 expression in GFP, Skp2, or mAkt expressing cells. H. Fas expression in GFP, Skp2 or mAkt expressing cells. I. Illustration depicting the mechanism by which Akt inhibition promotes cells death in p53−/− thymic lymphoma.
Since it was recently shown that D cyclins promote cell survival in hematopoietic cells by the inhibition of FasL and Fas receptor (Fas) expression (Choi et al., 2014), and since we did not find significant changes in BIM protein expression by Skp2 (Fig. S6B), we analyzed cyclin D1 level in Skp2 overexpressing cells and found that it is significantly elevated (Fig. 7G). The mechanism by which D cyclins inhibit FasL and Fas expression appears to be through the induction and activation of E2F1 (Choi et al., 2014). Therefore the inhibition of p27 and the induction of cyclin D1 by Skp2 raise the possibility that Skp2 inhibits FasL mRNA via the phosphorylation of Rb and the activation of E2F. It is therefore expected that Skp2 overexpression would also reduce the expression of Fas. Indeed, we found that Skp2 overexpression also markedly reduced the expression of Fas after Akt inhibition (Fig. 7H). The results suggest a mechanism by which Skp2 antagonizes Akt inhibition mediated cell death in p53−/− thymic lymphoma (Fig, 7I). Furthermore, the results uncovered a new mechanism by which Skp2 could promote cell survival.
We clearly showed that the predominant mechanism, which selectively sensitizes p53−/− thymic lymphoma to cell death by Akt inhibition, is mediated by FAS and FASL. However, as we indicated above, it is possible that p53 deficiency in general sensitizes cells to cell death by Akt inhibition as was shown for the isogenic colorectal cancer cells (Fig. 5F). Akt inhibition could induce cell death by multiple mechanisms and the mechanism that we have uncovered in the thymic lymphoma cells may not apply to the colorectal cancer cell lines. We found that Akt inhibition, despite inducing higher levels of apoptosis in the p53-deficient human cancer cells, inhibits FOXO phosphorylation to a similar extent in p53-proficient and p53-deficient human cancer cells (Fig. S7A). Unlike what was observed in the p53−/− thymic lymphoma cells, we did not find any significant and consistent changes in MCL-1 protein level upon Akt inhibition (Fig. S7A). BIM and FAS protein levels were also did not consistently change upon Akt inhibition, although in the p53−/− HCC116 cells we found a modest elevation of FAS and BIM proteins (Fig. S7A). We also did not find consistent changes in FasL expression. Only a modest increase of FasL mRNA was found in p53−/− SW40, and p53−/− DLD-1 cells when compared to the isogenic p53-proficient cells (Fig. S7B). Nevertheless caspase 3 cleavage was markedly induced in the p53-deficient cells when compared to the p53-proficient cells (Fig. S7C). These results support the notion that regardless of the mechanism p53 deficiency sensitizes cells to cell death induced by the inhibition of Akt.
Discussion
It is known that germ line deletion of Akt1 is sufficient to inhibit tumor development in several mouse models of cancer, which are associated with Akt activation. However, these results do not address whether Akt1 inhibition is sufficient to inhibit or regress tumors after tumor onset. In addition, it is not clear whether Akt inhibitors could be therapeutic for cancers in which activation of Akt is not implicated. Akt hyperactivation is perhaps one of the most frequently occurring events in human cancers, and therefore these cancers are rationale candidates for Akt inhibition therapy, but it is not known whether Akt inhibition is valuable in cancers, which do not display activation of Akt. Finally, it is not clear what are the potential mechanisms that could be developed by cancer cells to resist Akt inhibition therapy. In the current studies we addressed these three questions, and showed that systemic deletion of Akt1 in mice is therapeutic for p53−/− thymic lymphoma that does not display Akt activation.
The effect of systemic Akt1 deletion on the lifespan extension of p53−/− tumor bearing mice is profound, but is not sufficient to reach normal lifespan. Although the MRI studies showed that the systemic Akt1 deletion either halt the growth or regress the p53−/− thymic lymphoma, it is possible that other tumors developed in p53−/− mice caused the eventual mortality. Usually p53−/− mice also develop high incidence of sarcoma, but we have not detected sarcomas within the time frame that the mice were treated possibly because of the C57Bl6 background of the mice used in our studies. We did observe, however, sarcomas in older mice, which could contribute to the mortality even after Akt1 deletion. Alternatively or additionally, thymic lymphoma cells that escaped the deletion could contribute to the mortality. Finally, the possibility that p53−/− mice have a shorter lifespan regardless of tumor development cannot be completely excluded. Future studies are required to determine if other types of tumors, which are driven by p53 deletion or by mutated p53, such as sarcoma, could be also targeted by Akt1 inhibition.
The germ line deletion of Akt1 in the mouse has only minor consequences, and in fact Akt1−/− mice live longer than wild type mice (Chen et al., 2006). The systemic deletion of Akt1 in adult mice does not induce any overt phenotype, and therefore our results strongly suggest that Akt1 specific inhibition could be well tolerated and it is an efficient cancer therapy strategy even if the cancer cells do not display Akt activation. The results provide a new paradigm that might expand the spectrum of cancer types that could be candidates for Akt inhibition therapy. Restoration of p53 had been suggested as a strategy for cancer therapy (Martins et al., 2006; Ventura et al., 2007; Wang et al., 2011). However, restoration of p53 is difficult to achieve, and thus our results suggest a new feasible strategy for therapy of cancers that are driven by the loss of p53. Moreover, our results could be extended to human cancer cells deficient for p53 as we found that p53 deficiency sensitizes human cancer cells to cell death induced by Akt inhibition.
As expected we found a decrease in FOXO phosphorylation and mTORC1 activity after Akt1 deletion or Akt inhibition in p53−/− thymic lymphoma. The activation of FOXO resulted an increase in its pro-apoptotic transcriptional targets Bim and FasL. The inhibition of Akt also reduced the protein levels of Mcl-1. Akt inhibition could reduce the steady state level of Mcl-1 through inhibition of its mRNA translation, which is highly dependent on eIF4E (Wendel et al., 2007) or through its destabilization by GSK3, which is phosphorylated and inactivated by Akt (Maurer et al., 2006). The combination of the elevated Bim and FasL with the reduced Mcl-1 level could explain, the cell death induced by Akt inhibition in p53−/− thymic lymphoma. However, our results strongly indicated that the predominant cause of cell death is FasL mediated. Skp2 expression was also reduced after Akt inhibition. The reduced Skp2 expression is likely due to reduced Skp2 mRNA translation since it is highly dependent on Akt and on its downstream effectors mTORC1 and eIF4E, as we had shown previously (Nogueira et al., 2012). However, we observed that in the thymic lymphoma cells exogenous Skp2 protein is also reduced upon Akt inhibition, which cannot be explained only by mRNA translation. Other results showed that Skp2 is directly phosphorylated by Akt, which in turn maintains its cytoplasmic localization and stability (Gao et al., 2009; Lin et al., 2009), but these results were challenged by others (Bashir et al., 2010). We expected that ectopic Skp2 expression would override the inhibition of cell proliferation induced by Akt inhibition in p53−/− thymic lymphoma. Surprisingly we found that high level of ectopic Skp2 expression in p53−/− thymic lymphoma exerts resistance to cell death induced by Akt inhibition. Indeed previous reports documented that silencing of Skp2 in a variety of cancer cell lines elicits cell death (Jiang et al., 2005; Lee and McCormick, 2005). In particular pRb-deficient cancer cells are sensitized to apoptosis induced by Skp2 silencing with no effect on proliferation. These results suggest that Skp2 has a role in cell survival, which is independent from its role in cell proliferation (Wang et al., 2010a). Interestingly, high Skp2 expression was implicated in T cell lymphoma in mice and in high-grade lymphomas in human (Latres et al., 2001). Expression of Skp2 in T cells in mice profoundly increased the incidence of T cell lymphoma induced by oncogenic N-Ras (Latres et al., 2001), and mice lacking CREB Binding Protein (CBP) develop T cell lymphomagenesis with concomitant elevated Skp2 expression (Kang-Decker et al., 2004). We found that Skp2 decreased the induction of FasL and Fas by MK2206 treatment. Our results showed that the predominant mechanism by which Akt inhibition promotes cell death in p53−/− thymic lymphoma is via FASL and FAS. Both FAS silencing and caspase 8 inhibition inhibited the cell death induced by Akt inhibition. Consistently, it was reported that the deficiency of FasL accelerates development of tumors in p53+/− mice (Embree-Ku and Boekelheide, 2002). Skp2 overexpression negates apoptosis induced by Akt inhibition by prohibiting the induction of FasL and by reducing the expression of FAS. As was recently reported D cyclins promote hematopoietic cell survival through inhibition of FasL and Fas expression via E2F1 (Choi et al., 2014). Since we found that Skp2 overexpression induced cyclin D1 expression and inhibits p27 expression, which eventually could activate E2F1, we proposed a mechanism by which Skp2 negates Fas-mediated apoptosis induced by FOXO (Fig. 7I). Thus, Skp2 exerts resistance to Akt inhibition and could override both inhibition of mTORC1 and activation of FOXO, and could predict resistance to Akt inhibition therapy. Consistently, it was reported that resistance to rapamycin is linked to high Skp2 expression (Totary-Jain et al., 2012). Although Skp2 had been previously implicated in inhibition of apoptosis, the mechanism by which its exerts resistance to apoptosis is unclear. Our results provided a mechanism by which Skp2 could promote resistance to FAS-mediated cell death. The hypersensitivity to Akt inhibition exerted by p53 deficiency, although was demonstrated only in one particular mouse model of p53−/− thymic lymphoma could be potentially generalized to other cell types in future work. However, the mechanism by which Akt inhibition sensitizes p53−/− human epithelial cancer cells to cell death appears to be different from the mechanism uncovered in the thymic lymphoma cells, and could be dependent on the cells’ milieu.
In summary the results presented in this study provided a new concept showing that systemic Akt1 inhibition alone is sufficient to selectively regress and inhibit tumor progression even in the absence of Akt activation in this tumor. It was also emerged from these studies that deregulation of Skp2 expression is a potential mechanism for resistance to Akt inhibition.
Experimental Procedures
Reagent and antibodies
Roswell Park Memorial Institute medium-1640 (RPMI-1640) was purchased from Gibco and Hyclone. Fetal bovine serum (FBS) was purchased from Gemini. MK2206 was purchased from Selleckchem and ChemieTek. Captisol was purchased from Captisol, A Ligand Company. Antibodies against phospho-Akt (S473), Akt, phospho-S6, S6, phospho-FoxO3a, phospho-4EBP1, 4EBP1, cleaved caspase 3 and Gadph from Cell Signaling. Antibodies to actin from Sigma; Skp2, BIM, Cyclin D1, and cleaved caspase 8 antibodies are from Cell Signaling and abcam; FoxO3 antibodies from Upstate, p27 antibodies from BD bioscience, and Mcl-1 and FAS antibodies are from Santa Cruz Biotechnology. BrdU was purchased from Dako and fluorescein mouse second antibody was from Vector.
Mice
C57Bl/6 Akt1f/f mice were generated by Ozgene (Australia) as described in Fig S1. The Rosa26CreERT2 strain (Strain 01XAB) was obtained from Tyler Jacks’ laboratory (Massachusetts Institute of Technology, USA). The C57Bl/6 p53+/− strain was obtained from the Jackson Laboratories (Bar Harbor, ME). The double compound R26CreERT2; p53−/− mice and triple compound R26CreERT2; Akt1f/f;p53−/− mice were generated by mating the three individual strains. NU/NU nude mice were obtained from Charles River Laboratories (Wilmington, MA).
Genotyping
DNA samples from tail of mice or thymic lymphoma cells were analyzed by PCR using primer sets that can detect wild type, deleted p53, Akt1 floxed, and Rosa26CreERT2 alleles. Primers sequences are: For Akt1 floxed allele, 5′ – TCACAGAGATCCACCTGTGC-3′(A1-3loxp), 5′-GGGCCTCCATACACTCAAGA-3′(A1-i4-5R); p53, ACAGCGTGGTGGTACCTTAT, TCCTCGTGCTTTACGGTATC, TATACTCAGAGCCGGCCT; CreERT2, CCTGATCCTGGCAATTTCG (CRE-R1), GGAGCGGGAGAAATGGATATG (CRE-wtR) and AAAGTCGCTCTGAGTTGTTAT (CRE-uniF).
Isolation of primary thymocytes and establishment of p53 null thymic lymphoma cell lines
Primary thymocytes were isolated by gently pressing the thymus between two sterile glass slides, then washed with PBS and resuspended in RPMI medium supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin. For the generation of thymic lymphoma cell lines, tumors were excised and minced. Thymic lymphoma cells grew in suspension and they were maintained in RPMI medium supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin.
Isogenic colorectal cell lines
The cell lines HCT116, HCT116-p53−/−, DLD1, DLD-p53−/−, SW48, and SW48-p53−/− were previously described (Sur et al., 2009), and were kindly provided by Dr. Bert Vogelstein. Cells were grown in DMEM medium supplemented with 10% FBS. For treatment with MK2206 cells (300,000) were plated on 6cm and 12h later were exposed to either MK2206 or DMSO. Apoptosis was quantified with DAPI attaining as previously described (Kennedy et al., 1997).
Quantitative PCR
Total RNA was extracted from cells at the indicated treatment and time point by using TRIzol reagent (Invitrogen). First strand cDNA was produced with SuperScript III reverse transcriptase (Invitrogen). Quantitative Real-time PCR was performed with iQ SYBR green supermix (BIO-RAD) with the iQ5 real-time PCR detecting system. Relative levels of mRNA were compared by using actin or cyclophilin A primers to normalize for total RNA input. The reaction conditions were as follows: 5 minutes of denaturation at 95°C, followed by 45 cycles of denaturation for 15 seconds at 95°C, and annealing and elongation for 1 minute at 55°C.The mouse primer se quences are: Bim, 5′-CGACAGTCTCAGGAGGAACC-3′ (forward) and 5′-CCTTCTCCATACCAGACGGA-3′ (reverse); FasL, 5′-TCCGTGAGTTCACCAACCAAA-3′ (forward) and 5′-GGGGGTTCCCTGTTAAATGGG-3′ (reverse); Cycophilin A, 5′-TTCACAAACCACAATGGCACAGGG-3′ (forward) and 5′-TGCCGTCCAGCCAATCTGTCTTAT-3′ (reverse); Actin, 5′-GGCCGAGGAGCAAGAATGG-3′ (forward) and 5′-CATGCACTCTGCGATACGCT-3′ (reverse).
Western Blot analysis
Cells were collected and lysed in lysis buffer (1% Triton X-100, 20 mM HEPES, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA) containing phosphatase inhibitors (5 mM IAA, 20 nM OA, 10 mM sodium pyrophosphate, 20 mM β-glycero-glycerophosphate,100 mM NaF) and protease inhibitor cocktail (Roche Applied Science). Solubilized proteins were collected by centrifugation and quantified using a protein assay reagent (Bio-Rad). Equal amounts of protein of each sample were resolved by electrophoresis in 8%, 10% or 12% gel and transferred to polyvinylidene difluoride membranes (Bio-Rad).
Retroviral plasmids
The retroviral vector p-MSCV-IRES-GFP was obtained from Addgene (Addgene plasmid 20672). Myristoylated Akt1 (mAkt) was previously described (Skeen et al., 2006) and was inserted in the Sal1 and Xho1 of p-MSCV-IRES-GFP. The retroviral vector p-MSCV-Skp2-IRES-GFP was kindly provided by Michael Deininger and was previously described (Agarwal et al., 2008). The retroviral vector p-MSCV-p53-IRES-GFP was kindly provided by Dean Felsher and was previously described (Giuriato et al., 2006).
Retrovirus transfection and infection
Retroviruses were produced in phoenix-ecotropic cells by lipofectamine 2000 transfection. Thymic lymphoma cells were infected by using MagnetoFection™. Before infection, viruses mixed with ViroMag R/L and incubated at room temperature for 15 min. Cells mixed with CombiMag then plated in 12-well plate and incubated at magnetic plate for 20 min. After incubation, ViroMag R/L/viruses mixture were added to the CombiMag/cells mixture and incubated for 30–60 min followed by manufacture’s instructions. Seven days after infection, GFP-positive cells were sorted using MoFlo Legacy cell sorter to establish stably expressing cell lines.
Transfection with siRNA
Cells were transfected using MagnetoFection™. Two siRNAs pool were used: non-targeting control siRNA (ON-TARGETplus non-targeting siRNA SMARTpool, Dharmacon) or a siRNA pool consisting of four siRNAs specific for mouse Fas (ON-TARGETplus Fas siRNA SMARTpool, Dharmacon). Before infection, siRNA were mixed with SilentMag and incubated at room temperature for 15 min. In the meantime, cells were mixed with CombiMag, plated in a 12-well plate and incubated on the magnetic plate for 20 min. After incubation, siRNA/SilentMag mixture was added to the CombiMag/cells mixture on the magnetic plate for 30 min as recommended by manufacturer’s instructions. Three days post transfection cells were split for experiment and treated with DMSO or MK2206 for 18h. Flow cytometry was then performed to estimate cell death (PI staining), and samples were collected for immunoblotting.
Flow Cytometry, Cell cycle analysis, BrdU incorporation and Cell sorting
Cells were incubated with bromodeoxyuridine (BrdU) (3ug/ml) for 2 hours before the indicated time point for collection. Cells were resuspended in 1ml PBS after centrifugation and fixed by adding 3ml of 100% ethanol drop wise. and stored at 4°C. The day before analysis, cells were pelleted by centrifugation at 2,000 rpm for 5 min. Then cells were solubilized in 2N-HCl with 0.5% Triton-X followed by neutralization with 0.1M sodium borate, pH8.5. Cells were then incubated with anti- BrdU antibody (Dako) overnight at 4°C. Next day, cells were washed twice by washing buffer (PBS+0.5%BSA) and incubated with Fluorescein anti-mouse secondary antibody (Vector). Before analysis, the cells were stained with propidium iodide containing RNase and Triton X-100. Flow cytometry was performed using Beckman Coulter EPICS Elite ES (Beckman, Hialeh, FL) and analyzed using Cell lab QuantaTM SC. Cell sorting was performed using MoFlo Legacy cell sorter.
Assessment of FasL cell surface expression
Cells were incubated with DMSO or MK-2206 (1μM) for 18h before collection to assess FasL surface expression by FACS. For FasL staining, cell pellets were resuspended in cell staining buffer (BioLegend) and incubated with 1μg/3×106 cells PE anti-mouse FasL antibody (BioLegend) for 1h at 4°C in the dark. The cells were then washed twice in cell staining buffer and counterstained with 7-AAD (500ng/3×106 cells) for another 15 min at 4°C in the dark to assess viability. As a negative control, cells were stained with PE IgG isotype control antibody. Flow cytometry was then performed using Cell lab QuantaTM SC and both FasL expression (PE positive cells) and apoptosis (7-AAD positive cells) were quantified.
Caspase-8 inhibition
Cells were incubated with DMSO or MK-2206 (1μM) in absence or presence of 20μM caspase-8 inhibitor (Z-LETD-FMK, Biorad) for 18h before collection. The cells were then washed twice in PBS/5mM EDTA and stained with 7-AAD (500ng/3×106 cells) for 15 min at 4°C in the dark to assess viability. Flow cytometry was then performed using Cell lab QuantaTM SC and apoptosis (7-AAD positive cells) was quantified.
Xenograft Assay
Cells were counted and suspended in cold PBS. Cells (1.5×106) were injected subcutaneously into the rear flank of the nude mice. After palpable tumor formation, mice were randomized into two groups. Either corn oil or tamoxifen (1mg/100ul/per injection) were injected into the nude mice intraperitoneally every other day for two weeks. MK2206 was dissolved in 30% captisol per manufacturer’s instructions and delivered by oral gavage. Tumor sizes were measured with a caliper and calculated by length*height*width*0.5.
Magnetic Resonance Imaging (MRI) and Tumor Volume Analysis
All animals were sequentially imaged using an Agilent 9.4T preclinical MRI to screen for tumor growth and treatment response to tamoxifen. T1-weighted sequences were obtained in axial and coronal planes, using the following parameters: T1-weighted: TR/TE=900/14ms, matrix size 192X192, 1mm slice thickness, 20 sequences, 6 repetitions. Tumor volume measurements were performed using T1-weighted coronal and axial image stacks. The tumor volume was calculated by ImageJ measure stacks to obtain the tumor volume in mm3. For each time point, all measurements from the available sequences were used to calculate a mean volume +/− SEM in order to limit errors due to manual process.
Statistical analysis
Statistical analysis was done with unpaired Student’s t-tests. Survival curves were analyzed by log-rank test (Mantel-Cox) and data are expressed as SEM as indicated in the figure legends. Unless otherwise indicated all the experiments were performed for at least three times.
Supplementary Material
Systemic deletion of Akt1 is sufficient to inhibit and regress thymic lymphoma in p53−/− mice, emulating p53 restoration.
Akt inhibition could inhibit and regress tumors that are not driven by Akt activation
Skp2 inhibits apoptosis by inhibiting FasL and Fas expression
Overexpression of Skp2 could exert resistance to Akt inhibition
Acknowledgments
These studies were supported by NIH grants R01AG016927 and R01CA090764, and by VA Merit Award BX000733 to N.H..
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agarwal A, Bumm TG, Corbin AS, O’Hare T, Loriaux M, VanDyke J, Willis SG, Deininger J, Nakayama KI, Druker BJ, et al. Absence of SKP2 expression attenuates BCR-ABL-induced myeloproliferative disease. Blood. 2008;112:1960–1970. doi: 10.1182/blood-2007-09-113860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir T, Pagan JK, Busino L, Pagano M. Phosphorylation of Ser72 is dispensable for Skp2 assembly into an active SCF ubiquitin ligase and its subcellular localization. Cell Cycle. 2010;9 doi: 10.4161/cc.9.5.10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar PT, Hay N. The Two TORCs and Akt. Dev Cell. 2007;12:487–502. doi: 10.1016/j.devcel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- Chen ML, Xu PZ, Peng X, Chen WS, Guzman G, Yang X, Di Cristofano A, Pandolfi PP, Hay N. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/− mice. Genes Dev. 2006;20:1569–1574. doi: 10.1101/gad.1395006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T, Roninson I, Weng W, Suzuki R, Tobe K, et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15:2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YJ, Saez B, Anders L, Hydbring P, Stefano J, Bacon NA, Cook C, Kalaszczynska I, Signoretti S, Young RA, et al. D-cyclins repress apoptosis in hematopoietic cells by controlling death receptor Fas and its ligand FasL. Dev Cell. 2014;30:255–267. doi: 10.1016/j.devcel.2014.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embree-Ku M, Boekelheide K. FasL deficiency enhances the development of tumors in p53+/− mice. Toxicol Pathol. 2002;30:705–713. doi: 10.1080/01926230290166797. [DOI] [PubMed] [Google Scholar]
- Gao D, Inuzuka H, Tseng A, Chin RY, Toker A, Wei W. Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nat Cell Biol. 2009;11:397–408. doi: 10.1038/ncb1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuriato S, Ryeom S, Fan AC, Bachireddy P, Lynch RC, Rioth MJ, van Riggelen J, Kopelman AM, Passegue E, Tang F, et al. Sustained regression of tumors upon MYC inactivation requires p53 or thrombospondin-1 to reverse the angiogenic switch. Proc Natl Acad Sci U S A. 2006;103:16266–16271. doi: 10.1073/pnas.0608017103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9:1956–1967. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- Hollander MC, Maier CR, Hobbs EA, Ashmore AR, Linnoila RI, Dennis PA. Akt1 deletion prevents lung tumorigenesis by mutant K-ras. Oncogene. 2011;30:1812–1821. doi: 10.1038/onc.2010.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Caraway NP, Li R, Katz RL. RNA silencing of S-phase kinase-interacting protein 2 inhibits proliferation and centrosome amplification in lung cancer cells. Oncogene. 2005;24:3409–3418. doi: 10.1038/sj.onc.1208459. [DOI] [PubMed] [Google Scholar]
- Ju X, Katiyar S, Wang C, Liu M, Jiao X, Li S, Zhou J, Turner J, Lisanti MP, Russell RG, et al. Akt1 governs breast cancer progression in vivo. Proc Natl Acad Sci U S A. 2007;104:7438–7443. doi: 10.1073/pnas.0605874104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang-Decker N, Tong C, Boussouar F, Baker DJ, Xu W, Leontovich AA, Taylor WR, Brindle PK, van Deursen JM. Loss of CBP causes T cell lymphomagenesis in synergy with p27Kip1 insufficiency. Cancer Cell. 2004;5:177–189. doi: 10.1016/s1535-6108(04)00022-4. [DOI] [PubMed] [Google Scholar]
- Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, Hay N. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes and Development. 1997;11:701–713. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- Latres E, Chiarle R, Schulman BA, Pavletich NP, Pellicer A, Inghirami G, Pagano M. Role of the F-box protein Skp2 in lymphomagenesis. Proc Natl Acad Sci U S A. 2001;98:2515–2520. doi: 10.1073/pnas.041475098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, McCormick F. Downregulation of Skp2 and p27/Kip1 synergistically induces apoptosis in T98G glioblastoma cells. J Mol Med (Berl) 2005;83:296–307. doi: 10.1007/s00109-004-0611-7. [DOI] [PubMed] [Google Scholar]
- Lin HK, Wang G, Chen Z, Teruya-Feldstein J, Liu Y, Chan CH, Yang WL, Erdjument-Bromage H, Nakayama KI, Nimer S, et al. Phosphorylation-dependent regulation of cytosolic localization and oncogenic function of Skp2 by Akt/PKB. Nat Cell Biol. 2009;11:420–432. doi: 10.1038/ncb1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkin D. Li-fraumeni syndrome. Genes & cancer. 2011;2:475–484. doi: 10.1177/1947601911413466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroulakou IG, Oemler W, Naber SP, Tsichlis PN. Akt1 ablation inhibits, whereas Akt2 ablation accelerates, the development of mammary adenocarcinomas in mouse mammary tumor virus (MMTV)-ErbB2/neu and MMTV-polyoma middle T transgenic mice. Cancer Res. 2007;67:167–177. doi: 10.1158/0008-5472.CAN-06-3782. [DOI] [PubMed] [Google Scholar]
- Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127:1323–1334. doi: 10.1016/j.cell.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. 2006;21:749–760. doi: 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Nagahama H, Minamishima YA, Miyake S, Ishida N, Hatakeyama S, Kitagawa M, Iemura S, Natsume T, Nakayama KI. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell. 2004;6:661–672. doi: 10.1016/s1534-5807(04)00131-5. [DOI] [PubMed] [Google Scholar]
- Nogueira V, Sundararajan D, Kwan JM, Peng XD, Sarvepalli N, Sonenberg N, Hay N. Akt-dependent Skp2 mRNA translation is required for exiting contact inhibition, oncogenesis, and adipogenesis. EMBO J. 2012;31:1134–1146. doi: 10.1038/emboj.2011.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano M. Control of DNA synthesis and mitosis by the Skp2-p27-Cdk1/2 axis. Mol Cell. 2004;14:414–416. doi: 10.1016/s1097-2765(04)00268-0. [DOI] [PubMed] [Google Scholar]
- Skeen JE, Bhaskar PT, Chen CC, Chen WS, Peng XD, Nogueira V, Hahn-Windgassen A, Kiyokawa H, Hay N. Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1-dependent manner. Cancer Cell. 2006;10:269–280. doi: 10.1016/j.ccr.2006.08.022. [DOI] [PubMed] [Google Scholar]
- Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA, Jr, Kinzler KW, Vogelstein B, Papadopoulos N. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci U S A. 2009;106:3964–3969. doi: 10.1073/pnas.0813333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totary-Jain H, Sanoudou D, Dautriche CN, Schneller H, Zambrana L, Marks AR. Rapamycin resistance is linked to defective regulation of Skp2. Cancer Res. 2012;72:1836–1843. doi: 10.1158/0008-5472.CAN-11-2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- Walker KS, Deak M, Paterson A, Hudson K, Cohen P, Alessi DR. Activation of protein kinase B beta and gamma isoforms by insulin in vivo and by 3-phosphoinositide-dependent protein kinase-1 in vitro: comparison with protein kinase B alpha. Biochem J. 1998;331(Pt 1):299–308. doi: 10.1042/bj3310299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Bauzon F, Ji P, Xu X, Sun D, Locker J, Sellers RS, Nakayama K, Nakayama KI, Cobrinik D, et al. Skp2 is required for survival of aberrantly proliferating Rb1-deficient cells and for tumorigenesis in Rb1+/− mice. Nat Genet. 2010a;42:83–88. doi: 10.1038/ng.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Suh YA, Fuller MY, Jackson JG, Xiong S, Terzian T, Quintas-Cardama A, Bankson JA, El-Naggar AK, Lozano G. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J Clin Invest. 2011;121:893–904. doi: 10.1172/JCI44504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel HG, Silva RL, Malina A, Mills JR, Zhu H, Ueda T, Watanabe-Fukunaga R, Fukunaga R, Teruya-Feldstein J, Pelletier J, et al. Dissecting eIF4E action in tumorigenesis. Genes Dev. 2007;21:3232–3237. doi: 10.1101/gad.1604407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu PZ, Chen ML, Jeon SM, Peng XD, Hay N. The effect Akt2 deletion on tumor development in Pten(+/−) mice. Oncogene. 2012;31:518–526. doi: 10.1038/onc.2011.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.