

Abstract
Repetitive brain trauma is associated with a progressive neurological deterioration, now termed as chronic traumatic encephalopathy (CTE). Most instances of CTE occur in association with the play of sports, but CTE has also been reported in association with blast injuries and other neurotrauma. Symptoms of CTE include behavioral and mood changes, memory loss, cognitive impairment and dementia. Like many other neurodegenerative diseases, CTE is diagnosed with certainty only by neuropathological examination of brain tissue. CTE is a tauopathy characterized by the deposition of hyperphosphorylated tau (p‐tau) protein as neurofibrillary tangles, astrocytic tangles and neurites in striking clusters around small blood vessels of the cortex, typically at the sulcal depths. Severely affected cases show p‐tau pathology throughout the brain. Abnormalities in phosphorylated 43 kDa TAR DNA‐binding protein are found in most cases of CTE; beta‐amyloid is identified in 43%, associated with age. Given the importance of sports participation and physical exercise to physical and psychological health as well as disease resilience, it is critical to identify the genetic risk factors for CTE as well as to understand how other variables, such as stress, age at exposure, gender, substance abuse and other exposures, contribute to the development of CTE.
Keywords: Alzheimer's disease, chronic traumatic encephalopathy, tauopathy, TDP‐43, traumatic brain injury
Introduction
Over the last decade, there has been increased awareness regarding the long‐term consequences of mild traumatic brain injury (mTBI), including concussion and subconcussion, associated with sports participation and exposure to explosive blast 59. Although the concept that brain trauma from boxing might induce a chronic neurodegenerative condition was recognized nearly a century ago, it is only recently that modern sports, such as American football, soccer, ice hockey and rugby 57, 59, 74, 75, have been associated with a similar deterioration. The first report in the medical literature was made by Harrison Martland, a New Jersey pathologist and medical examiner, who described a clinical syndrome consisting of poor balance, floppiness of the extremities, mental confusion and slowing of muscular action that developed in “nearly half of the fighters who have stayed in the game long enough” 53. Moreover, Martland also noted that some affected boxers went on to develop a severe and progressive syndrome manifest as Parkinsonism and mental deterioration that necessitated a “commitment to an asylum” 53. Although Martland referred to this condition as “punch drunk” 53, other terms were gradually introduced to describe the condition, including “traumatic progressive encephalopathy” 79 and “dementia pugilistica” 63. From the 1940s onward, the broader designation “chronic traumatic encephalopathy” or “CTE” was in common use, recognizing that the condition could arise from brain trauma from a variety of sources in addition to boxing 22, 27, 43.
The neuropathological aspects of CTE were first reported as case reports or small case series of boxers who developed Parkinsonism and dementia 9, 18, 21, 32, 55, 70. Gross neuropathological findings included cerebral atrophy, enlarged ventricles, cavum septum pellucidum and depigmentation of the substantia nigra, with microscopic evidence of neurofibrillary tangles (NFT) in the cortex and brainstem, sometimes associated with senile plaques or neuronal loss. In 1973, Corsellis et al published a large clinicopathological series of CTE in 15 boxers, ranging in age from 57 to 91 years 19. This landmark paper further described enlarged lateral and third ventricles, thinning of the corpus callosum, cavum septum pellucidum with fenestrations and scarring of the cerebellar tonsils as important gross neuropathological findings. Microscopically, Corsellis et al used cresyl violet stains and Von Braunmühl's silver stains to characterize neuronal loss in the cerebral cortex, substantia nigra and cerebellar tonsils, neurofibrillary degeneration of the substantia nigra and cerebral cortex and senile plaques in 27% of cases 19. Subsequent re‐examination of Corsellis' original series of boxers and additional cases using beta‐amyloid (Aß) immunohistochemistry determined that 95% of CTE cases showed widespread diffuse Aß deposits 82, 96.
In 1990, Roberts et al reported the neuropathological findings of CTE in the first female subject, a nonboxer. The case involved a woman who has been physically abused for decades who developed dementia with prominent memory loss and confusion several years before her death at age 76 years 81. Necropsy revealed the “cauliflower ears” of pugilists and old rib fractures, while brain examination revealed cerebral atrophy, ventricular dilatation, septal cavum with fenestrated leaves and multiple lacunes. NFT were present in the hippocampus and brainstem; senile plaques, primarily of the diffuse type, were found in the frontal and temporal cortex.
In 1991, Hof et al reported CTE in a young woman, 24 years old, who was autistic and displayed prominent head‐banging behaviors 38. Neuropathological examination was notable for cerebral atrophy, cavum septum, NFT in the perirhinal and entorhinal cortex, amygdala, prepiriform and orbitofrontal cortex in the absence of Aß deposits.
Five young men, ranging in age from 23 to 28 years with neuropathological alterations of early CTE were reported by Geddes et al 27. The subjects included a soccer player, a “mentally subnormal” individual with a long history of head banging, an epileptic who frequently hit his head during seizures and two boxers. There were no grossly detectable changes to the brain, however, microscopic examination demonstrated argyrophilic, phosphorylated tau (p‐tau)‐positive NFTs and neuropil threads (NT) “strikingly arranged in groups, sited predominantly around small intracortical blood vessels, often found at the depths of the cortical sulci” 27. NFT were not found in any of the 21 age‐matched control cases that Geddes et al examined. Geddes et al also noted that the arrangement and distribution of NFT was very unlike the early p‐tau pathology of Alzheimer's disease (AD) 27. Furthermore, no hemosiderin, astrocytosis or microglial proliferation was evident around the vessels surrounded by p‐tau pathology nor was there any evidence of any Aß and the hippocampus was normal in all cases.
Hof et al reported the neuropathological findings in three boxers with severe memory disturbance, cognitive abnormalities, dysphoria and Parkinsonism and drew attention to the superficial distribution of the NFT to layer II and the upper third of layer III of the neocortex, unlike the distribution of NFT in AD 39.
In 1996, Williams and Tannenberg 99 reported the findings in a 33‐year‐old achondroplastic dwarf who worked for 15 years as a clown in a circus, participated in dwarf‐throwing events and had been knocked unconscious “a dozen times.” Autopsy showed dilatation of the lateral and third ventricles and cerebral aqueduct, a thinned corpus callosum, fenestrated septum pellucidum, shrinkage of the medial dorsal nucleus of the thalamus and atrophy of the vermis of the cerebellum. Microscopic examination revealed occasional NFT in the hippocampus without Aß 99.
Omalu et al 74, 75 first described the clinical and pathological findings of two retired National Football League (NFL) football players. The men had played football for 14–24 years including 8–17 years in the NFL as offensive linemen. Both had developed behavioral and mood changes, memory loss and cognitive impairment. Histologically, in one subject, there were NFT and NT in neocortex, associated with sparse diffuse Aß plaques 74. The brain of the other subject showed NFT in the frontal, temporal, inferior parietal, insular and entorhinal cortices, hippocampus, amygdala, diencephalon, substantia nigra and locus coeruleus 75. Omalu et al also reported the finding of CTE in a 40‐year‐old professional wrestler characterized by NFT and NT in the cerebral cortex, deep nuclei and brainstem without Aß deposits 76.
By 2014, the list of sports whose participants had been diagnosed with CTE expanded to include rugby, professional baseball, soccer, ice hockey and American football at the high school, college and professional level 30, 59, 60.
Moreover, early stage CTE was reported in military personnel exposed to repetitive mTBI from explosive blast injuries and repetitive concussion 30, 56, 73. The early p‐tau changes associated with blast neurotrauma share similarities with the changes of early CTE reported in young American football and soccer players, boxers, head‐bangers and others 27, 30, 59, 60, although it is likely that there are additional brain pathologies associated with blast exposure that are not characteristic of impact injuries. Four of the five veterans with early stage CTE were also diagnosed with posttraumatic stress disorder (PTSD) suggesting that PTSD might share biological and pathological underpinnings with CTE. Advanced CTE has also been reported after TBI sustained during military service 56.
In a large case series, McKee et al reported the clinical and immunocytochemical characteristics of 68 men with CTE, ranging in age 17–98 years, and compared them to 18 age‐matched men without a history of repetitive mTBI 59. McKee et al also introduced pathological criteria for the neuropathological diagnosis of CTE (Table 1) and a staging system for grading pathological severity (Figure 1) 59.
Table 1.
Criteria for the diagnosis of CTE [adapted from McKee et al 59]. Abbreviations: ATs = astrocytic tangles; CTE = chronic traumatic encephalopathy; NFTs = neurofibrillary tangles
Supportive features
|
Figure 1.
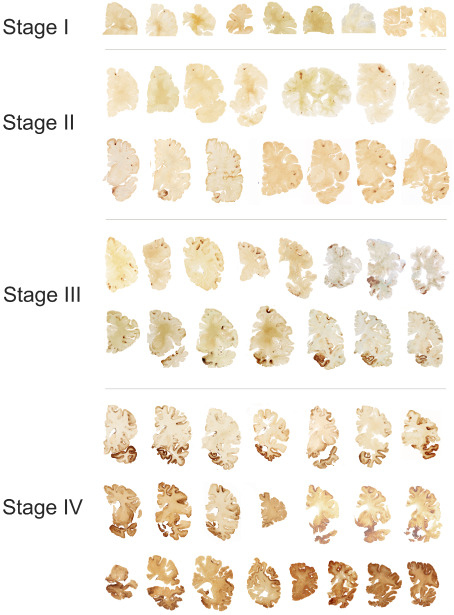
Stages of chronic traumatic encephalopathy (CTE) [adapted from McKee et al 59]. In stage I CTE, p‐tau pathology is found in discrete foci in the cerebral cortex, most commonly in the superior or lateral frontal cortices, typically around small vessels at the depths of sulci. In stage II CTE, there are multiple foci of p‐tau at the depths of the cerebral sulci and there is localized spread of neurofibrillary pathology from these epicenters to the superficial layers of adjacent cortex. The medial temporal lobe is spared neurofibrillary p‐tau pathology. In stage III CTE, p‐tau pathology is widespread; the frontal, insular, temporal and parietal cortices show widespread neurofibrillary degeneration with greatest severity in the frontal and temporal lobes, concentrated at the depths of the sulci. Also in stage III CTE, the amygdala, hippocampus and entorhinal cortex show substantial neurofibrillary pathology that is found in earlier CTE stages. In stage IV CTE, there is widespread severe p‐tau pathology affecting most regions of the cerebral cortex and the medial temporal lobe, sparing calcarine cortex in all but the most severe cases. All images, CP‐13 immunostained 50 μ tissue sections.
Neuropathology of CTE
Gross pathology
Grossly identifiable changes in the brain are unusual in early or mild CTE; if present, they are most often cavum septum pellucidum and mild enlargement of the frontal and temporal horns of the lateral ventricles. There may also be prominent perivascular spaces in the white matter, particularly in the temporal lobe. In intermediate and advanced CTE, macroscopic changes include a reduction in brain weight, gray and white matter atrophy (typically most severe in the frontal and anterior temporal lobes), enlargement of the lateral and third ventricles, cavum septum pellucidum, septal fenestrations, atrophy of the thalamus, hypothalamus and mammillary bodies, thinning of the isthmus of the septum corpus callosum and depigmentation of the locus coeruleus and substantia nigra (Figure 2). Although grossly identifiable cerebellar abnormalities were described in the initial reports of CTE affecting boxers, macroscopic cerebellar abnormalities are rarely present in CTE associated with other sports or activities 59.
Figure 2.
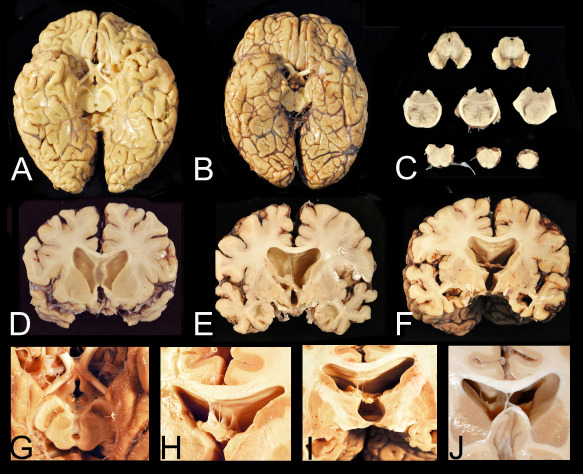
Gross neuropathological features of advanced chronic traumatic encephalopathy (CTE). A. CTE stage IV in a former professional football player. Despite the relatively large size of the brain, there is generalized, symmetrical brain atrophy that is most severe in the frontal and temporal lobes. There is also flattening of the hypothalamic floor, atrophy of the mammillary bodies and pallor of the substantia nigra. B. The brain of a subject with Alzheimer's disease, by contrast, also shows generalized, symmetrical brain atrophy but the hypothalamic floor, mammillary bodies and substantia nigra are normal. C. There is depigmentation of the substantia nigra and locus ceruleus and degeneration of the frontopontine tracts in the cerebral peduncle. D, E, F. Coronal sections show pronounced frontal and temporal lobe atrophy, marked ventricular dilatation, as well as cavum septum (A) and absence of the posterior septum (B) and (C). G. The floor of the hypothalamus is severely thinned and the mammillary bodies are markedly atrophic. H. Multiple septal perforations may be present. I. The posterior septum may be absent with only small blood vessels remaining. J. A large single septal perforation.
Microscopic pathology
In the mildest forms of disease, CTE is characterized by the deposition of p‐tau protein as NFT and NT in conspicuous clusters in the neocortex, often arranged along small blood vessels at the depths of the sulci. The NFT typically follow the penetrating small cortical vessels as linear accumulations extending from the surface of the brain to the lowest layers of the cortical gray matter, or when observed in cross‐section, as clusters NFT, pre‐tangles and dot‐shaped and thread‐like NT in a penumbra around small arterioles (Figure 3). Mild forms of the disease affect primarily the neurons, although there may be focal subpial p‐tau immunopositive astrocytes at the sulcal depths and occasional p‐tau immunoreactive glia around the affected vessel (Figure 3). The pathological significance of isolated subpial p‐tau astrocytes at the sulcal depths unaccompanied by perivascular NFT and NT in the underlying cortex is unknown, although subpial p‐tau astrocytes at the depths of the cortical sulci is not a phenomenon found in normal aging and has not been found in the brains of subjects without a history of repetitive brain trauma.
Figure 3.
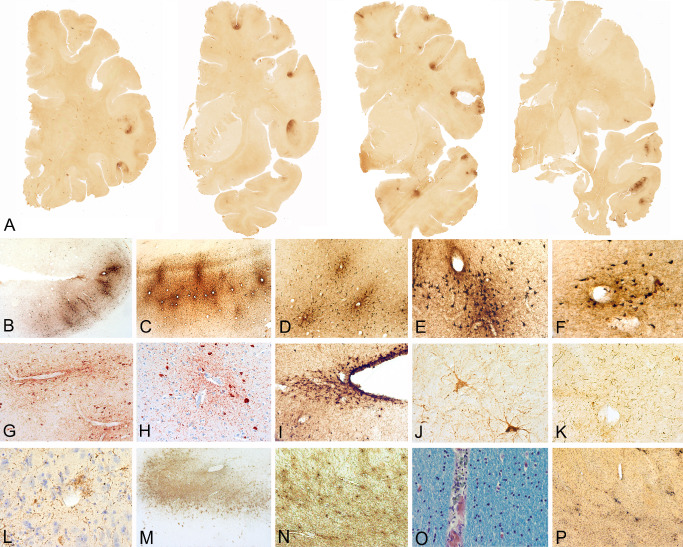
Patterns of 3R and 4R tau in chronic traumatic encephalopathy (CTE) [adapted from McKee et al 59]. A, D. 3R immunostaining shows scattered RD3 immunoreactive neurons in middle frontal cortex. B, E. 4R immunostaining shows many ET3 immunoreactive neurons and astrocytic tangles in the subpial region of the middle frontal cortex and at the depth of the sulcus. C, F. AT8 immunostaining shows 3R and 4R immunopositive neurons and astrocytic tangles in middle frontal cortex. G, J, M. 3R immunostaining shows scattered RD3 immunoreactive neurons in CA1 (G), CA2 (J) and CA4 hippocampus (M). H, K, N. 4R immunostaining shows many ET3 immunoreactive neurons in CA1 ( H ), CA2 (K) and CA4 hippocampus ( N ). I, L, O . I. AT8 immunostaining shows 3R and 4R immunopositive neurons in CA1 ( I ), CA2 (L) and CA4 hippocampus (O). All 10 μ paraffin embedded sections, magnification bars 50 μm.
Provisional criteria for the neuropathological diagnosis of CTE are listed in Table 1 59. These criteria are currently being validated by a panel of expert neuropathologists as part of a UO1 funded by the National Institute of Neurological Disorders and Stroke (NINDS) and National Institute of Biomedical Imaging and Bioengineering (NIBIB) (1U01NS086659‐01). The provisional criteria are founded on the hypothesis that the irregular, perivascular p‐tau pathology found in clusters at the depths of the cortical sulci distinguishes CTE from other tauopathies, including AD 27, 28, 57, 58, 59; this hypothesis will be formally assessed at the NINDS/NIBIB‐sponsored conference. One of the more difficult tasks will be deciphering the point at which the pathology is sufficient to make a diagnosis of CTE, as opposed to CTE‐related changes. This conundrum is reminiscent of the early phases of AD neuropathology when it was difficult to distinguish between AD‐related changes and a neuropathological diagnosis of low, intermediate and high likelihood AD 71. Similarly, stage I CTE is likely a preclinical pathology, sometimes asymptomatic, and sometimes associated with prolonged post‐concussive symptoms, but not necessarily progressive. Stage II CTE may occasionally be asymptomatic, although in many cases, it is associated with florid behavior and mood changes. Determining the degree and pattern of microscopic features necessary to diagnose comorbid CTE when another tauopathy is present will be another challenge for the future, such as when CTE is present in addition to AD, progressive supranuclear palsy, corticobasal degeneration, argyrophilic grain disease or many others.
The tau isoform profile and phosphorylation state in CTE are similar to AD 84 and the neuronal p‐tau pathology shows immunoreactivity to both 3 repeat and 4 repeat (4R) tau 59, 91. The 4R isoform of tau is predominantly expressed in astrocytes located in the subpial region of the sulcal depths 59, 91, although some of the neuronal abnormalities in the hippocampus also appear to be primarily 4R tau in CTE (Figure 4).
Figure 4.
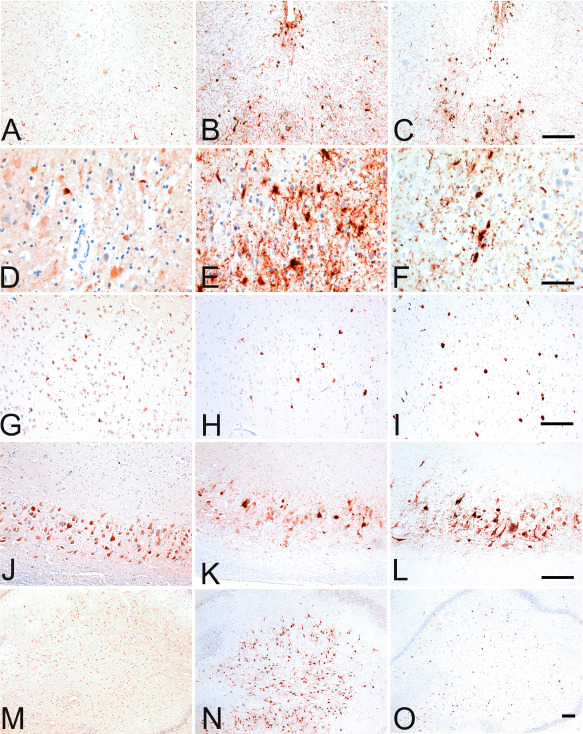
Microscopic findings in stage II chronic traumatic encephalopathy (CTE). A. Whole mount coronal sections in stage II CTE show multiple foci of p‐tau pathology primarily located at the depths of the cortical sulci of the frontal and temporal lobes (free floating 50 μ sections, AT8 immunostain). B–H. The p‐tau pathology consists of neurofibrillary tangles and dot‐like and thread‐like dystrophic neurites and is characteristically found around small blood vessels (B–F, free floating 50 μ sections, AT8 immunostain; G, H, 10 μ paraffin embedded sections, AT8 immunostain). (I). Subpial astrocytic tangles are also found at the cortical depths (free floating 50 μ sections, AT8 immunostain). Other pathologies include pretangles (J), dystrophic neurites in the white matter (K) and occasional p‐tau astrocytes (L) (free floating 50 μ sections, AT8 immunostain). There may be marked astrocytosis of the white matter (M, N) (free floating 50 μ sections, Glial fibrillary acidic protein immunostain). Hemosiderin‐laden macrophages (O) (10 μ paraffin section, Luxol fast blue hematoxylin and eosin stain) and multiple perivascular foci of reactive microglia (P) are found around small vessels in the cerebral white matter (free floating 50 μ sections, LN3 immunostain).
Staging of p‐tau pathology in CTE
Using the provisional criteria for CTE, it is possible to identify a relatively stereotyped sequence of pathological progression of p‐tau pathology that can be classified into four pathological stages 59. These stages of p‐tau pathology are accompanied by progressive macroscopic changes and progressive increases in axonal and 43 kDa TAR DNA‐binding protein (TDP‐43) pathologies and neuroinflammation. The staging of p‐tau pathology in CTE is adapted from the work of Braak and Braak who examined a series of 83 autopsy brains with AD for neurofibrillary changes and found that there was a characteristic distribution pattern of NFT and NT that permitted the differentiation of six stages 7. This staging system forms the basis for the neuropathological diagnosis of AD used by the National Institute on Aging 40 and similar staging schemes are now in use for Aß plaques in AD 95 and Lewy bodies in Parkinson's disease 8.
Using the pathological staging system for CTE, we found a significant correlation between the pathological stage of CTE severity and the duration of the football career, age at death and years since retirement from football among our series of former American football players 59.
Staging of pathology in CTE
Stage I
Brains with stage I CTE are grossly unremarkable but microscopically show rare, isolated perivascular focal epicenters of p‐tau NFTs and neuropil neurites (Figure 1). The NFTs and neuropil neurites predominate and the neurites are characteristically dot‐like as well as thread‐like. Occasional p‐tau immunoreactive glia and glial processes may be present. Tau pathology is most commonly localized to the depths of the cerebral sulci of the frontal, temporal, insular, septal and parietal cortices, although there may also be sparse NFT scattered throughout the adjacent cortex. There also may be subpial p‐tau astrocytic tangles in clusters at the depths of sulci. NFT and neurites are also found in the locus coeruleus. Approximately one‐half of cases with stage I p‐tau pathology show rare TDP‐43 neurites. Clusters of reactive microglia are found in the white matter with axonal swellings and distorted profiles in the subcortical U‐fibers. Aβ plaques are not found in subjects with stage I CTE, unless over the age of 50 years. Hemosiderin‐laden macrophages are characteristically found around small vessels in the white matter, usually not associated with p‐tau abnormalities.
Stage II
Subtle macroscopic changes may be found in stage II CTE cases including mild enlargement of the frontal horns of the lateral ventricles and third ventricle, cavum septum pellucidum and pallor of the locus coeruleus and substantia nigra. Microscopically, multiple epicenters of perivascular foci of p‐tau NFT and neurites are found at the depths of the frontal, temporal, parietal, insular and septal cortices. These cerebral epicenters typically consist of perivascular p‐tau NFT, pre‐tangles, neurites and occasional glia (Figure 3) and may be associated with p‐tau astrocytic tangles in the overlying subpial region. In the cortex adjacent to the focal epicenters, NFT are scattered throughout the cerebral layers with a tendency to be most numerous in the superficial cortical layers. There are also NFT in the locus coeruleus and substantia innominata. Mild TDP‐43 pathology may be found in stage II CTE cases as abnormal neurites and neuronal inclusions. Clusters of reactive microglia are often found in the subcortical white matter with axonal swellings and distorted profiles in the subcortical U‐fibers. Aβ plaques are found in 19% of subjects with stage II CTE, only if older than 50 years.
Stage III
Most brains with stage III CTE show macroscopic changes although the gross structural changes are minor relative to the changes found microscopically. Typically, there is a reduction in brain weight, mild frontal and temporal atrophy and enlargement of the lateral and third ventricles. One‐half of subjects have septal abnormalities including cavum septum pellucidum, septal perforations or fenestrations. Pallor of the locus coeruleus and substantia nigra, atrophy of the mammillary bodies, thalamus and hypothalamus and thinning of the corpus callosum are characteristic. Microscopically, confluent patches of NFT, NTs and astrocytic tangles are found around blood vessels at the sulcal depths, as well as in linear arrays in the superficial laminae of cortex. NFT are found in the superior, dorsolateral and inferior frontal, septal, insular, temporal pole, superior, middle and inferior temporal and inferior parietal cortices. NFT are also found in the olfactory bulbs, hippocampus, entorhinal cortex, amygdala, hypothalamus, mammillary bodies, nucleus basalis of Meynert, substantia nigra, dorsal and median raphe nuclei and locus coeruleus. Neurofibrillary degeneration in the hippocampus includes CA4 and CA2, as well as CA1.
Approximately one‐third of stage III cases have occasional NFT in the dentate nucleus of the cerebellum and the spinal cord gray matter. TDP‐43‐positive neurites and inclusions are found in the majority of cases. Thirteen percent of stage III CTE cases show sparse diffuse and neuritic Aβ plaques. White matter abnormalities, loss of myelinated nerve fibers, axonal dystrophy and axonal loss may be conspicuous (Figure 5).
Figure 5.
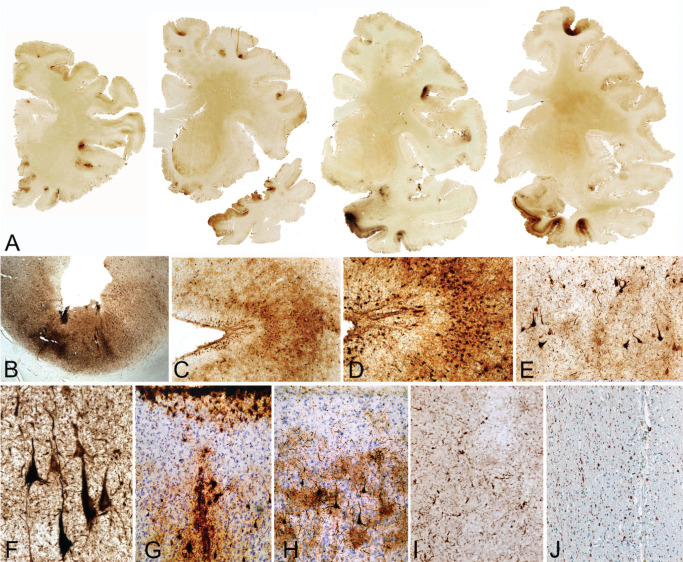
Microscopic changes in stage III chronic traumatic encephalopathy (CTE). (A) Whole mount coronal sections in stage III CTE show multiple cortical foci of p‐tau pathology throughout the frontal and temporal cortices. The cortical epicenters and depths of the sulci often consist of confluent masses of neurofibrillary tangles (NFT) and astrocytic tangles (ATs). (B–D) The p‐tau pathology consists of NFTs, ATs and dot‐like and thread‐like dystrophic neurites clustered around the penetrating cortical vessels. (E) Cortex adjacent to the cortical epicenters shows scattered NFT. (F ) The hippocampus shows dense neurofibrillary pathology. Subpial ATs (G) and p‐tau immunoreactive astrocytes may be prominent (H). There may be dystrophic neurites in the white matter (I). SMI‐34 immunostaining shows reduction in axonal staining and numerous large, irregular axonal varicosities (J). (A–I. free floating 50 μ sections, AT8 immunostain; J. SMI‐34 immunostain, 10‐μ paraffin section).
Stage IV
Brains with stage IV CTE show a decrease in brain weight, which in some instances may be substantial. Brain weights of 1000 g or less are not uncommon. In addition to generalized cerebral atrophy, there is pronounced atrophy of the frontal and temporal lobes, medial temporal lobe and anterior thalamus. There is also diffuse atrophy of the white matter and thinning of the corpus callosum, particularly the isthmus. There is typically severe thinning of the hypothalamic floor and atrophy of the mammillary bodies. Most cases of stage IV CTE show septal abnormalities including cavum septum, perforations, fenestrations or total absence of the posterior septum. The locus coeruleus and substantia nigra are pale. Microscopically, in stage IV CTE, there is widespread myelin loss, astrocytosis of the white matter and neuronal loss in the cerebral cortex, hippocampus and substantia nigra. Neuronal loss and astrocytosis may also be prominent in the frontal and temporal cortices associated with microvacuolation of layer II.
P‐tau pathology is densely distributed throughout the cerebrum, thalamus, hypothalamus, mammillary bodies, basal ganglia, brainstem, cerebellar dentate nucleus and occasionally, spinal cord. Primary visual cortex is typically spared. NFT, most of which are extracellular, are found throughout the hippocampal formation including the dentate gyrus, CA3, CA2 and CA4. CA1 typically shows severe loss of neurons with substantial numbers of extracellular tangles. P‐tau pathology also involves the cerebellum, including the dentate nucleus, granular cell layer, and sometimes, Purkinje cells. Irregular tau neurites are often found in the white matter of the anterior cerebellar vermis lining the fourth ventricle. Widespread TDP‐43 deposits are also found with dot‐like and thread‐like inclusions in neurites and intraneuronal cytoplasmic inclusions. There is marked loss of myelinated nerve fibers, axonal dystrophy and loss throughout the cerebral and cerebellar white matter (Figure 6).
Figure 6.
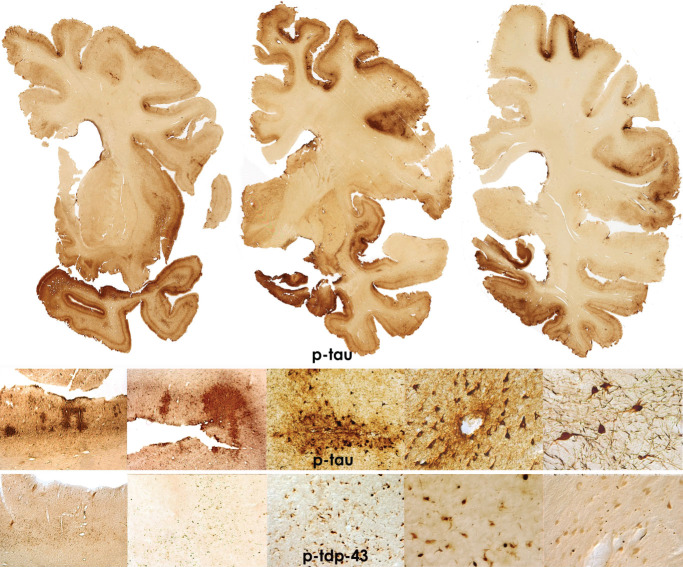
Microscopic changes in stage IV chronic traumatic encephalopathy (CTE). Whole mount coronal sections in stage IV CTE show widespread p‐tau pathology affecting most regions of the cerebral cortex and medial temporal lobe. Astrocytic tangles are prominent and there is marked neuronal loss in the cortex, amygdala and hippocampus. There are also widespread pTDP‐43 abnormalities. All images: 50 μ tissue sections, CP‐13 or p‐TDP‐43 immunostain.
TDP‐43 pathology
Abnormal TDP‐43 pathology is found in most cases of CTE. The pathology ranges from cytoplasmic neuronal inclusions that partially colocalize with p‐tau inclusions to glial inclusions and abnormal neurites 58. In stage I disease, TDP‐43 immunopositive neurites are found in the subcortical white matter and fornix. In stage II disease, TDP‐43 immunopositivity consists of isolated neurites or inclusions in the subcortical white matter, brainstem or medial temporal lobe, often in a subpial, periventricular or perivascular distribution. By stage III CTE, most cases show TDP‐43 immunoreactive neurites in the cerebral cortex, medial temporal lobe or brainstem. TDP‐43 immunoreactivity is found in nearly all stage IV cases, as TDP‐43‐positive, rounded and thread‐like neurites, intraglial and intraneuronal inclusions in cerebral cortex, medial temporal lobe, diencephalon, basal ganglia, brainstem and occasionally, spinal cord. In cases with the most severe TDP‐43 deposition, dense accumulations of TDP‐43 inclusions and neurites are found in all layers of the neocortex, particularly layer II, as well as occasional TDP‐43‐positive inclusions in the dentate fascia of the hippocampus, a distribution pattern that overlaps with the distribution of TDP‐43 found in frontotemporal lobar degeneration (FTLD) with TDP‐43 11.
As TDP‐43 is an RNA‐binding protein that regulates RNA metabolism 85, 86 and binds to tau 83, TDP‐43 dysregulation might play an influential role in tau isoform expression and the development of tau pathology 68. Diseases caused by abnormal TDP‐43 metabolism have shown altered tau metabolism including hyperphosphorylation, tau phosphatase resistance and deposition of p‐tau intracellular aggregates 93. Furthermore, animal models have shown TDP‐43 to be upregulated and relocated from the neuronal nucleus to the neuronal cytoplasm 65, 66 after traumatic injury.
Aß in CTE
Aß‐containing plaques, especially diffuse Aß plaques, are present in some cases of CTE, although they are not a consistent feature of CTE, are not found in early stage disease and are significantly associated with age at death 59. Compared with a normal aging population, Aß deposition occurs at an earlier age and at an accelerated rate in CTE and is associated with increased clinical and pathological severity (T. Stein, pers. comm.).
CTE with comorbid degenerative disease
Similar to most other neurodegenerative diseases 31, 42, 45, 88, 98, CTE is associated with the development of other neurodegenerations, especially as a function of increased age 37, 58, 59. Of 142 neuropathologically confirmed cases of CTE, 15 (10.56%) had coexistent motor neuron disease (MND), 15 (10.56%) had AD, 10 (7.04%) had Lewy Body disease, 4 (2%) had FTLD and 7 (4.51%) had two or more of these comorbidities; 89 (63.16%) of the cases were diagnosed only with CTE (Figure 7 ).
Figure 7.
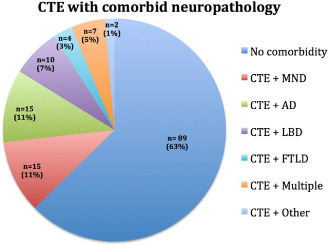
Chronic traumatic encephalopathy (CTE) with comorbid neuropathology. Of 142 neuropathologically confirmed cases of CTE at the VA‐BU‐SLI CTE Brain Bank, 15 (10.56%) had coexistent motor neuron disease (MND), 15 (10.56%) had Alzheimer's disease (AD), 10 (7.04%) had Lewy Body disease, 4 (2.%) had frontotemporal lobar degeneration (FTLD) and 7 (4.51%) had two or more of these comorbidities; 89 (63.16%) cases were diagnosed only with CTE.
CTE with MND
Approximately 10% of individuals with CTE develop a progressive MND characterized by weakness, atrophy, spasticity and fasciculations that appears clinically indistinguishable from Amyotrophic lateral sclerosis (ALS) 58, 59. In addition, a recent screen of ALS cases from the Mayo Clinic Jacksonville ALS brain bank and the Boston Veterans Affairs (VA) ALS brain bank found that six of the 91 (6.6%) and five of the 113 (4.4%) cases, respectively, had pathological features of CTE (all Caucasian males) 5. Most individuals with CTE and MND present with symptoms of ALS and develop mild cognitive and behavioral symptoms several years after the onset of motor weakness and fasciculations. Individuals with CTE and MND often die from respiratory failure at younger ages and in earlier stages of CTE (stage II–III) compared with CTE subjects without MND. Approximately one‐third of subjects with CTE and MND present with depression, behavioral or cognitive changes related to CTE years before developing MND symptoms. Subjects with CTE and MND usually show more severe TDP‐43 pathology than subjects with CTE alone. The marked accumulation of pTDP‐43 aggregates in advanced stages of CTE, the partial immunohistochemical colocalization of p‐tau with pTDP‐43, and the development of MND in some individuals with CTE suggests that CTE might share some pathogenic mechanisms with FTLD 20, 49.
Relationship of tau pathology to trauma
Although all cases of neuropathologically verified CTE have been associated with repetitive mild brain trauma, the pathophysiological mechanisms critical to developing a latent and progressive neurodegeneration and tauopathy after repetitive mTBI, including concussion and subconcussion, are only beginning to be identified. When the semisolid brain, suspended in cerebrospinal fluid (CSF) inside a boney skull, is subjected to the rapid acceleration, deceleration and rotational forces associated with impact injury, the brain elongates and deforms, stretching the microstructural elements, including neurons, glial cells and blood vessels. These stretch forces predominantly affect long fibers, specifically axons and blood vessels 54, 61 and are most severe where the direction of axons changes or where a change in tissue‐density occurs (e.g. around blood vessels or at the gray‐white matter interface) 16, 26, 80. Experimental studies of acceleration‐deceleration injuries in gyrencephalic animals further demonstrate that axonal injury and vascular disruption is most severe at focal stress points of the brain—particularly the perivascular region and the depths of the cerebral sulci 17, 87. Moreover, the early and predominant involvement of the superior and dorsolateral frontal lobes in CTE affecting former football players parallels the high frequency of impacts to the top of the head compared with those to the front, back and side of the head 35, 62. Functional MRI data in football players also shows activation impairments in dorsolateral prefrontal cortex that is associated with significantly higher numbers of head collisions to the top‐front of the head 94.
Pathological studies of concussion and post‐concussion syndrome in human subjects have found that multifocal traumatic axonal injury is often perivascular 6, 60, 77. Other changes found in acute and subacute concussion are microvascular injury and microhemorrhages 60, 77, astrocytosis, most severe in the cerebral white matter and brainstem white matter tracts, and clusters of activated microglia, most prominent in the white matter around small vessels 59, 77. Less common changes include focal clusters of p‐tau in NFTs, pre‐tangles and neurites or stage I CTE 60.
It is known from the experimental literature that acceleration‐deceleration injury causes tau protein, normally associated with microtubules in axons, to become abnormally phosphorylated, misfolded, aggregated and cleaved, all of which generate neurotoxic tau peptide fragments 1, 12, 46, 47, 59, 101. Nevertheless, how this combination of factors: axonal injury, breach of blood–brain barrier, neuroinflammation and aggregation of abnormally truncated and hyperphosphorylated proteins, including p‐tau and TDP‐43, develop into a progressive deterioration remains to be determined.
There are most likely additional factors that play a role in the initiation of p‐tau pathology in CTE, including genetic susceptibility or resistance, gender, physiological stress, additional environmental exposures and age at exposure. Studies have suggested that children and adolescents experience prolonged recovery rates after TBI compared with adults 24 as well as poorer outcomes 29. Animal models have shown that stress can lead to increased tau phosphorylation 72, 90. Environmental influences, such as alcohol, opiates or performance‐enhancing drugs, could also play a role in tau aggregation, deposition and propagation. P‐tau immunoreactivity in opiate user brains has been shown to be significantly higher than in controls 2. Glucocorticoids have also been associated with increased tau phosphorylation and greater cognitive deficits in rodent models 33, 89.
Spread of tau pathology
Tau phosphorylation and misfolding are potentially reversible processes 92, 97, 100; correspondingly, stage I or even stage II CTE might represent a reversible or nonprogressive pathology, at least in some subjects. However, repeated traumatic injuries and progressively greater accumulations of toxic p‐tau fragments have the potential to induce a self‐perpetuating process. We know that athletes who develop CTE develop a progressive neurodegeneration that typically produces clinical symptoms years to decades after retiring from the sport. This progressive post‐traumatic neurodegeneration may involve spreading of tau pathology intercellularly and extracellularly throughout the brain. Direct and indirect evidence for interneuronal tau transmission in animal models suggests that the transfer of toxic tau species between neurons might be caused by the interneuronal spreading of tau mediated by a prion‐like templated misfolding of tau 14, 15, 23, 34, 36, 48, 52. Other possible modes of transmission involve oligomeric or toxic N‐terminal tau in the receiving neuron with dysregulation of intracellular calcium 25, 78. Spreading of tau pathology may occur between neuronal synapses, but may also involve astrocytes, microglia and CSF pathways. CSF is present in the Virchow–Robin spaces surrounding penetrating arteries and brain interstitial fluid is cleared along paravenous drainage pathways, allowing the possibility that p‐tau transmission might involve this clearance pathway as has been shown for Aß 41.
Axonal pathology
Axonal injury, axonal degeneration, myelinated fiber loss and white matter atrophy are constant features of CTE. Moreover, axonal injury most likely plays a critical role in initiating the p‐tau pathology 59, 69. The degree of axonal dysintegrity parallels the severity of the neurodegeneration. In early stage CTE, stages I and II, scattered axonal varicosities are found in the deep layers of the frontal and temporal cortices, subcortical white matter and deep white matter tracts of the diencephalon and axons in the white matter are occasionally immunoreactive for p‐tau (Figure 3). In more advanced disease, stages III and IV, axonal loss is severe with frequent distorted axonal profiles in the cortex and subcortical white matter, macrophages and widespread p‐tau abnormalities in white matter.
Imaging and biomarkers
Although there are no validated imaging techniques or biomarkers to confirm the diagnosis of CTE in living subjects, potentially useful imaging and biomarkers include positron emission tomography (PET) ligands for p‐tau and neuroinflammation 13, 64, diffusion tensor imaging 4, 50, magnetic resonance spectroscopy 51 and CSF markers 10. P‐tau‐specific PET ligands, such as T‐807 and T‐808 reported by Chien et al 13 might prove to identify primary tauopathy, such as CTE, and p‐tau PET imaging combined with PET imaging for Aß might be useful to differentiate between CTE and AD 3, 44, 67. Recently, this technique was used to evaluate a retired NFL player with clinical symptoms suggestive of AD. He was found to have a negative florbetapir scan for Aß and a positive scan for T‐807, results supportive of CTE, although the pattern of T‐807 positivity most likely represented non‐specific off‐target labelling 64. Additionally, more conventional imaging techniques and biomarkers might be useful in identifying some of the pathological features of “probable CTE” in vivo, such as abnormalities of the septum pellucidum, cortical thinning and/or cortical atrophy (Figure 8) 67. Other potential biomarkers for “probable CTE” include normal Aβ CSF fluid levels and an elevated CSF p‐tau/tau ratio 67.
Figure 8.
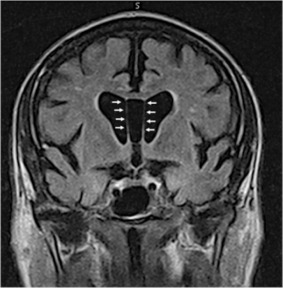
Coronal T1‐weighted Magnetic resonance imaging shows a cavum septum pellucidum, cortical and white matter atrophy in former professional football player with neuropathologically confirmed chronic traumatic encephalopathy stage IV (arrows indicate the location of the cavum septum).
CTE in the Military Setting
Most individuals with neuropathologically verified CTE were exposed to repetitive brain trauma through athletic participation. Although CTE has also been reported in association with military exposures 56, typically CTE affects veteran athletes with considerable traumatic exposure from sports. Sports‐related injuries are relatively stereotyped and predictable, whereas military‐related mTBI is acquired in widely heterogeneous ways, including recreational activities, physical training practices, falls, motor vehicle accidents, athletics and exposure to explosive blasts. Injury from explosive blasts varies depending on the strength of the explosive, whether the injury occurs in an open field, in or near buildings or in a motor vehicle. Military mTBI is also random; an individual may experience a single injury or many thousands of injuries over a similar time period depending on chance exposure.
There have been case reports of CTE occurring in young veterans of the recent conflict in Iraq and Afghanistan, five of the veterans were exposed to blast and one veteran experienced concussive injury 30, 56, 73. In the cases of CTE reported in young military veterans (n = 5 males; ages 22 to 45 years; mean, 31.0 years), the changes of CTE were mild and ranged from stage I to II CTE 30, 56. In all five, there was also evidence of myelinated fiber loss, axonal degeneration, microvascular degeneration and neuroinflammation. Symptoms included headaches, verbal and physical violence, irritability, attentional deficits, difficulty in sleeping, short‐term memory loss and depression. Four of the five were also diagnosed with PTSD.
The outcome of military‐related mTBI tends to be less predictable and less homogeneous than sports‐related mTBI and the neurodegenerative pathologies that occur after pure military mTBI remain largely unknown. It is likely that military‐related mTBI will give rise to neurodegenerations other than CTE and that post‐traumatic neurodegenerations are variable. Axonal injury after military mTBI is likely more severe than apparent pathologically, as mild axonal damage and loss are not well visualized histologically and are difficult to quantitate. Neuroimaging of white matter tracts, such as with diffusion tensor imaging and related modalities, may prove to be the best strategy for assessing the contribution of axonal injury to military‐related post‐traumatic neurodegeneration.
Conclusion
CTE is a distinct neurodegenerative disease that currently can only be diagnosed with certainty by post‐mortem neuropathological examination. Given the importance of sports participation and physical exercise to physical and psychological health as well as disease resilience, it is critical to identify the genetic risk factors for CTE as well as the contribution of exposure variables, stress, alcohol, substance abuse and other factors to the development of CTE. The acute consequences of mTBI, including concussion, subconcussion and blast injuries, are known to produce axonal injury, microvascular disruption, breach of the blood–brain barrier, neuroinflammation and, in some cases, deposition of small amounts of p‐tau 60. Further research is needed to identify the pathogenetic mechanisms responsible for developing a long‐term, often progressive, neurodegeneration after acute, repetitive mTBI. The neuropathology of CTE is characterized by a focal perivascular deposits of p‐tau in the neocortex that appear to spread to affect the superficial layers of adjacent cortex, and eventually, the medial temporal lobe, diencephalon and brainstem in susceptible individuals. The mechanisms underlying the spread of tau and the accumulation of TDP‐43 in the brain remain to be defined. Clinical and pathological diagnostic criteria have been proposed for CTE but remain to be validated. In vivo biomarkers and neuroimaging techniques hold the promise for developing an in vivo diagnostic technique for CTE. Additional prospective, longitudinal, epidemiological and animal studies will be essential to establishing the incidence and prevalence of CTE and to identifying underlying pathogenic mechanisms that will help target future preventative and therapeutic strategies (102–105).
Acknowledgments
The authors gratefully acknowledge the use of the resources and facilities at the Edith Nourse Rogers Memorial Veterans Hospital (Bedford, MA, USA). We also gratefully acknowledge the help of all members of the Chronic Traumatic Encephalopathy Program at Boston University School of Medicine, the Boston VA, as well as the individuals and families whose participation and contributions made this work possible. This work was supported by the National Institute of Neurological Disorders and Stroke (1UO1NS086659‐01), Department of Veterans Affairs, the Veterans Affairs Biorepository (CSP 501), the Translational Research Center for Traumatic Brain Injury and Stress Disorders (TRACTS), Veterans Affairs Rehabilitation Research and Development Traumatic Brain Injury Center of Excellence (B6796‐C), the National Institute of Aging Boston University Alzheimer's Disease Center (P30AG13846; supplement 0572063345–5), the National Institute of Aging Boston University Framingham Heart Study R01 (AG1649), the National Operating Committee on Standards for Athletic Equipment and the Sports Legacy Institute. This work was also supported by unrestricted gifts from the Andlinger Foundation, the WWE and the National Football League.
References
- 1. Amadoro G, Ciotti MT, Costanzi M, Cestari V, Calissano P, Canu N (2006) NMDA receptor mediates tau‐induced neurotoxicity by calpain and ERK/MAPK activation. Proc Natl Acad Sci U S A 103:2892–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anthony IC, Norrby KE, Dingwall T, Carnie FW, Millar T, Arango JC et al (2010) Predisposition to accelerated Alzheimer‐related changes in the brains of human immunodeficiency virus negative opiate abusers. Brain 133(Pt 12):3685–3698. [DOI] [PubMed] [Google Scholar]
- 3. Baugh CM, Robbins CA, Stern RA, McKee AC (2014) Current Understanding of Chronic Traumatic Encephalopathy. Curr Treat Options Neurol 16:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bazarian JJ, Zhu T, Zhong J, Janigro D, Rozen E, Roberts A et al (2014) Persistent, long‐term cerebral white matter changes after sports‐related repetitive head impacts. PLoS ONE 9:e94734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bieniek KF, Stein TD, Alvarez VE, Fry BT, Dickson DW, McKee AC (2014) Poster abstracts. Brain Pathol 24:39–103. [Google Scholar]
- 6. Blumbergs PC, Scott G, Manavis J, Wainwright H, Simpson DA, McLean A (1994) Staining of amyloid percursor protein to study axonal damage in mild head injury. Lancet 344:1055–1056. [DOI] [PubMed] [Google Scholar]
- 7. Braak H, Braak E (1991) Neuropathological staging of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 8. Braak H, Tredici KD, Rüb U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211. [DOI] [PubMed] [Google Scholar]
- 9. Brandenburg W, Hallervorden J (1954) [Dementia pugilistica with anatomical findings]. Virchows Arch 325:680–709. [DOI] [PubMed] [Google Scholar]
- 10. Buerger K, Ewers M, Pirttilä T, Zinkowski R, Alafuzoff I, Teipel SJ et al (2006) CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer's disease. Brain 129:3035–3041. [DOI] [PubMed] [Google Scholar]
- 11. Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ et al (2007) TDP‐43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol 171:227–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen LJ, Wang YJ, Tseng GF (2010) Compression alters kinase and phosphatase activity and tau and MAP2 phosphorylation transiently while inducing the fast adaptive dendritic remodeling of underlying cortical neurons. J Neurotrauma 27:1657–1669. [DOI] [PubMed] [Google Scholar]
- 13. Chien DT, Bahri S, Szardenings AK, Walsh JC, Mu F, Su M‐Y et al (2013) Early clinical PET imaging results with the novel PHF‐tau radioligand [F‐18]‐T807. J Alzheimers Dis 34:457–468. [DOI] [PubMed] [Google Scholar]
- 14. Clavaguera F, Tolnay M, Goedert M (2013) Prion‐like properties of assembled tau protein. In: Proteopathic Seeds and Neurodegenerative Diseases. Jucker M, Christen Y (eds), pp. 87–95. Springer: Heidelberg, New York, Dordrecht, London. [Google Scholar]
- 15. Clavaguera F, Hench J, Goedert M, Tolnay M (2015) Prion‐like transmission and spreading of tau pathology. Neuropathol Appl Neurobiol 41:47–58. [DOI] [PubMed] [Google Scholar]
- 16. Cloots R, Van Dommelen J, Nyberg T, Kleiven S, Geers M (2011) Micromechanics of diffuse axonal injury: influence of axonal orientation and anisotropy. Biomech Model Mechanobiol 10:413–422. [DOI] [PubMed] [Google Scholar]
- 17. Cloots RJ, Gervaise HM, van Dommelen JA, Geers MG (2008) Biomechanics of traumatic brain injury: influences of the morphologic heterogeneities of the cerebral cortex. Ann Biomed Eng 36:1203–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Constantinidis J, Tissot R (1967) [Generalized Alzheimer's neurofibrillary lesions without senile plaques. (Presentation of one anatomo‐clinical case)]. Schweiz Arch Neurol Neurochir Psychiatr 100:117–130. [PubMed] [Google Scholar]
- 19. Corsellis J, Bruton C, Freeman‐Browne D (1973) The aftermath of boxing. Psychol Med 3:270–303. [DOI] [PubMed] [Google Scholar]
- 20. Costanza A, Weber K, Gandy S, Bouras C, Hof PR, Giannakopoulos P, Canuto A (2011) Review: Contact sport‐related chronic traumatic encephalopathy in the elderly: clinical expression and structural substrates. Neuropathol Appl Neurobiol 37:570–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Courville CB (1962) Punch drunk. Its pathogenesis and pathology on the basis of a verified case. Bull Los Angel Neuro Soc 27:160–168. [PubMed] [Google Scholar]
- 22. Critchley M (1949) Punch‐drunk syndromes: the chronic traumatic encephalopathy of boxers. In: Hommage a Clovis Vincent. Maloine: Paris. [Google Scholar]
- 23. de Calignon A, Polydoro M, Suárez‐Calvet M, William C, Adamowicz DH, Kopeikina KJ et al (2012) Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 73:685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Field M, Collins MW, Lovell MR, Maroon J (2003) Does age play a role in recovery from sports‐related concussion? A comparison of high school and collegiate athletes. J Pediatr 142:546–553. [DOI] [PubMed] [Google Scholar]
- 25. Frost B, Jacks RL, Diamond MI (2009) Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 284:12845–12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gaetz M (2004) The neurophysiology of brain injury. Clin Neurophysiol 115:4–18. [DOI] [PubMed] [Google Scholar]
- 27. Geddes J, Vowles G, Nicoll J, Revesz T (1999) Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol 98:171–178. [DOI] [PubMed] [Google Scholar]
- 28. Geddes JF, Vowles GH, Robinson SF, Sutcliffe JC (1996) Neurofibrillary tangles, but not Alzheimer‐type pathology, in a young boxer. Neuropathol Appl Neurobiol 22:12–16. [PubMed] [Google Scholar]
- 29. Giza CC, Griesbach GS, Hovda DA (2005) Experience‐dependent behavioral plasticity is disturbed following traumatic injury to the immature brain. Behav Brain Res 157:11–22. [DOI] [PubMed] [Google Scholar]
- 30. Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA et al (2012) Chronic traumatic encephalopathy in blast‐exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med 4:134ra60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gorell JM, Johnson CC, Rybicki BA (1994) Parkinson's disease and its comorbid disorders: an analysis of Michigan mortality data, 1970 to 1990. Neurology 44:1865–1868. [DOI] [PubMed] [Google Scholar]
- 32. Grahmann H, Ule G (1956) [Diagnosis of chronic cerebral symptoms in boxers (dementia pugilistica & traumatic encephalopathy of boxers).]. Psychiatr Neurol (Basel) 134:261–283. [PubMed] [Google Scholar]
- 33. Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM (2006) Glucocorticoids increase amyloid‐beta and tau pathology in a mouse model of Alzheimer's disease. J Neurosci 26:9047–9056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guo JL, Lee VM‐Y (2011) Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer‐like tangles. J Biol Chem 286:15317–15331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guskiewicz KM, Mihalik JP, Shankar V, Marshall SW, Crowell DH, Oliaro SM et al (2007) Measurement of head impacts in collegiate football players: relationship between head impact biomechanics and acute clinical outcome after concussion. Neurosurgery 61:1244–1253. [DOI] [PubMed] [Google Scholar]
- 36. Hall GF, Patuto BA (2012) Is tau ready for admission to the prion club? Prion 6:223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hazrati L‐N, Tartaglia MC, Diamandis P, Davis KD, Green RE, Wennberg R et al (2013) Absence of chronic traumatic encephalopathy in retired football players with multiple concussions and neurological symptomatology. Frontiers Hum Neurosci 7:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hof P, Knabe R, Bovier P, Bouras C (1991) Neuropathological observations in a case of autism presenting with self‐injury behavior. Acta Neuropathol 82:321–326. [DOI] [PubMed] [Google Scholar]
- 39. Hof PR, Bouras C, Buee L, Delacourte A, Perl DP, Morrison JH (1992) Differential distribution of neurofibrillary tangles in the cerebral cortex of dementia pugilistica and Alzheimer's disease cases. Acta Neuropathol 85:23–30. [DOI] [PubMed] [Google Scholar]
- 40. Hyman BT, Trojanowski JQ (1997) Editorial on consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol 56:1095–1097. [DOI] [PubMed] [Google Scholar]
- 41. Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA et al (2012) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med 4:147ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jicha GA, Carr SA (2010) Conceptual evolution in Alzheimer's disease: implications for understanding the clinical phenotype of progressive neurodegenerative disease. J Alzheimers Dis 19:253–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jordan BD (1993) Chronic neurologic injuries in boxing. Med Aspects Boxing 1:177–185. [Google Scholar]
- 44. Jordan BD (2013) The clinical spectrum of sport‐related traumatic brain injury. Nat Rev Neurol 9:222–230. [DOI] [PubMed] [Google Scholar]
- 45. Kalaria RN (2000) The role of cerebral ischemia in Alzheimer's disease. Neurobiol Aging 21:321–330. [DOI] [PubMed] [Google Scholar]
- 46. Kanaan NM, Morfini GA, LaPointe NE, Pigino GF, Patterson KR, Song Y et al (2011) Pathogenic forms of tau inhibit kinesin‐dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J Neurosci 31:9858–9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Khlistunova I, Biernat J, Wang Y, Pickhardt M, von Bergen M, Gazova Z et al (2006) Inducible expression of Tau repeat domain in cell models of tauopathy Aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem 281:1205–1214. [DOI] [PubMed] [Google Scholar]
- 48. Kim W, Lee S, Jung C, Ahmed A, Lee G, Hall GF (2010) Interneuronal transfer of human tau between Lamprey central neurons in situ. J Alzheimers Dis 19:647–664. [DOI] [PubMed] [Google Scholar]
- 49. King A, Sweeney F, Bodi I, Troakes C, Maekawa S, Al‐Sarraj S (2010) Abnormal TDP‐43 expression is identified in the neocortex in cases of dementia pugilistica, but is mainly confined to the limbic system when identified in high and moderate stages of Alzheimer's disease. Neuropathology 30:408–419. [DOI] [PubMed] [Google Scholar]
- 50. Koerte IK, Ertl‐Wagner B, Reiser M, Zafonte R, Shenton ME (2012) White matter integrity in the brains of professional soccer players without a symptomatic concussion. JAMA 308:1859–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lin A, Liao H, Merugumala S, Prabhu S, Meehan IIIW, Ross B (2012) Metabolic imaging of mild traumatic brain injury. Brain Imaging Behav 6:208–223. [DOI] [PubMed] [Google Scholar]
- 52. Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K (2012) Trans‐synaptic spread of tau pathology in vivo. PLoS ONE 7:e31302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Martland HS (1928) Punch drunk. J Am Med Assoc 91:1103–1107. [Google Scholar]
- 54. Maxwell WL, Povlishock JT, Graham DL (1997) A mechanistic analysis of nondisruptive axonal injury: a review. J Neurotrauma 14:419–440. [DOI] [PubMed] [Google Scholar]
- 55. McCown IA (1959) Protecting the boxer. J Am Med Assoc 169:1409–1413. [DOI] [PubMed] [Google Scholar]
- 56. McKee AC, Robinson ME (2014) Military‐related traumatic brain injury and neurodegeneration. Alzheimers Dement 10:S242–S253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McKee AC, Cantu RC, Nowinski CJ, Hedley‐Whyte ET, Gavett BE, Budson AE et al (2009) Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 68:709–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW et al (2010) TDP‐43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 69:918–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH et al (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136(Pt 1):43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. McKee AC, Daneshvar DH, Alvarez VE, Stein TD (2014) The neuropathology of sport. Acta Neuropathol 127:29–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Medana I, Esiri M (2003) Axonal damage: a key predictor of outcome in human CNS diseases. Brain 126:515–530. [DOI] [PubMed] [Google Scholar]
- 62. Mihalik JP, Bell DR, Marshall SW, Guskiewicz KM (2007) Measurement of head impacts in collegiate football players: an investigation of positional and event‐type differences. Neurosurgery 61:1229–1235. [DOI] [PubMed] [Google Scholar]
- 63. Millspaugh J (1937) Dementia pugilistica. US Naval Med Bull 35:297Y303. [Google Scholar]
- 64. Mitsis E, Riggio S, Kostakoglu L, Dickstein D, Machac J, Delman B et al (2014) Tauopathy PET and amyloid PET in the diagnosis of chronic traumatic encephalopathies: studies of a retired NFL player and of a man with FTD and a severe head injury. Transl Psychiatr 4:e441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Moisse K, Mepham J, Volkening K, Welch I, Hill T, Strong MJ (2009) Cytosolic TDP‐43 expression following axotomy is associated with caspase 3 activation in NFL^(‐/‐) mice: Support for a role for TDP‐43 in the physiological response to neuronal injury. Brain Res 1296:176–186. [DOI] [PubMed] [Google Scholar]
- 66. Moisse K, Volkening K, Leystra‐Lantz C, Welch I, Hill T, Strong MJ (2009) Divergent patterns of cytosolic TDP‐43 and neuronal progranulin expression following axotomy: implications for TDP‐43 in the physiological response to neuronal injury. Brain Res 1249:202–211. [DOI] [PubMed] [Google Scholar]
- 67. Montenigro PH, Baugh CM, Daneshvar DH, Mez J, Budson AE, Au R et al (2014) Clinical subtypes of chronic traumatic encephalopathy: literature review and proposed research diagnostic criteria for traumatic encephalopathy syndrome. Alzheimers Res Ther 6:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Morales R, Green KM, Soto C (2009) Cross currents in protein misfolding disorders: interactions and therapy. CNS Neurol Disord Drug Targets 8:363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mouzon BC, Bachmeier C, Ferro A, Ojo JO, Crynen G, Acker CM et al (2014) Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann Neurol 75:241–254. [DOI] [PubMed] [Google Scholar]
- 70. Neubuerger KT, Sinton DW, Denst J (1959) Cerebral atrophy associated with boxing. AMA Arch Neurol Psychiatry 81:403–408. [DOI] [PubMed] [Google Scholar]
- 71. Newell KL, Hyman BT, Growdon JH, Hedley‐Whyte ET (1999) Application of the National Institute on Aging (NIA)‐Reagan Institute criteria for the neuropathological diagnosis of Alzheimer disease. J Neuropathol Exp Neurol 58:1147–1155. [DOI] [PubMed] [Google Scholar]
- 72. Okawa Y, Ishiguro K, Fujita SC (2003) Stress‐induced hyperphosphorylation of tau in the mouse brain. FEBS Lett 535:183–189. [DOI] [PubMed] [Google Scholar]
- 73. Omalu B, Hammers JL, Bailes J, Hamilton RL, Kamboh MI, Webster G, Fitzsimmons RP (2011) Chronic traumatic encephalopathy in an Iraqi war veteran with posttraumatic stress disorder who committed suicide. Neurosurg Focus 31:E3. [DOI] [PubMed] [Google Scholar]
- 74. Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH (2005) Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 57:128–134. [DOI] [PubMed] [Google Scholar]
- 75. Omalu BI, DeKosky ST, Hamilton RL, Minster RL, Kamboh MI, Shakir AM, Wecht CH (2006) Chronic traumatic encephalopathy in a national football league player: part II. Neurosurgery 59:1086–1093. [DOI] [PubMed] [Google Scholar]
- 76. Omalu BI, Fitzsimmons RP, Hammers J, Bailes J (2010) Chronic traumatic encephalopathy in a professional American wrestler. J Forensic Nurs 6:130–136. [DOI] [PubMed] [Google Scholar]
- 77. Oppenheimer D (1968) Microscopic lesions in the brain following head injury. J Neurol Neurosurg Psychiatry 31:299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Park SY, Ferreira A (2005) The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta‐amyloid‐induced neurodegeneration. J Neurosci 25:5365–5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Parker HL (1934) Traumatic encephalopathy (“punch drunk”) of professional pugilists. J Neurol Psychopatho 15:20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Povlishock JT (1993) Pathobiology of traumatically induced axonal injury in animals and man. Ann Emerg Med 22:980–986. [DOI] [PubMed] [Google Scholar]
- 81. Roberts G, Whitwell H, Acland PR, Bruton C (1990) Dementia in a punch‐drunk wife. Lancet 335:918–919. [DOI] [PubMed] [Google Scholar]
- 82. Roberts GW, Allsop D, Bruton C (1990) The occult aftermath of boxing. J Neurol Neurosurg Psychiatry 53:373–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sato T, Takeuchi S, Saito A, Ding W, Bamba H, Matsuura H et al (2009) Axonal ligation induces transient redistribution of TDP‐43 in brainstem motor neurons. Neuroscience 164:1565–1578. [DOI] [PubMed] [Google Scholar]
- 84. Schmidt M, Zhukareva V, Newell K, Lee V, Trojanowski J (2001) Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer's disease. Acta Neuropathol 101:518–524. [DOI] [PubMed] [Google Scholar]
- 85. Sephton CF, Cenik C, Kucukural A, Dammer EB, Cenik B, Han Y‐H et al (2010) Identification of neuronal RNA targets of TDP‐43‐containing ribonucleoprotein complexes. J Biol Chem 286:1204–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sephton CF, Cenik B, Cenik BK, Herz J, Yu G (2012) TDP‐43 in central nervous system development and function: clues to TDP‐43‐associated neurodegeneration. Biol Chem 393:589–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, Saatman KE et al (1999) Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol 58:982–992. [DOI] [PubMed] [Google Scholar]
- 88. Sonnen JA, Larson EB, Crane PK, Haneuse S, Li G, Schellenberg GD et al (2007) Pathological correlates of dementia in a longitudinal, population‐based sample of aging. Ann Neurol 62:406–413. [DOI] [PubMed] [Google Scholar]
- 89. Sotiropoulos I, Catania C, Riedemann T, Fry JP, Breen KC, Michaelidis TM, Almeida OF (2008) Glucocorticoids trigger Alzheimer disease‐like pathobiochemistry in rat neuronal cells expressing human tau. J Neurochem 107:385–397. [DOI] [PubMed] [Google Scholar]
- 90. Sotiropoulos I, Catania C, Pinto LG, Silva R, Pollerberg GE, Takashima A et al (2011) Stress acts cumulatively to precipitate Alzheimer's disease‐like tau pathology and cognitive deficits. J Neurosci 31:7840–7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Stein TD, Alvarez VE, McKee AC (2014) Chronic traumatic encephalopathy: a spectrum of neuropathological changes following repetitive brain trauma in athletes and military personnel. Alzheimers Res Ther 6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Stieler JT, Bullmann T, Kohl F, Tøien Ø, Brückner MK, Härtig W et al (2011) The physiological link between metabolic rate depression and tau phosphorylation in mammalian hibernation. PLoS ONE 6:e14530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Strong MJ, Yang W (2011) The frontotemporal syndromes of ALS. Clinicopathological correlates. J Mol Neurosci 45:648–655. [DOI] [PubMed] [Google Scholar]
- 94. Talavage TM, Nauman EA, Breedlove EL, Yoruk U, Dye AE, Morigaki KE et al (2014) Functionally‐detected cognitive impairment in high school football players without clinically‐diagnosed concussion. J Neurotrauma 31:327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Thal DR, Capetillo‐Zarate E, Del Tredici K, Braak H (2006) The development of amyloid beta protein deposits in the aged brain. Sci Aging Knowledge Environ 2006:re1. [DOI] [PubMed] [Google Scholar]
- 96. Tokuda T, Ikeda S, Yanagisawa N, Ihara Y, Glenner G (1991) Re‐examination of ex‐boxers' brains using immunohistochemistry with antibodies to amyloid β‐protein and tau protein. Acta Neuropathol 82:280–285. [DOI] [PubMed] [Google Scholar]
- 97. Van der Jeugd A, Hochgräfe K, Ahmed T, Decker JM, Sydow A, Hofmann A et al (2012) Cognitive defects are reversible in inducible mice expressing pro‐aggregant full‐length human Tau. Acta Neuropathol 123:787–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Waite LM, Broe GA, Creasey H, Grayson DA, Cullen JS, O'Toole B et al (1997) Neurodegenerative and other chronic disorders among people aged 75 years and over in the community. Med J Aust 167:429–432. [DOI] [PubMed] [Google Scholar]
- 99. Williams DJ, Tannenberg AE (1996) Dementia pugilistica in an alcoholic achondroplastic dwarf. Pathology 28:102–104. [DOI] [PubMed] [Google Scholar]
- 100. Wolozin B (2012) Regulated protein aggregation: stress granules and neurodegeneration. Mol Neurodegener 7:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zilka N, Filipcik P, Koson P, Fialova L, Skrabana R, Zilkova M et al (2006) Truncated tau from sporadic Alzheimer's disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett 580:3582–3588. [DOI] [PubMed] [Google Scholar]
- 102. Margulies SS, Kilbaugh T, Sullivan S, Smith C, Propert K, Byro M et al (2015) Establishing a clinically relevant large animal model platform for TBI therapy development: using cyclosporin a as a case study. Brain Pathology 25:289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. McKee AC, Stein T, Kiernan P, Alvarez V (2015) The neuropathology of chronic traumatic encephalopathy. Brain Pathology 25:336–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Montenigro PH, Bernick C, Cantu, RC (2015) Clinical features of repetitive traumatic brain injury and chronic traumatic encephalopathy. Brain Pathology 25:351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Smith C (2015) The study and consequences of repetitive traumatic brain injury. Brain Pathology 25:287–288. [DOI] [PMC free article] [PubMed] [Google Scholar]