

Abstract
The direct decarboxylative arylation of α-oxo acids has been achieved via synergistic visible light-mediated photoredox and nickel catalyses. This method offers rapid entry to aryl and alkyl ketone architectures from simple α-oxo acid precursors via an acyl radical intermediate. Significant substrate scope is observed with respect to both the oxo acid and arene coupling partners. This mild decarboxylative arylation can also be utilized to efficiently access medicinal agents, as demonstrated by the rapid synthesis of fenofibrate.
Keywords: photoredox catalysis, nickel catalysis, decarboxylation, arylation, α-oxo acid
Photoredox catalysis employing visible light has recently emerged as a valuable platform for the design of unique one-electron transfer pathways that allow the invention of valuable new chemical reactions.[1] In this vein, our laboratory has described the decarboxylative coupling of α-amino, α-oxy and alkyl carboxylic acids with aryl halides, a protocol that enables broad access to Csp3–Csp2 bonds using abundant and inexpensive starting materials.[2] This new fragment coupling relies on the capacity of photoredox catalysts to simultaneously modulate the oxidation states of organometallic intermediates while generating open shell organic species that can interface with transition metal catalysts (e.g. Pd, Ni, Cu).[2–3] Recently, we questioned whether this synergistic catalysis pathway might provide a direct and mild route to ketones via the radical decarboxylative coupling of simple α-oxo acids and aryl halides, a transformation that to our knowledge has not previously been described.[4] Herein we detail the successful execution of these ideals, and present a new mechanism for the production of diaryl, alkyl–aryl, and dialkyl carbonyls at room temperature without the requirement for CO, strong bases, or organometallic reagents.
Ketones have long been established as a linchpin functionality in organic chemistry due to their innate capacity to function as electrophiles across a tremendous array of bond forming reactions (e.g. to form C–C, C=C, C–N and RO–C=O bonds). Moreover, ketones are a common structural element found in a wide range of agrochemicals, bioactive natural products, pharmaceuticals, and electronic materials (including photovoltaics).[5] Common protocols for ketone synthesis currently include (i) organometallic additions to Weinreb amides,[6] (ii) Stille couplings between acyl chlorides and stannanes,[7] (iii) metal-catalyzed carbonylations between aryl halides and prefunctionalized transmetallation reagents (e.g. boronic acids),[8] and (iv) alkene hydroacylations.[9] While the synthetic value of these coupling strategies is self-evident, the development of new catalytic transformations that provide access to structurally diverse ketones using simple, inexpensive substrates would be welcomed by synthetic chemists.
Within the realm of open-shell chemistry, acyl radicals derived from acyl selenides and tellurides have long been used to initiate cyclization cascades to generate complex ketones via formal hydroacylation reactions.[10] However, the synthetic utility of acyl radicals has been somewhat limited due to their innate nucleophilicity[11] along with the immoderate conditions required for their generation (typically entailing high temperatures, UV light, or stoichiometric tin reagents). As a critical advantage, we postulated that the implementation of photoredox-mediated decarboxylation[2,12] would allow for a broad range of acyl radicals to be accessed from α-oxo acids such as pyruvic acid, thereby allowing ketone production from an abundant, non-metal based source. As a key design element, this photoredox approach to nickel-acyl complex formation would allow facile generation of a series of carbonyl products using mild conditions (room temperature), and without the need for toxic reagents or stoichiometric oxidants.[13]
A detailed mechanism for the proposed metallaphotoredox aryl cross-coupling with α-oxo acids is shown in Scheme 1. It is well established that photoredox catalyst Ir[dF(CF3)ppy]2-(dtbbpy)+ 1[14] readily absorbs photons upon visible light irradiation to generate the oxidizing excited state *Ir[dF(CF3)-ppy]2(dtbbpy)+ 2 [E1/2III*/II = +1.21 V vs. saturated calomel electrode (SCE) in CH3CN].[15] Base-mediated deprotonation of an α-oxo acid substrate (such as pyruvic acid (3), shown) and subsequent single-electron oxidation of the resulting carboxylate functionality (E1/2red = +1.03 V vs. SCE in DMSO)[13d] by the excited photocatalyst 2 should generate the reduced photocatalyst 4 and a corresponding carboxyl radical species. At this stage, we presumed that this open-shell dicarbonyl intermediate would rapidly extrude CO2 to deliver the critical acyl radical species 5. Within the same time frame, the second catalytic cycle would initiate via oxidative addition of the Ni0 catalyst 6[16] into the aryl halide (e.g. 4-iodotoluene (7), as shown) to generate NiII-aryl complex 8. The resulting electro-philic metal species 8 would then rapidly trap the nucleophilic acyl radical 5 to produce nickel-acyl complex 9. At this stage, reductive elimination from this NiIII complex would be expected to forge the requisite Csp2–Csp2 bond of compound 10, while expelling the corresponding NiI complex 11. Finally, single-electron transfer (SET) from the photocatalyst IrII species 4 to the NiI-dtbbpy complex 11 would return the metal catalyst to the required Ni0 oxidation state in an exergonic process. Indeed, the thermodynamic requirements of the two electron reduction of NiII to Ni0 are favorable (E1/2II/0 = −1.2 V vs. SCE in DMF)[17] given the corresponding reduction potential of the IrII complex 4 (E1/2III/II = −1.37 V vs. SCE in CH3CN).[15] It should be noted that this second photoredox-mediated SET event regenerates the ground state IrIII catalyst 1 while reconstituting the requisite Ni0 complex 6, completing the photoredox and nickel cycles simultaneously.
Scheme 1.

Merged photoredox and nickel catalytic cycles.
We began our investigations into this new acyl cross-coupling protocol using phenylglyoxylic acid and para- iodotoluene in the presence of Ir[dF(CF3)ppy]2(dtbbpy)+ 1, NiCl2·dtbbpy 12, Cs2CO3, and DMF. Given the findings from our previous studies into the decarboxylative nickel-catalyzed arylation of amino acids, we presumed that a strongly oxidizing photocatalyst (e.g. 1) would be required to oxidize the carboxylate and initiate the CO2 extrusion step. Indeed, we were delighted to achieve proof of concept using the optimized catalytic conditions from our earlier studies; however, the efficiency of ketone product formation was poor (Table 1, entry 1, 13% yield). Pleasingly, changing from Cs2CO3 to Li2CO3 led to a notable increase in reaction efficiency (entry 2, 38% yield). Moreover, implementation of a more powerful 34 Watt blue LED lamp in lieu of the standard blue LED strips provided further improvement (entry 3, 60% yield), highlighting that the reaction is likely photon-limited. The addition of two equivalents of water produced a modest boost in yield while adding further equivalents was detrimental, presumably due to protonolysis of the putative NiII-aryl complex at high H2O concentration (entries 5, 6).[18] Ultimately, increasing the photocatalyst loading to 2 mol% provided superior conditions, delivering the desired diaryl ketone adduct in excellent yield (entry 7, 88%).
Table 1.
Optimization of decarboxylative α-oxo acid arylation.[a]
| ||||
|---|---|---|---|---|
| entry | base | light source | H2O | yield |
| 1 | Cs2CO3 | Blue LED strips | 0 equiv. | 13% |
| 2 | Li2CO3 | Blue LED strips | 0 equiv. | 38% |
| 3 | Li2CO3 | 34 W Blue LED | 0 equiv. | 60% |
| 4b | Li2CO3 | 34 W Blue LED | 0 equiv. | 74% |
| 5b | Li2CO3 | 34 W Blue LED | 2 equiv. | 84% |
| 6b | Li2CO3 | 34 W Blue LED | 8 equiv. | 54% |
| 7b,c | Li2CO3 | 34 W Blue LED | 2 equiv. | 88% |
Yield determined by 1H NMR using an internal standard.
Reaction time 72 hours.
2 mol% photocatalyst.
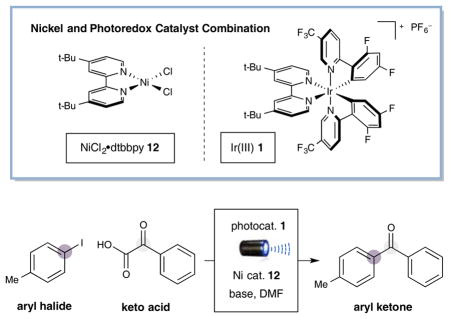
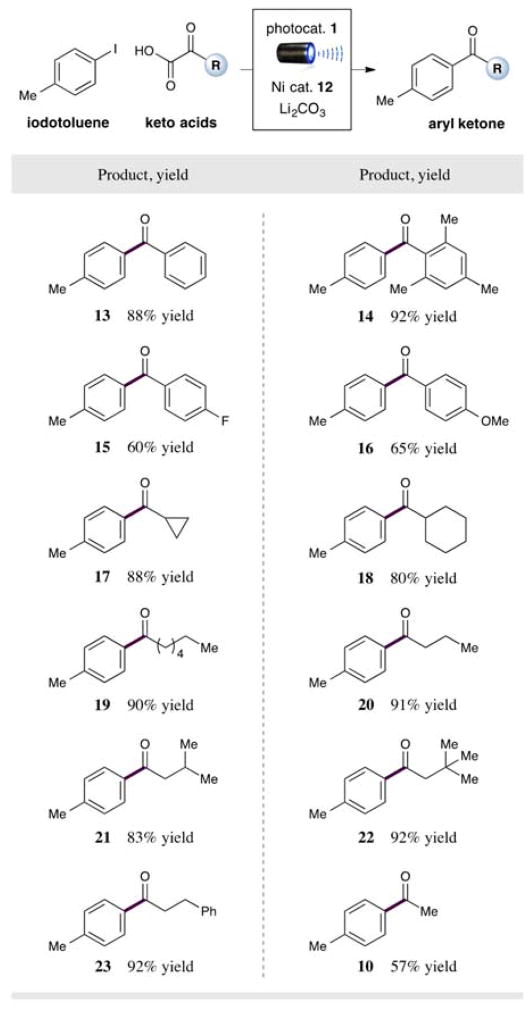
With optimal conditions in hand, we next examined the scope of the α-oxo acid component in this new cross-coupling protocol. As described in Table 2, a wide range of α-keto acids bearing aromatic and aliphatic substituents are amenable to this CO2 extrusion mechanism. Pleasingly, this open shell pathway allows for the ready implementation of an ortho-substituted aryl ring on the keto acid component (product 14, 92% yield), a significant limitation for many previous cross-coupling systems due to the accompanying steric demands.[4] Surprisingly, electron-deficient arenes can be tolerated on the keto acid, despite the inherent difficulty of the carboxylate oxidation step (product 15, 60% yield). Electron-rich aryl glyoxylic acids readily serve as efficient nucleophiles, generating the product ketones in good yields (products 14 and 16, 92% and 65% yield).
Table 2.
Decarboxylative coupling: scope of the α-oxo acid.[a]
|
Reaction performed using the optimized conditions from Table 1 (see Supporting Information). All cited yields are isolated.
As further detailed in Table 2, a range of aliphatic keto acids can also serve as efficient coupling partners. Particularly notable are cyclic systems such as cyclopropyl and cyclohexyl glyoxylic acids (products 17 and 18, 88% and 80% yield). Moreover, a variety of acyclic alkyl-substituted ketones are readily accessible using the appropriate α-oxo acids (products 19–23, 83–92% yield). Perhaps most pleasing, pyruvic acid, which is available from biomass, can be readily harnessed to generate aryl acylation adducts directly (product 10, 57% yield).
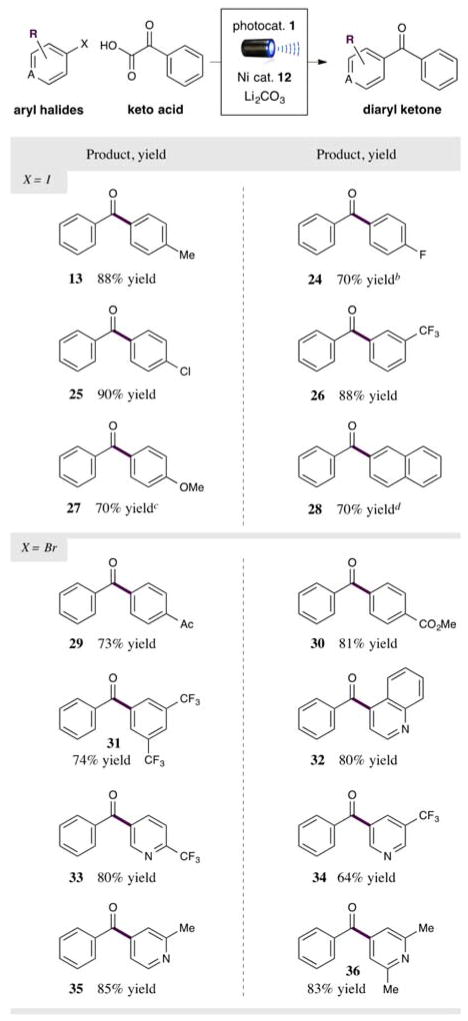
With respect to the electrophilic coupling partner, the mild conditions employed with this photoredox-Ni technology allow for a wide range of aryl halides to be employed (Table 3). For example, iodoarenes bearing fluoride and chloride substituents can be used to generate halogenated ketones without the formation of dehalogenation adducts (products 24 and 25, 70% and 90% yield). The enhanced rate of oxidative addition to aryl iodides relative to aryl chlorides allows for chemoselectivity in the cross-coupling of 1-chloro-4-iodobenzene, which allows the chloride group to be retained as a handle for further synthetic manipulations or as a structural element for medicinal chemistry. In this context, it is important to note that a trifluoromethyl group can also be incorporated on the arene ring (product 26, 88% yield). Electron-rich arenes can also serve as electrophiles, albeit with increased reaction times (products 27 and 28, both 70% yield). Not surprisingly, electron-deficient arenes are highly competent in this cross-coupling protocol, enabling the use of bromoarenes as electrophiles in these cases (products 29–36, 64–85% yield). As a critical requirement for the broad scale implementation of this technology, heteroarenes can be successfully employed to rapidly build nitrogen-containing aryl ketone adducts (products 32–36, 64–85% yield). Specifically, 3-bromopyridines can be coupled effectively (products 33 and 34, 80% and 64% yield). Last, we have further determined that 4-bromopyridines are valuable coupling partners, and that the steric environment around the nitrogen is inconsequential with respect to reaction yield (products 32, 35, and 36, 80–85% yield).
Table 3.
Decarboxylative coupling: scope of the aryl halide.[a]
|
Reaction performed using the optimized conditions from Table 1 (see Supporting Information). All cited yields are isolated.
Reaction performed using 5.0 equiv. of acid and Li2CO3.
84 hours. [d] 90 hours.
To further demonstrate the utility and generality of this novel cross-coupling reaction, we sought to employ electrophiles beyond the realm of aromatic halides. As shown in equation 3, hindered vinyl halides can be readily utilized in the procedure to build α,β-unsaturated ketones directly. Perhaps more importantly, alkyl halides such as bromocyclopentane have succesfully been utilized in this new protocol to generate dialkyl ketones [Eq. (4)]. This latter example highlights the use of nickel catalysis to allow oxidative insertion into Csp3-bearing halides without the intervention of detrimental β-hydride elimination pathways.
Finally, we sought to highlight the value of this new technology for medicinal chemistry via the rapid production of a biologically active small molecule incorporating a diaryl ketone. Within this context, fenofibrate, a cholesterol modulating pharmaceutical of the fibrate class, currently ranked 47th in American retail sales,[5c] was synthesized via a single decarboxylation step (three steps overall) in 71% yield [Eq. (5)].
Supplementary Material
Figure 1.

Metallaphotoredox decarboxylative arylation of α-oxo acids.
Scheme 2.

Metallaphotoredox decarboxylation and its application.
Footnotes
The authors are grateful for financial support provided by the NIH General Medical Sciences (Grant NIHGMS (R01 GM093213-01) and gifts from Merck, AbbVie, and Bristol Myers Squibb. L. C. acknowledges a postdoctoral fellowship from the Shanghai Institute of Organic Chemistry.
References
- 1.a) Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tucker JW, Stephenson CRJ. J Org Chem. 2012;77:1617. doi: 10.1021/jo202538x. [DOI] [PubMed] [Google Scholar]; c) Narayanam JMR, Stephenson CRJ. Chem Soc Rev. 2011;40:102. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]; d) Nicewicz D, MacMillan DWC. Science. 2008;322:77. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Ischay MA, Anzovino ME, Du J, Yoon TP. J Am Chem Soc. 2008;130:12886. doi: 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]
- 2.Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC. Science. 2014;345:437. doi: 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Yu WY, Sit WN, Zhou Z, Chain ASC. Org Lett. 2009;11:3174. doi: 10.1021/ol900756g. [DOI] [PubMed] [Google Scholar]; b) Kalyani D, McMurtrey KB, Neufeldt SR, Sanford MS. J Am Chem Soc. 2011;133:18566. doi: 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ye Y, Sanford MS. J Am Chem Soc. 2012;134:9034. doi: 10.1021/ja301553c. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Neufeldt SR, Sanford MS. Adv Synth Catal. 2012;354:3517. doi: 10.1002/adsc.201200738. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Tellis JC, Primer DN, Molander GA. Science. 2014;345:433. doi: 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Noble A, McCarver SJ, MacMillan DWC. J Am Chem Soc. 2015;137:624. doi: 10.1021/ja511913h. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Primer DN, Karakaya I, Tellis JC, Molander GA. J Am Chem Soc. 2015;137:2195. doi: 10.1021/ja512946e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For Cu-catalyzed decarboxylative couplings of α-oxo acids at 170 °C see: Gooβen LJ, Rudolphi F, Oppel C, Rodríguez N. Angew Chem Int Ed. 2008;47:3043. doi: 10.1002/anie.200705127.
- 5.a) Walter MW. Nat Prod Rep. 2002;19:278. doi: 10.1039/b100919m. [DOI] [PubMed] [Google Scholar]; b) McDaniel R, Thamchaipenet A, Gustafsson C, Fu H, Betlach M, Betlach M, Ashley G. Proc Natl Acad Sci USA. 1999;96:1846. doi: 10.1073/pnas.96.5.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) McGrath NA, Brichacek M, Njardarson JT. J Chem Educ. 2010;87:1348. [Google Scholar]; d) Kamat PV. Chem Rev. 1993;93:267. [Google Scholar]
- 6.a) Nahm S, Weinreb SM. Tetrahedron Lett. 1981;22:3818. [Google Scholar]; b) Singh J, Satyamurthi N, Aidhen IS. J Prakt Chem. 2000;342:340. [Google Scholar]
- 7.Stille JK. Angew Chem Int Ed. 2003;25:508. [Google Scholar]
- 8.a) Miyaura N, Suzuki A. Chem Rev. 1995;95:2457. [Google Scholar]; b) Wu XF, Neumann H, Beller M. Chem Soc Rev. 2011;40:4986. doi: 10.1039/c1cs15109f. [DOI] [PubMed] [Google Scholar]
- 9.Willis MC. Chem Rev. 2010;110:725. doi: 10.1021/cr900096x. [DOI] [PubMed] [Google Scholar]
- 10.Chatgilialoglu C, Crich D, Komatsu M, Ryu I. Chem Rev. 1999;99:1991. doi: 10.1021/cr9601425.For hydroacylation of α,β-unsaturated esters via aerobic C-H activation, see: Chudasama V, Fitzmaurice RJ, Caddick S. Nat Chem. 2010;2:592. doi: 10.1038/nchem.685.
- 11.Citterio A, Minisci F, Porta O, Sesana G. J Am Chem Soc. 1977;99:7960. [Google Scholar]
- 12.a) Rueda-Becerril M, Mahé O, Drouin M, Majewski MB, West JG, Wolf MO, Sammis GM, Paquin JF. J Am Chem Soc. 2014;136:2637. doi: 10.1021/ja412083f. [DOI] [PubMed] [Google Scholar]; b) Zuo Z, MacMillan DWC. J Am Chem Soc. 2014;136:5257. doi: 10.1021/ja501621q. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chu L, Ohta C, Zuo Z, MacMillan DWC. J Am Chem Soc. 2014;136:10886. doi: 10.1021/ja505964r. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Noble A, MacMillan DWC. J Am Chem Soc. 2014;136:11602. doi: 10.1021/ja506094d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Caronna T, Frona G, Minisci F, Porta O. J Chem Soc, Perkin Trans. 1972;2:2035. [Google Scholar]; b) Fontana F, Minisci F, Barbosa MCN, Vismara E. J Org Chem. 1991;56:2866. [Google Scholar]; c) Liu J, Liu Q, Yi H, Qin C, Bai R, Qi X, Lan Y, Lei A. Angew Chem Int Ed. 2014;53:502. doi: 10.1002/anie.201308614. [DOI] [PubMed] [Google Scholar]; d) Majek M, von Wangelin AJ. Angew Chem Int Ed. 2015;54:2270. doi: 10.1002/anie.201408516. [DOI] [PubMed] [Google Scholar]
- 14.dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-(trifluoromethyl)pyridine, dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine.
- 15.Lowry MS, Goldsmith JI, Slinker JD, Rohl R, Pascal RA, Jr, Malliaras GG, Bernhard S. Chem Mater. 2005;17:5712. [Google Scholar]
- 16.To reduce the Ni(II) catalyst to Ni(0), a sacrificial amount of acid must be oxidized by 2 to turn over the photocatalytic cycle. See ref 2, 11d.
- 17.Rolling Y, Troupel M, Tuck DG, Perichon J. J Organomet Chem. 1986;303:131. [Google Scholar]
- 18.Ueda M, Saitoh A, Oh-tani S, Miyaura N. Tetrahedron. 1998;54:13079. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.