

Abstract
Rationale
Regulation of striated muscle contraction is achieved by Ca2+-dependent steric modulation of myosin cross-bridge cycling on actin by the thin filament troponin-tropomyosin complex. Alterations in the complex can induce contractile dysregulation and disease. For example, mutations between or near residues 112–136 of cardiac troponin-T, the crucial N-terminal TnT1 tropomyosin-binding region, cause cardiomyopathy. The Drosophila up101 Glu/Lys amino acid substitution lies C-terminally adjacent to this phylogenetically conserved sequence.
Objective
Using a highly integrative approach, we sought to determine the molecular trigger of up101 myofibrillar degeneration, to evaluate contractile performance in the mutant cardiomyocytes, and to examine the effects of the mutation on the entire Drosophila heart to elucidate regulatory roles for conserved TnT1 regions and provide possible mechanistic insight into cardiac dysfunction.
Methods and Results
Live video imaging of Drosophila cardiac tubes revealed the troponin-T mutation prolongs systole and restricts diastolic dimensions of the heart, due to increased numbers of actively cycling myosin cross-bridges. Elevated resting myocardial stiffness, consistent with up101 diastolic dysfunction, was confirmed by an atomic force microscopy-based nanoindentation approach. Direct visualization of mutant thin filaments via electron microscopy and three-dimensional reconstruction resolved destabilized tropomyosin positioning and aberrantly exposed myosin binding sites under low Ca2+ conditions.
Conclusions
As a result of troponin-tropomyosin dysinhibition, up101 hearts exhibit cardiac dysfunction and remodeling comparable to that observed during human restrictive cardiomyopathy. Thus, reversal of charged residues about the conserved tropomyosin-binding region of TnT1 may perturb critical intermolecular associations required for proper steric regulation, which likely elicits myopathy in our Drosophila model.
Keywords: Drosophila, steric regulation, tropomyosin, thin filament, myosin, diastolic dysfunction cardiomyopathy, animal model contractile proteins
INTRODUCTION
Muscle contraction results from an orchestrated series of molecular events that culminate in transient interactions between myosin-containing thick and actin-containing thin filaments. The high propensity for cyclic ATP-dependent acto-myosin associations indicates that without regulation, muscle would remain in a continuous state of contraction, and hence non-functional. Striated muscle regulation is primarily achieved by Ca2+-dependent modulation of myosin cross-bridge binding on actin by the thin filament troponin-tropomyosin complex.1–3 The complex consists of an elongated coiled-coil tropomyosin (Tm) dimer that extends along 7 monomers of the long-pitch actin helix and three troponin (Tn) subunits: troponin-C (TnC, the Ca2+-binding subunit), troponin-I (TnI, the inhibitory subunit), and troponin-T (TnT, the Tm-binding subunit). These elements (7 actin monomers, 1 Tn complex, and 1 Tm molecule) comprise “regulatory units”, which run continuously in series along thin filaments.
It is generally acknowledged that regulatory units adopt various structural “states” characterized by different Tm positions that govern contraction.1–5 At rest regulatory units are maintained predominantly in the “blocked (B-) state”.5–7 Here, Tm influenced by TnI (and TnT-see below) inhibits contraction by sterically occluding myosin binding sites on actin. Upon activation, Ca2+ binding to TnC promotes a release of TnI-mediated inhibition and the movement of Tm azimuthally from subdomains 1 and 2 and toward subdomains 3 and 4 of F-actin resulting in the “closed (C-) state”.5–7 Initial myosin binding to actin subsequently has allosteric effects on thin filaments such that there is further displacement of Tm and spread of accessible myosin binding sites along regulatory units to establish the “open (M-) state” and promote the cooperative activation of contraction.4,5,8 Thus, as more cross-bridges are recruited there is a proportional increase in active tension generation and transmission of contractile force. The positional states adopted by Tm appear to represent average locations of the coiled-coil as Tm is thought to oscillate dynamically between the B-, C- and M-states at all Ca2+ levels, and it is the average azimuthal location of this equilibrium that actually is controlled by Tn, Ca2+ and myosin.4,9,10
The TnT subunit of the Tn complex consists of two domains that can be separated by limited proteolysis.11,12 The C-terminal TnT2 domain binds TnI and TnC. Together they form the globular head, core domain of the Tn complex and act as the Ca2+ sensor of the thin filament. The N-terminal TnT1 domain is highly helical, terminates in the head-to-tail Tm-Tm overlap region, enhances the cooperativity of myosin S1 binding to the thin filament, and by binding to Tm increases its affinity for actin.13–17 Moreover, roles for TnT1 in cardiac myofilament regulation are supported by several findings including a report that describes the N-terminal domain of TnT, in the absence of all other Tn components, inhibits myosin interaction with actin-Tm filaments as a result of the TnT1 tail promoting the B-state.18 These results suggest the Tn tail may directly contribute to the inhibition of muscle contraction in the absence of Ca2+ and plays a critical role in modulating the average position of Tm under different physiological, and potentially pathological, conditions.
TnT is the transducer that links the effects of Ca2+ binding interactions of the globular head of Tn to the physical location of Tm along the thin filament. This fundamental property is likely compromised by disease-causing mutations. Alterations in the cardiac TnT (cTnT) gene account for roughly 15% of documented familial hypertrophic cardiomyopathy (HCM)-causing mutations with nearly 70% of these localized to cTnT1 (Fig 1).19–21 cTnT1 mutations are also linked to dilated (DCM) and restrictive cardiomyopathies (RCM) (Fig 1).22–26 Many of these mutations have been investigated in vivo, in vitro and in silico and have been shown to result in a wide range of complex molecular and physiological effects.20,21
Figure 1. Multiple sequence alignment of TnT1 segments.

Clustal Omega multiple sequence alignment of the Tm binding region of TnT1 from various species: Hs-Homo sapiens; Bt-Bos taurus; Oa-Ovis aries; Rn-Rattus norvegicus; Mm-Mus musculus; Gg-Gallus gallus; Dm-Drosophila melanogaster. TRT1-slow skeletal TnT isoform; TRT2-cardiac TnT isoform; TRT3-fast skeletal TnT isoform; Dm cTRT-upheld isoform K, the predominant isoform identified in the Drosophila heart.28 Residues are shaded based on degree of conservation. An (*) indicates positions that have identical residues, a (:) indicates conservation with strongly similar residues and a (.) indicates weakly similar residues. The loci of human cardiomyopathy-causing mutations are highlighted: (red) hypertrophic, (blue) dilated, (green) restrictive cardiomyopathy mutations. Residues 113–136 of human cTnT1 are highly conserved across analyzed sequences (encompassed by black box), and are believed to be critical for Tm associations throughout the animal kingdom.44 The Drosophila up101 residue (E89K of upheld isoform K) is highlighted in yellow and indicated by ↓. For reference, the Mus musculus cardiac TnT1 region modeled by Ertz-Berger et al.55, the purported Tm-binding residues of human fast skeletal TnT as described by Jin and Chong45 and the chicken fast skeletal TnT1 fragment resolved by Murakami et al.78 (PDB: 2Z5H) are underlined.
The diverse consequences of cTnT1 mutations correlate with the clinical heterogeneity of the myopathic responses.20,21 A detailed understanding of such disorders can benefit from model systems that facilitate integrative interrogation of disease pathogenesis from the level of the whole organ down to the molecular machinery. Drosophila melanogaster is a powerful genetic model for investigating the structure and function of striated muscle hierarchically. The indirect flight (IFM) and cardiac muscles of insects are, in fact, comprised of myofibrillar proteins that are highly homologous to those expressed in vertebrates.27,28 Direct structural evidence indicates that the steric Tn-Tm-based regulatory mechanism of contraction operates in insects.29 Furthermore, Drosophila myofibrillar mutants show disparate cardiomyopathic responses analogous to those observed in humans.30,31 Drosophila has a single TnT gene. Specific constitutively expressed TnT point mutations do not perturb assembly of IFM myofibrils but cause their deterioration within days after utilization.32,33 The up101 amino acid substitution, originally described as Glu88Lys, localizes to the TnT1 domain.33 The corresponding IFM degenerative syndrome was ascribed to abnormal acto-myosin interactions.33 Since TnT1 forms crucial Tm associations, modulates the average Tm position along regulatory units and accumulates numerous cardiomyopathy-causing mutations, we tested the hypothesis that the up101 amino acid substitution induces cardiomyopathy in flies due to contractile dysinhibition and disrupted steric regulation. Thus, we sought to resolve the initial molecular step that triggers up101 IFM myofibrillar degeneration, to evaluate cardiomyocyte contractile performance and to examine the effects of the lesion on the entire Drosophila heart in order 1) to ascertain critical regulatory roles for conserved TnT1 regions and 2) to provide a possible mechanistic basis for cardiac dysfunction. We show the up101 charge reversal induces cardiomyopathy, prolonged periods of muscle activation and excessive cross-bridge formation even at rest. We demonstrate these changes are associated with altered diastolic dimensions and mechanical properties of the heart and its constituent myocytes, in vivo. Direct visualization of up101 thin filaments reveals the mutation strongly promotes formation of the C-state in the absence of Ca2+, when a B-state is expected, which likely serves as the initial molecular factor for the cardiac pathology. Similarly, human cardiomyopathy mutations dispersed throughout this conserved region likely disrupt critical TnT1-Tm interactions needed to establish Ca2+-dependent thin filament regulatory states.
METHODS
Fly lines, culture conditions and husbandry
upheld101 (up101) mutant flies, originally recovered from a mutagenesis screen of Canton-S Drosophila32 were obtained from the Bloomington Drosophila Stock Center at Indiana University. The Canton-S strain served as the wildtype control line.
For up101 IFM thin filament isolation, up101 f car; b el Mhc12 cn double mutants were generated by standard mating procedures. The genetic alteration in the Mhc12 strain prevents myosin heavy chain accumulation in the IFM. Thus thick filament assembly and up101-induced IFM degeneration does not occur.33
All up101 analyses were performed on the hearts and thin filaments of homozygous animals.
Confocal microscopy
Confocal microscopy was performed as detailed by Alayari et al. (2009)34 with an Olympus FluoView FV10i Confocal Microscope system at 10× magnification.
Cardiac image analysis of semi-intact Drosophila preparations
Cardiac tubes of three-week old male and female adult flies (n=40–45) were surgically exposed under oxygenated artificial hemolymph (AH) according to Vogler and Ocorr (2009).35
Image analysis of heart contractions was performed using high speed movies of semi-intact Drosophila preparations as previously.31,36 30 second movies were taken at ~120 frames per second using a Hamamatsu Orca Flash 2.8 CMOS camera on a Leica DM5000B TL microscope with a 10× (0.30 NA) immersion lens. M-mode kymograms were generated using a MATLAB-based image analysis program.37 Measurements of cardiac dimensions and contractile dynamics were obtained as output from the MATLAB-based program. Two-way ANOVAs were employed to test if the effects of gender or genotype (or if an interaction exists) for each cardiac parameter were significant. When measured values were not normally distributed data were logarithmically transformed and significance confirmed via two-way ANOVA. Large sample sizes mitigated concern regarding infrequent and mild evidence of variance heterogeneity. Significance was assessed at p<0.05.
Imaging of blebbistatin-induced changes in cardiac dimensions
Beating hearts from three-week old female Canton-S (n=15) and up101 (n=20) flies were imaged as described above using a 20× (0.50 NA) immersion objective lens. The hearts were recorded at various focal depths to resolve clear cardiac edges along the length of the tube. After filming, hearts were treated with 100μmol/L blebbistatin (Cayman chemical, Ann Arbor, MI) in AH for ~30 minutes at room temperature. Following complete blebbistatin-induced cessation of beating, cardiac tubes were fixed (8% formaldehyde in 1× PBS) for 20 minutes at 25°C and rinsed three times for 10 minutes in 1× PBS with continuous shaking. The hearts were filmed again post-treatment at various focal depths.
Movies of individual hearts, pre- and post-blebbistatin treatment were opened in HCImage Live software. Diastolic and “blebbistatin-relaxed” diameters were measured at identical longitudinal distances and focal depths, which permitted multiple clear edge views, along the tubes. Three distinct diameter measures were recorded across opposing cardiomyocytes of each heart tube and averaged for each fly. The effect of blebbistatin treatment on cardiac diameters was evaluated using a paired t-test of the means of the matched groups before and after blebbistatin incubation. An unpaired t-test was used to identify significant differences in the cardiac response to blebbistatin between genotypes. Significance was assessed at p<0.05.
Nanoindentation by atomic force microscopy (AFM)
Nanoindentation, to determine the transverse stiffness at the cellular seam of the conical chamber, was performed with an Asylum Research MFP-3D Bio Atomic Force Microscope mounted on a Nikon Ti-U fluorescence inverted microscope with a 120pN/nm silicon nitride cantilever pre-mounted with a two μm-radius borosilicate sphere (Novascan Technologies, Ames, IA) as previously described.38 Prior to indentation, up to three semi-intact heart preparations were immobilized on glass coverslips and submerged in AH. Myogenic contractions were confirmed and then arrested with administration of 10mmol/L EGTA in AH. Eight force curves per conical chamber were obtained from discrete locations at the ventral midline from three-week old female Canton-S (n=14) and up101 (n=15) flies. Following indentation in EGTA, hearts were washed with fresh AH and restoration of myogenic contraction was confirmed. Hearts were then incubated in 100μmol/L blebbistatin in AH. Inhibition of contraction was confirmed within 30 minutes of incubation. Indentation was repeated at the same cardiac locations. Following final indentation, blebbistatin was photo-inactivated and resumption of myogenic contraction was visually confirmed to ensure myocardium remained viable. Force-indentation curves were analyzed with automated, custom-written software in MATLAB to calculate myocardial elastic modulus or “stiffness” (E, measured in Pascal; Pa) as described previously.38 All force curves recorded from each heart tube were averaged for each fly. The effect of blebbistatin treatment on cardiac stiffness was evaluated using a paired t-test of the means of the matched groups before and after incubation. Unpaired t-tests were used to identify significant differences 1) in stiffness between genotypes under each chemical condition and 2) in the cardiac response to blebbistatin among genotypes. Significance was assessed at p<0.05.
Electron microscopy and three-dimensional reconstruction
IFM thin filaments were isolated from the thoraces of up101 f car; b el Mhc12 cn Drosophila according to Cammarato et al., (2004)29 with the following modifications: the “chemically skinned” thoraces were rinsed in 25ml of rigor solution and homogenized in 0.5ml of fresh buffer in a glass homogenizer. Homogenates were centrifuged at 16,000g for 60 minutes to sediment particulate material, including trace amounts of non-IFM thin filaments still bound in rigor to thick filaments derived from additional thoracic muscles. 0.3ml of the resultant supernatant, which contained IFM thin filaments, was diluted two to ten-fold with either rigor solution by itself or the buffer with 0.1mmol/L CaCl2 added in excess of the EGTA present, immediately prior to preparation for EM. Thin filaments were negatively stained and imaged as detailed previously.5,29,39
Helical reconstruction, which resolves actin monomer structure and Tm strands, but not Tn, was performed on IFM thin filament segments which encompassed four-seven regulatory units from 18 up101 filaments maintained in EGTA and 14 up101 filaments maintained in the presence of Ca2+ according to standard methods.5,39,40 The statistical significance of densities in reconstructions was computed from the standard deviations associated with contributing points as previously described.5,39,41,42
RESULTS
The homologous Tm binding region of TnT1 is associated with multiple disease mutations
The strongest association between Tm and Tn involves a C-terminal recognition site of striated muscle Tm and TnT1.43,44 Likewise, evolutionarily conserved sequences within TnT1 presumably mediate interaction with Tm.44–46 In particular, multiple sequence alignments identified a highly conserved and highly charged domain corresponding to residues 112–136 of human cTnT1 that likely comprise a critical Tm-binding element (Fig 1).44 The importance of this TnT1 domain is further underscored by the high number of disease-inducing mutations that reside within and closely flank this region (Fig 1). The Drosophila up101 TnT glutamic acid to lysine amino acid substitution is located just downstream of this element and likely perturbs TnT1-Tm interactions.
The Drosophila up101 TnT mutation alters cardiac morphology
Adult Drosophila melanogaster possess a 1mm long pulsatile heart tube, which is located in the abdomen along the insect’s body axis and is composed of a single layer of contractile cardiomyocytes (Fig 2A).47 The myocytes form bilateral rows that join together through specialized cell junctions to create a simple linear tube that extends down the dorsal body wall.47,48
Figure 2. Drosophila heart tube morphology is sensitive to the up101 TnT1 mutation.
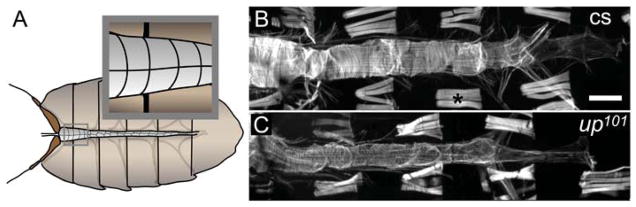
A) Illustration depicting the Drosophila heart. The cardiac tube consists of a single layer of contractile cardiomyocytes. Inset: the anterior conical chamber (CC) is ~130μm wide and tapers gradually. The cardiomyocytes of the CC exhibit a characteristic rectangular structure similar to vertebrates and are densely populated with parallel bundles of myofibrils that run the length of the cells. They are symmetrically aligned such that the bipolar ends of the myocytes form prominent “seams” along the midventral and middorsal surface.48 As a result of this arrangement, the junctional seams of cell-cell contact bear the brunt of contractile stress from opposing myocytes. Depending on gender and on genotype, the remainder of wildtype heart tubes are roughly 70μm in diameter. B) Alexa594-TRITC phalloidin stained Canton-S (cs) control and C) up101 mutant heart tubes. Note obvious differences in cardiac diameter. Furthermore, up101 hearts exhibited reduced F-actin staining compared to Canton-S hearts as suggested by diminished fluorescence intensity in the cardiomyocytes relative to that in the retractors of tergite muscles (*). Scale bar = 100μm.
Drosophila cardiac morphology is sensitive to mutation. Alexa594-phalloidin staining of fixed Canton-S Drosophila heart tubes revealed typical spiraling myofibrillar arrangements within the cardiomyocytes (Fig 2B and C). The fibers were tightly packed with myofilamentous F-actin. Compared to Canton-S hearts, up101 TnT1 mutant hearts exhibited an apparent loss of myofilaments as noted by reduced F-actin staining relative to that expressed in the retractors of tergite muscles. The mutant heart tube also appeared substantially narrower than that of the wildtype control. The overall morphological effects of the up101 mutation on the heart, however, seem less severe than the auto-destructive effects of the lesion on the IFM33.
The Drosophila up101 TnT mutation induces restrictive physiology and diastolic dysfunction
Beating Drosophila heart tubes were imaged to further investigate the pathophysiological effects of the up101 TnT1 mutation on live cardiac muscle. M-mode traces, which illustrate the positions of the heart wall edges over time, reveal cardiac dimensions and cardiac contraction dynamics (Fig 3A). up101 hearts were characterized by reduced diameters and sustained periods of systole relative to control hearts.
Figure 3. up101 Drosophila display perturbed cardiac dimensions and contractile properties.

A) M-mode kymograms generated from high speed videos of beating Canton-S (cs) wildtype and up101 heart tubes. Vertical arrows delineate diastolic (DD) and systolic (SD) diameters. Horizontal arrows demarcate complete heart periods (HP) and systolic intervals (SI). Relative to Canton-S hearts, up101 cardiac tubes displayed reduced diameters and prolonged periods of systole. B) Semi-automated optical heartbeat analysis of Canton-S and up101 hearts. Regardless of gender, up101 Drosophila revealed highly significant alterations in several cardiac structural and functional parameters relative to control flies (also see Online Table I). Significant reductions in cardiac dimensions, in fractional shortening and in heart period as well as prolonged systolic intervals were observed in the mutant flies. The genotype and gender effects on cardiac variables were determined by two-way ANOVA. Together, these data suggest the up101 TnT mutation induces restrictive cardiac physiology and incomplete diastolic relaxation.
We quantified the effects of altered TnT1 on cardiac dimensions and contractile performance and compared the data to those obtained from Canton-S flies (Fig 3B and Online Table I). Mutant TnT1 expression resulted in significantly reduced diastolic and systolic diameters for both male and female up101 relative to control flies. Consistent with ~6% (Canton-S) and 4% (up101) gender-related differences in body size (Online Figure I), female Drosophila had significantly larger cardiac diameters relative to male flies. A significant interaction effect was determined between genotype and gender for diastolic and systolic diameter measurements. This suggests the up101 mutation differentially perturbs male versus female heart dimensions. The effect of the TnT1 lesion on diastolic diameters was greater than that on systolic diameters, which resulted in a significant decrease in mutant fractional shortening relative to control. The mutation appeared to differentially influence male versus female cardiac shortening (interaction p value<0.0001). Overall up101 hearts appeared restricted and unable to adequately relax during diastole.
We additionally evaluated inherent myogenic contractile dynamics using motion analysis software (Fig 3B and Online Table I).37 The heart period, which is the combined length of time required for a single diastolic and subsequent systolic event of the cardiac cycle, was significantly shorter for up101 mutant flies relative to control. Therefore, the mutants display a faster myogenic heart rate. Moreover, a significant interaction effect was determined between genotype and gender, which suggests the TnT1 lesion differentially perturbs the heart rate of males relative to female flies. The up101 systolic interval was significantly prolonged relative to that of Canton-S. The degree of prolonged contractile events of the mutant hearts was determined to be gender dependent (interaction p=0.003). Dividing the systolic interval by the heart period demonstrates the extent of time during the cardiac cycle the hearts are contracting. Regardless of gender, the up101 TnT1 mutation promoted a significant increase in SI/HP ratio.
Thus, as observed in humans exhibiting restrictive cardiac physiology and diastolic dysfunction, the up101 TnT1 mutation seemingly perturbs the ability of the myocardium to reestablish resting diastolic diameters and it prolongs systolic contractile events.
The up101 TnT1 mutation reduces cardiac tube diameters during diastole by increasing actively cycling cross-bridges
Blebbistatin is a small molecule inhibitor of several striated muscle myosins and impedes acto-myosin interaction in cardiac preparations from multiple species.49,50 Blebbistatin treatment of wildtype Drosophila cardiac tubes completely prevented heart wall motion within 15–30 minutes (Fig 4A). Interestingly the diameter across the heart wall in the presence of blebbistatin appeared slightly larger than the maximum diameter across the wall during peak diastole. This implies that in Drosophila cardiac muscle not all regulatory units are sterically preventing acto-myosin interactions at diastolic Ca2+ levels. To quantify the extent of basal, diastolic shortening wildtype Canton-S fly hearts were filmed before and after incubation with blebbistatin. The average diameter following drug treatment was ~3% greater than that determined during diastole (Fig 4B). This difference was highly significant. These data suggest a small population of residual cross-bridges is actively cycling, generating force, reducing myocyte length and establishing basal mechanical tone in wildtype myocardium during diastole. Interestingly, a paired analysis of up101 cardiac diameters during diastole and following blebbistatin treatment revealed a highly significant increase of roughly 13% (Fig 4B). Comparing the change in diameters for each genotype revealed the response to blebbistatin was significantly greater for up101 hearts relative to that for control hearts (Fig 4B). These observations are consistent with diastolic dysfunction and restrictive physiology due to excessively dysinhibited cross-bridge cycling, enhanced mechanical tone and incomplete relaxation during diastole for the up101 TnT mutant hearts relative to control hearts.
Figure 4. Blebbistatin has differential effects on diameters of control versus up101 TnT1 mutant hearts.
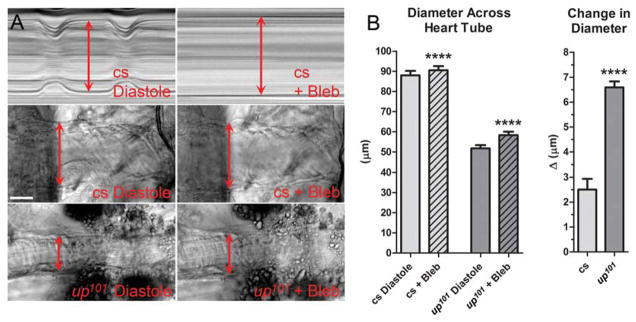
Beating hearts were treated with 100μmol/L blebbistatin (Bleb) to identify a potential contribution of disproportionate, strong acto-myosin interactions to up101-mediated diastolic dysfunction. A) Top: M-Modes generated from an identical region of the same Canton-S (cs) control heart during its cardiac cycle and following blebbistatin-induced relaxation. Note complete, blebbistatin-generated cessation of cardiomyocyte movement and a slight increase in diameter across the heart tube relative to that during diastole. Bottom: Individual frames from movies of Canton-S and up101 hearts during peak diastolic and post blebbistatin treatment time points. The cell-lengthening response to blebbistatin appears greater for mutant hearts relative to that of control. B) The diameter across discrete locations of Canton-S hearts increased from 87.9±2.2μm (mean±SEM) during diastole to 90.5±2.1μm post blebbistatin incubation while that for up101 hearts increased from 51.7±1.6μm to 58.3±1.7μm. A paired t-test revealed these responses were highly significant (****p<0.0001). The average change in diameter for up101 hearts (6.60±.29μm) was significantly greater (unpaired t-test, ****p<0.0001) than that determined for Canton-S hearts (2.50±.43μm). This is consistent with a greater number of dysinhibited strong acto-myosin associations during diastole, which promote enhanced myocyte shortening for up101 hearts relative to control.
The Drosophila up101 TnT mutation enhances myocardial stiffness under low Ca2+
To gain direct insight into altered mechanical properties of the mutant hearts during diastole, we exploited the unique geometry of the cardiac tube, in particular the conical chamber, to resolve basal tension differences, in vivo at the level of individual cardiomyocytes. The transverse stiffness at the cellular seams of the conical chamber is directly related to the amount of contractile stress exerted by, and transmitted to, the adjoined ends of opposing cardiomyocytes. Nanoindentation at the ventral cell–cell junction, where cytoskeletal elements converge, was performed on each genotype under different chemical conditions (Fig 5A). The transverse stiffness of EGTA-inhibited Canton-S cardiomyocytes within the conical chamber was 2kPa and subsequent incubation in blebbistatin reduced stiffness ~25% (Fig 5B). These data corroborate our optical assessment and provide direct mechanical evidence that, at rest, a small number of unimpeded cross-bridges are actively generating force. The transverse stiffness of EGTA-maintained up101 fibers was 3.3kPa more than 50% higher than that of EGTA-inhibited control cardiomyocytes (Fig 5B). Addition of blebbistatin significantly reduced up101 transverse stiffness, which more closely resembled that of blebbistatin-treated control flies. The mechanical response to blebbistatin was significantly greater for up101 myocytes compared to Canton-S myocytes (Fig 5B). These data are consistent with elevated stress at the ends of the mutant cells at rest due to a larger population of uninhibited actively cycling myosin cross-bridges in up101 cardiomyocytes relative to that in control.
Figure 5. Excessive cross-bridge cycling increases the transverse stiffness of up101 cardiomyocytes.
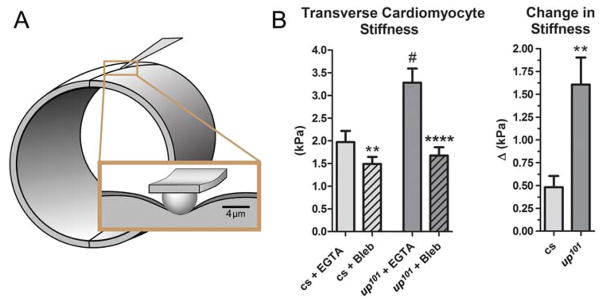
An atomic force microscopy-based nano-indentation technique was employed to measure basal myocardial stress and to assess residual active cross-bridges, in vivo. A) Illustration portraying the nano-indentation scheme of Drosophila myocardium. Indentations were made at the ventral midline of the heart tubes under 10mmol/L EGTA-relaxed and 100μmol/L blebbistatin-relaxed (Bleb) conditions. Longitudinal stress, derived from active and unimpeded force-generating acto-myosin associations is transmitted to the midventral seam and can be detected as elevated transverse stiffness via nano-indentation. B) The stiffness at identical midventral locations of Canton-S (cs) conical chambers decreased from 1.97±.25kPa (mean±SEM) in EGTA to 1.49±.16kPa post blebbistatin incubation while that for up101 hearts decreased from 3.28±.32kPa to 1.68±.19kPa respectively. A paired t-test revealed these responses were significant for both Canton-S (**p<0.01) and for up101 (****p<0.0001) cardiomyocytes. Furthermore, the transverse cardiomyocyte stiffness for up101 hearts in EGTA was significantly greater (unpaired t-test, #p<0.01) than that determined for Canton-S control hearts. The average blebbistatin-induced change in stiffness for up101 hearts (1.61±.3kPa) was significantly greater (unpaired t-test, **p<0.01) than that for Canton-S hearts (.48±.12kPa). This is consistent with a higher number of basally dysinhibited acto-myosin interactions, which promote increased myocyte transverse stiffness under low Ca2+ conditions, in up101 hearts relative to control.
The Drosophila up101 TnT mutation promotes the C-state even in the absence of Ca2+
To directly determine the primary consequence of the up101 glutamic acid to lysine amino acid substitution and to provide a mechanistic basis for myopathy, we generated three-dimensional reconstructions of purified thin filaments expressing the TnT mutation. Thin filaments were isolated from the IFM of up101 Drosophila and either maintained in EGTA or treated with Ca2+ prior to negative staining. Electron micrographs showed that the thin filaments were not obviously different from control filaments and possessed the characteristic double helical array of actin subunits and additionally displayed periodic Tn bulges and elongated Tm strands (not shown).6,29 Three-dimensional reconstructions of the mutant filaments were determined by helical analysis.40 Surface views showed that Ca2+-dependent Tm strand movement, typical of control filaments29, was not a general feature of the mutants (Fig 6A). The majority of IFM thin filaments from up101 flies revealed Tm strands that contacted subdomains 3 and 4 of successive actin monomers in both the absence and presence of Ca2+. However, 17% of individual Ca2+-free mutant thin filaments differed and exhibited Tm strands that contacted actin monomers along the inner edge of subdomains 1 and 2 of F-actin. When the two sets of Ca2+-free data were combined, the Tm density in helical projection can be seen to contact only the inner domains of actin (Fig 6B). Therefore, Tm is most associated with the inner aspect of the up101 IFM thin filaments, distal to known myosin binding sites and hence unlikely to sterically prevent contraction. This is likely responsible for the destructive hypercontraction of the IFM and for the excessive residual cross-bridge cycling in relaxed hearts.
Figure 6. Three-dimensional reconstructions of up101 thin filaments reveal perturbed steric regulation.

A) Surface views of reconstructed up101 IFM thin filaments showing the position of Tm strands on actin. The four subdomains of a single actin monomer are labeled on the F-actin control reconstruction (subdomains-1 and -2 = outer domain and subdomains-3 and -4 = inner domain of F-actin). Note the presence of helically wound, continuous strands of Tm density (double-headed black arrows) running longitudinally along the up101 thin filaments that are not seen on the F-actin control. Under both chemical conditions Tm clearly associated with the inner domains (subdomain-3) of successive actin monomers (triple gray arrows) on up101 thin filaments. In the absence of Ca2+ however, the surface view reveals a small amount of extra density extending from Tm (black arrowheads) to the extreme inner edge of the outer domains (subdomain-1). This additional density is due to Tm on 17% of mutant thin filaments within the average structure remaining in the B-state. Under both chemical conditions however, mutant IFM thin filaments were primarily in the C-state with the outer domains of actin exposed. The average phase residuals (±SD), a measurement of the agreement among thin filaments generating reconstructions in each data set (EGTA and Ca2+) were 61.6°±5.8° and 62.0°±7.8° respectively. The average up-down phase residuals, a measure of the filament polarity were 19.4°±5.1° and 20.4°±8.6° respectively, values comparable to those previously reported. The average number of actin subunits per turn of the genetic helix of the IFM thin filaments were 2.155±.007 and 2.156±.008 respectively, consistent with a 28/13 helix. B) Helical projections for F-actin and up101 IFM thin filaments. Helical projections are formed by projecting component densities of the reconstructions down the long-pitch actin helices onto a plane perpendicular to the filament axis. Thus, the projections reveal the axially averaged positions of F-actin and Tm, which appear symmetrical on either side of the filament. The up101 TnT1 mutation causes Tm to associate primarily with the inner domains of actin (Ai) in the absence of Ca2+. The helical projections revealed no difference in Tm positioning in the absence or presence of Ca2+, and thus steric regulation is disrupted as a result of the mutation. Densities in all maps were shown to be significant over background noise at greater than the 99.95% confidence levels.
DISCUSSION
TnT makes extensive associations with multiple thin filament components including the TnI-TnC binary complex, Tm and actin and therefore is central to thin filament-mediated regulation of striated muscle contraction. TnT mutations, which predominately localize to the N-terminal TnT1 domain, are frequently associated with a host of myopathic responses (Fig 1). A number of investigative efforts have shown cardiomyopathic cTnT lesions induce a range of complex effects including altered myofilament in vitro sliding velocity, disturbed Ca2+ sensitivity of force generation, decreased cTnT-Tm affinity, impaired ability to stabilize the Tm overlap, perturbed efficacy of promoting Tm binding to actin, disrupted folding stability and secondary structure of cTnT1 and mutation-specific changes in peptide flexibility, all of which likely contribute to disease pathogenesis.44,51–55
Drosophila muscles are also sensitive to TnT alterations. Previously, constitutive TnT1 mutations were shown to drastically disrupt both the structure and function of IFMs within days of adulthood.33 We speculate that since the up101 mutation resides in the N-terminal region of TnT and it induces myosin-dependent IFM degeneration, the lesion likely causes cardiomyopathy in flies due to contractile dysinhibition and perturbed steric regulation.
TnT-binding Tms from vertebrates and invertebrates share a well-conserved amino acid segment believed to be a critical TnT1 recognition site.43 Similarly, multiple sequence alignments illustrate a high degree of evolutionary conservation among stretches of TnT1 postulated to be critical for Tm associations (Fig 1).44 Residues 112–136 of human cTnT were found to be 70% homologous across analyzed sequences, which suggests an essential Tm binding role for this element throughout the animal kingdom. This region is highly charged and intermolecular electrostatic associations likely dictate proper function. Importantly, engineered Tn constructs bind tightly to the thin filament only if they contain the entire 112–136 TnT1 domain.15,17,44,56 The Drosophila up101 mutation lies just downstream of this region. Introduction of basic charges could disturb conserved interactions immediately at the Tm-TnT1 interface that are required for proper steric regulation. Similarly cTnT cardiomyopathy mutations located in and adjacent to this region (Fig 1) are also expected to influence Tm-TnT1 associations. Interestingly, our data illustrate that the up101 charge reversal mutation results in a cardiac phenotype reminiscent of human RCM.
The up101 amino acid substitution may also promote molecular pathogenesis by potentially altering overall TnT performance, including mutation-driven propagated effects that could influence TnT function at a distance. These effects may involve 1) changes in helical stability of TnT1 and subsequently the flexibility of this region, which could compromise effective interactions with Tm53; 2) alterations in local helical electrostatic compaction and consequently distal helical expansion that drives unwinding and flexibility changes at remote distances along TnT55,57; and 3) disruptions that possibly propagate through a complex structural pathway to perturb the affinity of cardiac TnC for Ca2+.58 The latter could potentially contribute to changes in myoflament Ca2+ sensitivity, as frequently observed in other models of TnT-based cardiomyopathies.21,54,58,59
Regardless of gender, the up101 Drosophila mutation markedly reduced diastolic volumes and extended systole in mutant relative to control hearts (Fig 3B). These changes are consistent with diastolic dysfunction, a hallmark of HCM and RCM. Diastolic dysfunction is characterized by impaired relaxation, decreased distensibility and increased myocardial stiffness, which can result from excessive acto-myosin interactions.60,61 Thus, up101 Drosophila serve as a unique model to investigate the root of these pathological alterations in myocyte properties.
The diameter changes across the Drosophila heart, in response to the myosin-specific inhibitor blebbistatin, suggest that diastole in flies is accompanied by a small but significant population of residual, force generating cross-bridges that actively shorten the cardiomyocytes and impact diastolic tone (Fig 4). Detection of cycling myosin cross-bridges at submaximal, diastolic levels of Ca2+, which affect cardiomyocyte mechanical properties, is not unprecedented.62,63 Although beyond the scope of the current study, these basal acto-myosin associations could bolster the prominent stretch activation response common to insect muscle and may have important implications in myofilament length dependent activation of cardiomyocytes. Interestingly, specific TnI-inducing RCM and Tm-causing HCM mutations are associated with excessive cross-bridge cycling during diastole.64,65 These thin filament mutations were shown to reduce basal sarcomere length and elevate diastolic tension of cardiomyocytes. Likewise, the reduced diastolic diameter of up101 hearts is due, in part, to excessive, less inhibited force generating acto-myosin interactions that promote enhanced cell shortening relative to control hearts. Application of the myosin inhibitor however did not restore mutant heart diameter to that of blebbistatin-treated control flies (Fig 4B). Thus, additional remodeling events must transpire over the three week pre-analysis period that influence up101 myocyte dimensions. Changes in cellular dimensions are consistent with pathological responses and tissue disorganization that accompany cardiac disease. For example, pathological stimuli associated with several cardiomyopathies induce changes in myocyte geometry and shape that help determine contractile function and whole heart morphology.66
The myosin-dependent changes in up101 cardiac dimensions are accompanied by differences in resting myocardial stiffness. We employed a novel AFM-based technique to resolve Drosophila myocardial tension disparities, in vivo with single cell resolution (Fig 5A).38,67 The geometric nature of the cardiac tube allows indentation and transverse stiffness determination from discrete cellular loci in the intact organ with no mechanical artifacts due to myocyte isolation, seeding or plating. By determining the transverse stiffness at cell junctions, we can directly assess the degree of active longitudinal tension generated and transmitted to the connections between the ends of coupled conical chamber myocytes. Thus, we can quantify the relative extent of contractile dysinhibition by comparing the transverse stiffness at the midventral seam from different genotypes before and after incubation with blebbistatin. Control cardiomyocytes showed roughly a 25% drop in transverse stiffness at the midline upon incubation with the myosin inhibitor, consistent with a small number of residual force-generating acto-myosin associations under low Ca2+, diastole-like conditions (Fig 5B). The transverse stiffness of the up101 cardiomyocyte junction was over 60% greater than that of control and showed an ~50% decrease following blebbistatin treatment. The large discrepancies in resting transverse stiffness and in response to the myosin-specific inhibitor indicate the TnT1 mutation promotes a greater number of actively cycling, force generating cross-bridges in up101 myocytes relative to Canton-S myocytes and these contribute to the diastolic dysfunction observed under relaxing conditions.
TnT is pivotal in modulating the average position of Tm between the B-, C- and M-states along thin filaments.21 Disease-causing mutations in Tn subunits may lead to changes in the distribution of these states and therefore disrupt regulation of contractile force.68 To directly assess a mutation-specific redistribution of regulatory states we purified and imaged up101 IFM thin filaments. In the complete absence of Ca2+, surface views of three-dimensional reconstructions of up101 thin filaments revealed Tm stands, on average, making contact with the inner domains of successive actin monomers along the long pitch helices of the filament (Fig 6). However, a small amount of density could be seen extending from the extreme inner edge of the outer domains of actin to the Tm strands on these filaments (Fig 6A, black arrowheads). This extra density is the result of a small population of up101 thin filaments, which in the absence of Ca2+ exhibited Tm in the B-state, as seen with wildtype filaments lacking Ca2+.29 Nonetheless, the vast majority of the up101 thin filaments in the absence of Ca2+ were shown to be in the C-state. Thus, TnT products of the up101 allele may alter the equilibrium of the Tm position at rest such that at any given time the majority of thin filament regulatory units are in the C-state and not the B-state. This is consistent with the mutation disrupting the inherent, and possibly vital, B-state promoting property of the TnT1 tail18, a property that appears dependent upon a TnT1 subdomain that includes the up101 locus. When the up101 mutation is expressed in vivo and in the presence of thick filaments, excessive cross-bridge cycling would occur in a dysregulated fashion. Over time this could lead to destruction of the IFM and drive restrictive remodeling in the heart.
Analysis of myocytes from several TnT-based cardiomyopathy models has revealed significant alterations in cellular Ca2+ handling.69,70 Moreover, a Drosophila TnI mutant, which is characterized by an enlarged cardiac chamber and cardiac contractile dysfunction, displays abnormalities in the cytosolic Ca2+ transient as well as changes in transcription of proteins associated with Ca2+ handling.71 Therefore, the up101 TnT mutant may similarly exhibit downstream alterations in Ca2+ kinetics and homeostasis that potentially contribute to the observed cardiac phenotype. Quantitative polymerase chain reaction analysis was performed to assay possible changes in the Ca2+-handling biosignature of up101 relative to Canton-S hearts (Online Figure II).71 No significant differences were identified between the lines in transcript levels of L-type Ca2+ channels, ryanodine receptors, SERCA, Na/Ca exchangers, or in inositol-3-phosphate receptors. Although this assay does not preclude possible post-transcriptional or post-translational modifications that could influence the encoded proteins, nor does it completely rule out all potential adaptive Ca2+ responses, the results suggest the cardiac phenotype we observe is primarily due to the direct, Ca2+-independent, C-state promoting effect the up101 TnT mutation exerts on thin filaments.
Our integrative data are consistent with a mechanism of diastolic dysfunction and restrictive cardiac pathology based on a fundamental inability of the homozygous, up101 TnT1 mutant thin filaments to properly block myosin cross-bridge cycling at rest (Fig 7). Here, a disproportionally large number of up101 regulatory units adopt the C-state under low Ca2+ conditions. This permits an exceedingly high number of strong stereospecific acto-myosin associations and excessive formation of the M-state that would promote decreased diastolic heart chamber volumes and elevated diastolic myocardial stiffness. Moreover, inordinate myosin binding increases the affinity of Tn for Ca2+.1,72,73 Thus, fewer mutant regulatory units required to undergo Ca2+-dependent unblocking combined with enhanced Ca2+ sensitivity “primes” the system for systole. As a result, for a given Ca2+ transient, systole would commence earlier and terminate later, which is consistent with the highly prolonged systolic intervals observed in up101 hearts relative to control.
Figure 7. Hierarchical modeling of the diastolic effects of the up101 TnT1 mutation.

Diastole is characterized by low levels of free Ca2+. Control thin filament regulatory units are predominantly maintained in the blocked “B”-state (red Tm). Consequently the majority of myosin heads are in an unbound or weakly bound, non-force generating state. However, a small population of cross-bridges, under basal conditions, forms strong stereo-specific force generating associations. These actively cycling cross-bridges generate a finite amount of contractile stress as characterized by elevated transverse stiffness and slightly shortened cardiomyocytes. Incubation with blebbistatin inhibits basal myosin cross-bridge cycling, relieves residual mechanical stress and restores cardiomyocyte length. However, under low Ca2+ conditions the up101 TnT1 mutation appears to aberrantly promote formation of the “C”-state (green Tm). An increase in the proportion of poorly impeded actively cycling cross-bridges stimulates a high degree of resting mechanical tone and excessive cardiomyocyte shortening. Thus, relative to control cardiac tubes, the subsequent myocardial responses of up101 cardiac tubes to blebbistatin are significantly elevated. These findings suggest diastolic performance in the mutant hearts is likely severely compromised due to contractile dysinhibition and enhanced myocardial stiffness.
We anticipate similar, however potentially less severe responses in up101 heterozygotes. Due to cooperativity of contractile activation and the continuous nature of regulatory units, the effects of the mutation could potentially be transmitted to neighboring, non-mutant regulatory units and thus influence the regulatory status of regions up- and down-stream of the lesion. These propagated effects along the thin filament may also be sufficient to promote myosin cross-bridge cycling, decreased diastolic chamber volumes and elevated diastolic myocardial stiffness, but potentially to a lesser extent than that found in homozygotes due to the presence of some normally functioning, wildtype regulatory units. Thus, as with other models of cardiomyopathy we expect a relationship between the number of mutant up101 alleles and phenotype severity.74–77
TnT1 is essential for proper transduction of Ca2+ signals and modulation of Tm position along regulatory units in striated muscle. Here we examined the effects of the constitutively expressed up101 TnT1 mutation at multiple levels in Drosophila. We provide the first, direct structural evidence of how a mutation, adjacent to the conserved Tm-binding element of TnT1, perturbs steric regulation, promotes contractile dysinhibition and diastolic dysfunction and drives cardiac remodeling. Our results emphasize the potential significance of Tm-TnT1 electrostatic associations for proper steric regulation throughout the animal kingdom. Numerous indices of up101 cardiac function suggest a shift in equilibrium status of thin filament regulatory units to a mutation-induced preponderance of the C-state even at rest. We propose human cardiomyopathy mutations located in and close to the homologous Tm-binding element of TnT1 may also alter the fundamental B-state promoting role of TnT118 and thereby activate pathological remodeling cascades. Overall our study indicates that Drosophila is a valuable tool for investigating the most proximal, direct effects of thin filament lesions and that flies are an effective and unique model to resolve, at multiple levels, how such mutations perturb the normal distribution of force generating regulatory states and elicit cardiac dysfunction.
Supplementary Material
Novelty and Significance.
What Is Known?
The N-terminal troponin-T tail promotes a conformational state of the thin filament that inhibits muscle contraction.
Mutations in and around the highly conserved tropomyosin-binding region of the N-terminal cardiac troponin-T tail result in diverse human cardiomyopathies.
The Drosophila melanogaster glutamic acid to lysine up101 troponin-T mutation resides just downstream of the homologous tropomyosin-binding element and results in skeletal muscle degeneration.
What New Information Does This Article Contribute?
up101 Drosophila exhibit a cardiac phenotype that is characterized by restrictive physiology and diastolic dysfunction.
Under relaxing conditions, up101 cardiomyocytes appear shorter and stiffer than control cardiomyocytes due to elevated numbers of basally cycling myosin cross-bridges.
The up101 troponin-T mutation disrupts the thin filament conformational state that suppresses myosin activity under low Ca2+ conditions.
Troponin-T mutations result in heterogeneous and diverse myopathies and in vivo models of such disorders could facilitate understanding of the underlying pathophysiology. This study describes an integrative approach to characterize the effects of the up101 mutation on the Drosophila heart. We show how a troponin-T lesion, adjacent to the evolutionarily conserved and highly charged N-terminal tropomyosin-binding element, perturbs muscle regulation, promotes contractile dysinhibition and diastolic dysfunction and drives cardiac remodeling. Our results highlight the potential significance of troponin-T-tropomyosin electrostatic associations for proper contractile regulation. Human cardiomyopathy mutations that localize in, and close to, the well-conserved tropomyosin-binding domain of troponin-T may also alter thin filament regulatory conformational states and thereby contribute to activation of pathological remodeling cascades.
Acknowledgments
We thank Douglas Deutschman, Ph.D. (San Diego State University) for help with statistical analysis, Georg Vogler, Ph.D. (Sanford Burnham Medical Research Institute) for artwork assistance and Anna Blice-Baum, Ph.D. (Johns Hopkins University) for technical assistance.
SOURCES OF FUNDING
This work was supported by NIH T32HL105373 and American Heart Association 13PRE14410037 (to GK), by NIH R21HL106529 and DP02OD006460 (to AJE), by NIH R37-036153 (to WL), and by American Heart Association 10SDG4180089 and an American Federation for Aging Research Grant (to AC).
Nonstandard Abbreviations and Acronyms
- Tn
troponin
- Tm
tropomyosin
- TnC
troponin-C
- TnI
troponin-I
- TnT
troponin-T
- TnT1
N-terminal fragment of troponin-T
- HCM
hypertrophic cardiomyopathy
- DCM
dilated cardiomyopathy
- RCM
restrictive cardiomyopathy
- Cs
Canton-S
- up101
upheld101
- IFM
indirect flight muscle
- AH
artificial hemolymph
- EGTA
ethylene glycol tetraacetic acid
- B-state
blocked state
- C-state
closed state
- M-state
open-state
- CC
conical chamber
- AFM
atomic force microscopy
Footnotes
Disclosures
None.
References
- 1.Tobacman LS. Thin filament-mediated regulation of cardiac contraction. Annu Rev Physiol. 1996;58:447–481. doi: 10.1146/annurev.ph.58.030196.002311. [DOI] [PubMed] [Google Scholar]
- 2.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 3.Lehman W, Craig R. Tropomyosin and the steric mechanism of muscle regulation. Adv Exp Med Biol. 2008;644:95–109. doi: 10.1007/978-0-387-85766-4_8. [DOI] [PubMed] [Google Scholar]
- 4.McKillop DF, Geeves MA. Regulation of the interaction between actin and myosin subfragment 1: Evidence for three states of the thin filament. Biophys J. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vibert P, Craig R, Lehman W. Steric-model for activation of muscle thin filaments. J Mol Biol. 1997;266:8–14. doi: 10.1006/jmbi.1996.0800. [DOI] [PubMed] [Google Scholar]
- 6.Lehman W, Craig R, Vibert P. Ca2+-induced tropomyosin movement in Limulus thin filaments revealed by three-dimensional reconstruction. Nature. 1994;368:65–67. doi: 10.1038/368065a0. [DOI] [PubMed] [Google Scholar]
- 7.Lehman W, Vibert P, Uman P, Craig R. Steric-blocking by tropomyosin visualized in relaxed vertebrate muscle thin filaments. J Mol Biol. 1995;251:191–196. doi: 10.1006/jmbi.1995.0425. [DOI] [PubMed] [Google Scholar]
- 8.Lehrer SS, Geeves MA. The muscle thin filament as a classical cooperative/allosteric regulatory system. J Mol Biol. 1998;277:1081–1089. doi: 10.1006/jmbi.1998.1654. [DOI] [PubMed] [Google Scholar]
- 9.Maytum R, Westerdorf B, Jaquet K, Geeves MA. Differential regulation of the actomyosin interaction by skeletal and cardiac troponin isoforms. J Biol Chem. 2003;278:6696–6701. doi: 10.1074/jbc.M210690200. [DOI] [PubMed] [Google Scholar]
- 10.Pirani A, Xu C, Hatch V, Craig R, Tobacman LS, Lehman W. Single particle analysis of relaxed and activated muscle thin filaments. J Mol Biol. 2005;346:761–772. doi: 10.1016/j.jmb.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 11.Perry SV. Troponin T: Genetics, properties and function. J Muscle Res Cell Motil. 1998;19:575–602. doi: 10.1023/a:1005397501968. [DOI] [PubMed] [Google Scholar]
- 12.Wei B, Jin JP. Troponin T isoforms and posttranscriptional modifications: Evolution, regulation and function. Arch Biochem Biophys. 2011;505:144–154. doi: 10.1016/j.abb.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson P, Amphlett GW, Perry SV. The primary structure of troponin T and the interaction with tropomyosin. Biochem J. 1975;151:85–97. doi: 10.1042/bj1510085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pearlstone JR, Smillie LB. The binding site of skeletal alpha-tropomyosin on troponin-T. Can J Biochem. 1977;55:1032–1038. doi: 10.1139/o77-154. [DOI] [PubMed] [Google Scholar]
- 15.Hill LE, Mehegan JP, Butters CA, Tobacman LS. Analysis of troponin-tropomyosin binding to actin. Troponin does not promote interactions between tropomyosin molecules. J Biol Chem. 1992;267:16106–16113. [PubMed] [Google Scholar]
- 16.Schaertl S, Lehrer SS, Geeves MA. Separation and characterization of the two functional regions of troponin involved in muscle thin filament regulation. Biochemistry. 1995;34:15890–15894. doi: 10.1021/bi00049a003. [DOI] [PubMed] [Google Scholar]
- 17.Hinkle A, Goranson A, Butters CA, Tobacman LS. Roles for the troponin tail domain in thin filament assembly and regulation. A deletional study of cardiac troponin T. J Biol Chem. 1999;274:7157–7164. doi: 10.1074/jbc.274.11.7157. [DOI] [PubMed] [Google Scholar]
- 18.Tobacman LS, Nihli M, Butters C, Heller M, Hatch V, Craig R, Lehman W, Homsher E. The troponin tail domain promotes a conformational state of the thin filament that suppresses myosin activity. J Biol Chem. 2002;277:27636–27642. doi: 10.1074/jbc.M201768200. [DOI] [PubMed] [Google Scholar]
- 19.Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O’Donoghue A, Spirito P, Matsumori A, Moravec CS, Seidman JG. Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med. 1995;332:1058–1064. doi: 10.1056/NEJM199504203321603. [DOI] [PubMed] [Google Scholar]
- 20.Willott RH, Gomes AV, Chang AN, Parvatiyar MS, Pinto JR, Potter JD. Mutations in troponin that cause HCM, DCM and RCM: What can we learn about thin filament function? J Mol Cell Cardiol. 2010;48:882–892. doi: 10.1016/j.yjmcc.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 21.Tardiff JC. Thin filament mutations: Developing an integrative approach to a complex disorder. Circ Res. 2011;108:765–782. doi: 10.1161/CIRCRESAHA.110.224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mogensen J, Murphy RT, Shaw T, Bahl A, Redwood C, Watkins H, Burke M, Elliott PM, McKenna WJ. Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2004;44:2033–2040. doi: 10.1016/j.jacc.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 23.Stefanelli CB, Rosenthal A, Borisov AB, Ensing GJ, Russell MW. Novel troponin T mutation in familial dilated cardiomyopathy with gender-dependant severity. Mol Genet Metab. 2004;83:188–196. doi: 10.1016/j.ymgme.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 24.Peddy SB, Vricella LA, Crosson JE, Oswald GL, Cohn RD, Cameron DE, Valle D, Loeys BL. Infantile restrictive cardiomyopathy resulting from a mutation in the cardiac troponin T gene. Pediatrics. 2006;117:1830–1833. doi: 10.1542/peds.2005-2301. [DOI] [PubMed] [Google Scholar]
- 25.Menon SC, Michels VV, Pellikka PA, Ballew JD, Karst ML, Herron KJ, Nelson SM, Rodeheffer RJ, Olson TM. Cardiac troponin T mutation in familial cardiomyopathy with variable remodeling and restrictive physiology. Clin Genet. 2008;74:445–454. doi: 10.1111/j.1399-0004.2008.01062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaski JP, Syrris P, Burch M, Tome-Esteban MT, Fenton M, Christiansen M, Andersen PS, Sebire N, Ashworth M, Deanfield JE, McKenna WJ, Elliott PM. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart. 2008 doi: 10.1136/hrt.2007.134684. [DOI] [PubMed] [Google Scholar]
- 27.Bernstein SI, O’Donnell PT, Cripps RM. Molecular genetic analysis of muscle development, structure, and function in Drosophila. Int Rev Cytol. 1993;143:63–152. doi: 10.1016/s0074-7696(08)61874-4. [DOI] [PubMed] [Google Scholar]
- 28.Cammarato A, Ahrens CH, Alayari NN, Qeli E, Rucker J, Reedy MC, Zmasek CM, Gucek M, Cole RN, Van Eyk JE, Bodmer R, O’Rourke B, Bernstein SI, Foster DB. A mighty small heart: The cardiac proteome of adult Drosophila melanogaster. PLoS One. 2011;6:e18497. doi: 10.1371/journal.pone.0018497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cammarato A, Hatch V, Saide J, Craig R, Sparrow JC, Tobacman LS, Lehman W. Drosophila muscle regulation characterized by electron microscopy and three-dimensional reconstruction of thin filament mutants. Biophys J. 2004;86:1618–1624. doi: 10.1016/S0006-3495(04)74229-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolf MJ, Amrein H, Izatt JA, Choma MA, Reedy MC, Rockman HA. Drosophila as a model for the identification of genes causing adult human heart disease. Proc Natl Acad Sci U S A. 2006;103:1394–1399. doi: 10.1073/pnas.0507359103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cammarato A, Dambacher CM, Knowles AF, Kronert WA, Bodmer R, Ocorr K, Bernstein SI. Myosin transducer mutations differentially affect motor function, myofibril structure, and the performance of skeletal and cardiac muscles. Mol Biol Cell. 2008;19:553–562. doi: 10.1091/mbc.E07-09-0890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Homyk T, Jr, Szidonya J, Suzuki DT. Behavioral mutants of Drosophila melanogaster. III. Isolation and mapping of mutations by direct visual observations of behavioral phenotypes. Mol Gen Genet. 1980;177:553–565. doi: 10.1007/BF00272663. [DOI] [PubMed] [Google Scholar]
- 33.Fyrberg E, Fyrberg CC, Beall C, Saville DL. Drosophila melanogaster troponin-T mutations engender three distinct syndromes of myofibrillar abnormalities. J Mol Biol. 1990;216:657–675. doi: 10.1016/0022-2836(90)90390-8. [DOI] [PubMed] [Google Scholar]
- 34.Alayari NN, Vogler G, Taghli-Lamallem O, Ocorr K, Bodmer R, Cammarato A. Fluorescent labeling of Drosophila heart structures. J Vis Exp. 2009 doi: 10.3791/1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vogler G, Ocorr K. Visualizing the beating heart in Drosophila. J Vis Exp. 2009 doi: 10.3791/1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ocorr K, Reeves NL, Wessells RJ, Fink M, Chen HS, Akasaka T, Yasuda S, Metzger JM, Giles W, Posakony JW, Bodmer R. KCNQ potassium channel mutations cause cardiac arrhythmias in Drosophila that mimic the effects of aging. Proc Natl Acad Sci U S A. 2007;104:3943–3948. doi: 10.1073/pnas.0609278104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fink M, Callol-Massot C, Chu A, Ruiz-Lozano P, Izpisua Belmonte JC, Giles W, Bodmer R, Ocorr K. A new method for detection and quantification of heartbeat parameters in Drosophila, zebrafish, and embryonic mouse hearts. Biotechniques. 2009;46:101–113. doi: 10.2144/000113078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaushik G, Fuhrmann A, Cammarato A, Engler Adam J. In situ mechanical analysis of myofibrillar perturbation and aging on soft, bilayered Drosophila myocardium. Biophysical journal. 2011;101:2629–2637. doi: 10.1016/j.bpj.2011.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vibert P, Craig R, Lehman W. Three-dimensional reconstruction of caldesmon-containing smooth muscle thin filaments. J Cell Biol. 1993;123:313–321. doi: 10.1083/jcb.123.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Owen CH, Morgan DG, DeRosier DJ. Image analysis of helical objects: The Brandeis helical package. J Struct Biol. 1996;116:167–175. doi: 10.1006/jsbi.1996.0027. [DOI] [PubMed] [Google Scholar]
- 41.Milligan RA, Flicker PF. Structural relationships of actin, myosin, and tropomyosin revealed by cryo-electron microscopy. J Cell Biol. 1987;105:29–39. doi: 10.1083/jcb.105.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trachtenberg S, DeRosier DJ. Three-dimensional structure of the frozen-hydrated flagellar filament. The left-handed filament of Salmonella typhimurium. J Mol Biol. 1987;195:581–601. doi: 10.1016/0022-2836(87)90184-7. [DOI] [PubMed] [Google Scholar]
- 43.Li Y, Mui S, Brown JH, Strand J, Reshetnikova L, Tobacman LS, Cohen C. The crystal structure of the C-terminal fragment of striated-muscle alpha-tropomyosin reveals a key troponin T recognition site. Proc Natl Acad Sci U S A. 2002;99:7378–7383. doi: 10.1073/pnas.102179999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hinkle A, Tobacman LS. Folding and function of the troponin tail domain. Effects of cardiomyopathic troponin T mutations. J Biol Chem. 2003;278:506–513. doi: 10.1074/jbc.M209194200. [DOI] [PubMed] [Google Scholar]
- 45.Jin JP, Chong SM. Localization of the two tropomyosin-binding sites of troponin T. Arch Biochem Biophys. 2010;500:144–150. doi: 10.1016/j.abb.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manning EP, Guinto PJ, Tardiff JC. Correlation of molecular and functional effects of mutations in cardiac troponin T linked to familial hypertrophic cardiomyopathy: An integrative in silico/in vitro approach. J Biol Chem. 2012;287:14515–14523. doi: 10.1074/jbc.M111.257436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller A. The internal anatomy and histology of the imago of Drosophila melanogaster. Biology of Drosophila. 1950 [Google Scholar]
- 48.Rizki TM. The circulatory system and associated cells and tissues. The genetics and biology of Drosophila. 1978;2b:397–452. [Google Scholar]
- 49.Limouze J, Straight AF, Mitchison T, Sellers JR. Specificity of blebbistatin, an inhibitor of myosin II. J Muscle Res Cell Motil. 2004;25:337–341. doi: 10.1007/s10974-004-6060-7. [DOI] [PubMed] [Google Scholar]
- 50.Fedorov VV, Lozinsky IT, Sosunov EA, Anyukhovsky EP, Rosen MR, Balke CW, Efimov IR. Application of blebbistatin as an excitation-contraction uncoupler for electrophysiologic study of rat and rabbit hearts. Heart Rhythm. 2007;4:619–626. doi: 10.1016/j.hrthm.2006.12.047. [DOI] [PubMed] [Google Scholar]
- 51.Lin D, Bobkova A, Homsher E, Tobacman LS. Altered cardiac troponin T in vitro function in the presence of a mutation implicated in familial hypertrophic cardiomyopathy. J Clin Invest. 1996;97:2842–2848. doi: 10.1172/JCI118740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morimoto S, Yanaga F, Minakami R, Ohtsuki I. Ca2+-sensitizing effects of the mutations at Ile-79 and Arg-92 of troponin T in hypertrophic cardiomyopathy. Am J Physiol. 1998;275:C200–207. doi: 10.1152/ajpcell.1998.275.1.C200. [DOI] [PubMed] [Google Scholar]
- 53.Palm T, Graboski S, Hitchcock-DeGregori SE, Greenfield NJ. Disease-causing mutations in cardiac troponin T: Identification of a critical tropomyosin-binding region. Biophys J. 2001;81:2827–2837. doi: 10.1016/S0006-3495(01)75924-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harada K, Potter JD. Familial hypertrophic cardiomyopathy mutations from different functional regions of troponin T result in different effects on the pH and Ca2+ sensitivity of cardiac muscle contraction. J Biol Chem. 2004;279:14488–14495. doi: 10.1074/jbc.M309355200. [DOI] [PubMed] [Google Scholar]
- 55.Ertz-Berger BR, He H, Dowell C, Factor SM, Haim TE, Nunez S, Schwartz SD, Ingwall JS, Tardiff JC. Changes in the chemical and dynamic properties of cardiac troponin T cause discrete cardiomyopathies in transgenic mice. Proc Natl Acad Sci U S A. 2005;102:18219–18224. doi: 10.1073/pnas.0509181102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fisher D, Wang G, Tobacman LS. NH2-terminal truncation of skeletal muscle troponin T does not alter the Ca2+ sensitivity of thin filament assembly. J Biol Chem. 1995;270:25455–25460. doi: 10.1074/jbc.270.43.25455. [DOI] [PubMed] [Google Scholar]
- 57.Guinto PJ, Manning EP, Schwartz SD, Tardiff JC. Computational characterization of mutations in cardiac troponin T known to cause familial hypertrophic cardiomyopathy. Journal of Theoretical and Computational Chemistry. 2007;06:413–419. doi: 10.1142/S0219633607003271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Manning EP, Tardiff JC, Schwartz SD. Molecular effects of familial hypertrophic cardiomyopathy-related mutations in the TnT1 domain of cTnT. J Mol Biol. 2012;421:54–66. doi: 10.1016/j.jmb.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chandra M, Tschirgi ML, Tardiff JC. Increase in tension-dependent ATP consumption induced by cardiac troponin T mutation. Am J Physiol Heart Circ Physiol. 2005;289:H2112–2119. doi: 10.1152/ajpheart.00571.2005. [DOI] [PubMed] [Google Scholar]
- 60.Kass DA, Bronzwaer JG, Paulus WJ. What mechanisms underlie diastolic dysfunction in heart failure? Circ Res. 2004;94:1533–1542. doi: 10.1161/01.RES.0000129254.25507.d6. [DOI] [PubMed] [Google Scholar]
- 61.Borbely A, Papp Z, Edes I, Paulus WJ. Molecular determinants of heart failure with normal left ventricular ejection fraction. Pharmacol Rep. 2009;61:139–145. doi: 10.1016/s1734-1140(09)70016-7. [DOI] [PubMed] [Google Scholar]
- 62.Campbell KS, Patel JR, Moss RL. Cycling cross-bridges increase myocardial stiffness at submaximal levels of Ca2+ activation. Biophys J. 2003;84:3807–3815. doi: 10.1016/S0006-3495(03)75108-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.King NM, Methawasin M, Nedrud J, Harrell N, Chung CS, Helmes M, Granzier H. Mouse intact cardiac myocyte mechanics: Cross-bridge and titin-based stress in unactivated cells. J Gen Physiol. 2011;137:81–91. doi: 10.1085/jgp.201010499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Davis J, Wen H, Edwards T, Metzger JM. Thin filament disinhibition by restrictive cardiomyopathy mutant R193H troponin I induces Ca2+-independent mechanical tone and acute myocyte remodeling. Circ Res. 2007;100:1494–1502. doi: 10.1161/01.RES.0000268412.34364.50. [DOI] [PubMed] [Google Scholar]
- 65.Bai F, Weis A, Takeda AK, Chase PB, Kawai M. Enhanced active cross-bridges during diastole: Molecular pathogenesis of tropomyosin’s HCM mutations. Biophys J. 2011;100:1014–1023. doi: 10.1016/j.bpj.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McCain ML, Parker KK. Mechanotransduction: The role of mechanical stress, myocyte shape, and cytoskeletal architecture on cardiac function. Pflugers Arch. 2011;462:89–104. doi: 10.1007/s00424-011-0951-4. [DOI] [PubMed] [Google Scholar]
- 67.Kaushik G, Zambon AC, Fuhrmann A, Bernstein SI, Bodmer R, Engler AJ, Cammarato A. Measuring passive myocardial stiffness in Drosophila melanogaster to investigate diastolic dysfunction. J Cell Mol Med. 2012;16:1656–1662. doi: 10.1111/j.1582-4934.2011.01517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chalovich JM. Disease causing mutations of troponin alter regulated actin state distributions. J Muscle Res Cell Motil. 2012;33:493–499. doi: 10.1007/s10974-012-9305-x. [DOI] [PubMed] [Google Scholar]
- 69.Haim TE, Dowell C, Diamanti T, Scheuer J, Tardiff JC. Independent FHC-related cardiac troponin T mutations exhibit specific alterations in myocellular contractility and calcium kinetics. J Mol Cell Cardiol. 2007;42:1098–1110. doi: 10.1016/j.yjmcc.2007.03.906. [DOI] [PubMed] [Google Scholar]
- 70.Guinto PJ, Haim TE, Dowell-Martino CC, Sibinga N, Tardiff JC. Temporal and mutation-specific alterations in Ca2+ homeostasis differentially determine the progression of cTnT-related cardiomyopathies in murine models. Am J Physiol Heart Circ Physiol. 2009;297:H614–626. doi: 10.1152/ajpheart.01143.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lin N, Badie N, Yu L, Abraham D, Cheng H, Bursac N, Rockman HA, Wolf MJ. A method to measure myocardial calcium handling in adult Drosophila. Circ Res. 2011;108:1306–1315. doi: 10.1161/CIRCRESAHA.110.238105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moss RL, Razumova M, Fitzsimons DP. Myosin crossbridge activation of cardiac thin filaments: Implications for myocardial function in health and disease. Circ Res. 2004;94:1290–1300. doi: 10.1161/01.RES.0000127125.61647.4F. [DOI] [PubMed] [Google Scholar]
- 73.Hinken AC, Solaro RJ. A dominant role of cardiac molecular motors in the intrinsic regulation of ventricular ejection and relaxation. Physiology (Bethesda) 2007;22:73–80. doi: 10.1152/physiol.00043.2006. [DOI] [PubMed] [Google Scholar]
- 74.Tardiff JC, Hewett TE, Palmer BM, Olsson C, Factor SM, Moore RL, Robbins J, Leinwand LA. Cardiac troponin T mutations result in allele-specific phenotypes in a mouse model for hypertrophic cardiomyopathy. J Clin Invest. 1999;104:469–481. doi: 10.1172/JCI6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.James J, Zhang Y, Osinska H, Sanbe A, Klevitsky R, Hewett TE, Robbins J. Transgenic modeling of a cardiac troponin I mutation linked to familial hypertrophic cardiomyopathy. Circ Res. 2000;87:805–811. doi: 10.1161/01.res.87.9.805. [DOI] [PubMed] [Google Scholar]
- 76.Du CK, Morimoto S, Nishii K, Minakami R, Ohta M, Tadano N, Lu QW, Wang YY, Zhan DY, Mochizuki M, Kita S, Miwa Y, Takahashi-Yanaga F, Iwamoto T, Ohtsuki I, Sasaguri T. Knock-in mouse model of dilated cardiomyopathy caused by troponin mutation. Circ Res. 2007;101:185–194. doi: 10.1161/CIRCRESAHA.106.146670. [DOI] [PubMed] [Google Scholar]
- 77.Ahmad F, Banerjee SK, Lage ML, Huang XN, Smith SH, Saba S, Rager J, Conner DA, Janczewski AM, Tobita K, Tinney JP, Moskowitz IP, Perez-Atayde AR, Keller BB, Mathier MA, Shroff SG, Seidman CE, Seidman JG. The role of cardiac troponin T quantity and function in cardiac development and dilated cardiomyopathy. PLoS One. 2008;3:e2642. doi: 10.1371/journal.pone.0002642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Murakami K, Stewart M, Nozawa K, Tomii K, Kudou N, Igarashi N, Shirakihara Y, Wakatsuki S, Yasunaga T, Wakabayashi T. Structural basis for tropomyosin overlap in thin (actin) filaments and the generation of a molecular swivel by troponin-T. Proc Natl Acad Sci U S A. 2008;105:7200–7205. doi: 10.1073/pnas.0801950105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.