

Abstract
Understanding the interaction between fear and reward at the circuit and molecular levels has implications for basic scientific approaches to memory and for understanding the etiology of psychiatric disorders. Both stress and exposure to drugs of abuse induce epigenetic changes that result in persistent behavioral changes, some of which may contribute to the formation of a drug addiction or a stress-related psychiatric disorder. Converging evidence suggests that similar behavioral, neurobiological and molecular mechanisms control the extinction of learned fear and drug-seeking responses. This may, in part, account for the fact that individuals with post-traumatic stress disorder have a significantly elevated risk of developing a substance use disorder and have high rates of relapse to drugs of abuse, even after long periods of abstinence. At the behavioral level, a major challenge in treatments is that extinguished behavior is often not persistent, returning with changes in context, the passage of time or exposure to mild stressors. A common goal of treatments is therefore to weaken the ability of stressors to induce relapse. With the discovery of epigenetic mechanisms that create persistent molecular signals, recent work on extinction has focused on how modulating these epigenetic targets can create lasting extinction of fear or drug-seeking behavior. Here, we review recent evidence pointing to common behavioral, systems and epigenetic mechanisms in the regulation of fear and drug seeking. We suggest that targeting these mechanisms in combination with behavioral therapy may promote treatment and weaken stress-induced relapse.
Keywords: Addiction, amygdala, behavior, DNA methylation, epigenetics, extinction, fear conditioning, histone acetylation, medial prefrontal cortex, PTSD, substance abuse
A large body of evidence shows that prolonged stress or even a single traumatic experience can lead to neurobiological and behavioral changes that are persistent. This has many deleterious long-term consequences and can lead to the development of post-traumatic stress disorder (PTSD). Post-traumatic stress disorder is associated with epigenetic changes that result in altered gene expression and subsequent protein synthesis. These epigenetic changes may account for the development of the disease itself in the form of exaggerated fear responses to neutral or mild stimuli and may render individuals more susceptible to the use and subsequent dependence on drugs of abuse. Substance use disorders (SUDs) similarly evoke persistent changes in gene expression that are mediated by epigenetic mechanisms. Because both PTSD and SUDs involve a memory component and an inability to extinguish unwanted behavioral responses, similar epigenetic changes may underlie long-term memory effects in both anxiety disorders and addiction.
Because of the overlap in neurobiological circuits and epigenetic mechanisms that mediate stress, fear and substance abuse, there is a high likelihood that activating this circuit even long after trauma may cause relapse of fear, anxiety or drug-seeking behavior. There is an emerging consensus that the best treatment strategies engage the circuits involved in behavioral inhibition, coupled with pharmacological manipulations designed to target those circuits, with the goal of creating lasting behavioral inhibition. Recent attention has focused on how these circuits and signals overlap during the reactivation and suppression of different memories. An ultimate goal of research on these processes is to understand how appetitive and aversive memories interact, and what causes stressful experiences to induce relapse even after long periods of abstinence. We focus this review on some of those interactions, emphasizing how an epigenetic approach to memory suppression may be a particularly useful avenue to pursue in designing treatments for disorders that involve failures of inhibition, such as PTSD and substance abuse.
Stress and post-traumatic stress disorder
Post-traumatic stress disorder is classified as an axis I anxiety disorder that develops after an individual experiences a chronic or acute stressor, one that usually involves the threat of serious injury or death to oneself or to someone nearby (American Psychiatric Association 2013). PTSD is accompanied by a constellation of symptoms including hyperarousal, avoidance, sleep disturbances and difficulty concentrating, the hallmark of which is a re-experiencing of the traumatic event during which the individual relives the traumatic experience despite no longer being in danger. At its simplest, PTSD can be conceptualized as a disturbance of the hypothalamic-pituitary-adrenal (HPA) axis, which governs the stress response.
The HPA axis has long been known to regulate cortisol (CORT) release in response to stressful stimuli. Classically, this has been described in the context of ‘fight or flight’ where an organism must make the decision to flee from a stressful stimulus (a predator, for example) or to stay and fight. In both scenarios, the organism requires a cascade of metabolic processes that will provide the necessary energy to fight or flee. Exposure to a stressful stimulus induces the release of corticotropin-releasing factor (CRF) from the paraventricular nucleus of hypothalamus, which in turn promotes the release of adrenocorticotropic hormone (ACTH) from the pituitary gland, which then acts peripherally on the adrenal glands to induce the release of glucocorticoids. In humans, the primary stress hormone is known as cortisol, whereas in rodents it is known as corticosterone. In both species, glucocorticoids activate both mineralocorticoid (MR) and glucocorticoid receptors (GR), which results in a cell signaling cascade that varies depending on which receptor has been activated. One of the primary roles of the glucocorticoid receptors is to regulate a negative feedback loop, whereby the activation of the receptors results in the termination of the production of CRF, and subsequently ACTH and cortisol itself.
The relationship between PTSD and the HPA is not as clear as was once presumed. Initial hypotheses postulated that a hyperactive stress response, in which encounters with mild stimuli resulted in a robust stress response, was the primary effector driving the disease. This idea has been countered by studies finding significantly decreased urinary levels of cortisol in individuals with PTSD (Glover & Poland 2002; Yehuda et al. 1990, 1995), significant increases in urinary cortisol (LeMieux & Coe 1995; Pitman & Orr 1990) or no significant changes in cortisol levels between individuals with PTSD and controls (Baker et al. 1999). These differences are not surprising when the individual variability associated with cortisol levels is considered. Factors such as age, weight, genetic background, circulating levels of cortisol-binding protein (CBP; which binds cortisol and renders it inactive), smoking status, age and even mood can affect circulating cortisol (Yehuda 2006). It is therefore not a simple hyperactivation of the HPA in response to stressful stimuli that leads to the development of PTSD.
One finding that does appear fairly consistent, however, is that individuals with PTSD have higher circulating and cerebrospinal fluid (CSF) levels of CRF (Bremner & Vermetten 2001; Bremner et al. 1997). This finding is consistent with other mood disorders, like depression, which is a component of/highly comorbid with PTSD, in which CRF is also elevated (Nemeroff et al. 1984). These findings suggest that PTSD may develop as a result of alterations in glucocorticoid receptor responsiveness (Yehuda 2006). A common endocrinological test of HPA activity is the dexamethasone (DEX) challenge. DEX is a synthetic cortisol, and administration activates the GRs that are responsible for negative feedback, which, in healthy individuals, suppresses cortisol levels. In response to DEX, individuals with PTSD show an increased suppression of cortisol, which suggests a hyperresponsiveness of GRs (Rinne et al. 2002; Stein et al. 1997; Yehuda 2002; Yehuda & Antelman 1993). This is in stark contrast to studies on both acute and chronic stress, where levels of CRF and cortisol are elevated, and individuals show a decrease in cortisol suppression in response to DEX (Holsboer 2003). The exact mechanism through which this increased GR responsiveness leads to or contributes to the pathology observed in PTSD, however, remains unclear. Some evidence suggests that alterations in the HPA precede the traumatic experience and may explain why certain individuals develop PTSD in response to a traumatic event while others do not (Yehuda 2002).
It is therefore apparent that one of the difficulties in treating PTSD is the underlying complexity in the response of the HPA axis in those patients. In patients that do develop PTSD, the underlying challenge is to determine why the emotional aftereffects of the memory persist and what can be done to weaken this long-term impact. Over time, PTSD can lead to persistent issues with stress and arousal that affect other systems. These include long-term alterations in responsiveness to stressful stimuli and memory reactivation by a non-specific stress response. Following a traumatic incident, individuals with PTSD re-experience the event, which in turn induces a re-experiencing of the stress and fear associated with the original trauma (Shvil et al. 2013). As a consequence of this experience, PTSD patients will often self-medicate or relapse even after long periods of abstinence (Bradizza et al. 2006).
The problem of addiction and relapse in PTSD
One of the major challenges in developing effective treatments for PTSD is the high level of comorbidity between PTSD and SUDs; among people with lifetime PTSD, lifetime SUD is estimated at 21–43%, compared with 8–25% in those without PTSD (Jacobsen et al. 2001). Individuals with PTSD are more likely to develop SUDs (Kofoed et al. 1993) and once addicted, are more likely to relapse (Brown et al. 1996) than are unaffected individuals. This is especially troubling because those circuits and cellular signals that are involved in stress and memory are also involved in substance abuse (reviewed in Tipps et al. 2014). Indeed, many authors have pointed out that addiction is a disorder of learning and memory, in which normal mechanisms in place to incorporate new information from the environment are hijacked by drugs of abuse (e.g. Hyman et al. 2006). This results in contexts evoking drug cravings, owing to the repeated intake of drugs in the presence of certain contextual cues (e.g. physical, social and temporal contexts). Drugs of abuse can also maintain anxious avoidance responses, preventing the extinction of anxiety in these situations, thus creating a vicious cycle in which problematic behaviors are maintained and even strengthened by both positive and negative reinforcement contingencies. As with treatment of the hyperemotional content of memories, the treatments for addiction are also difficult because the effects of drug abuse can persist even across long periods of abstinence. PTSD also produces persistent changes that result in unwanted behavioral responses. Individuals who experience a stressor or series of stressors in a specific environment demonstrate inappropriate fear responses in otherwise innocuous environments in response to stimuli that remind the individual of the traumatic event. For example, a veteran may hear the crash of a wrecking ball tearing down a nearby building and be reminded of the sounds associated with his or her experience in a combat zone. This often results in a re-experiencing of the memory associated with the original trauma, which leads to an exaggerated fear response. Therefore, both PTSD and addiction are examples of learning paradigms in which long-term memories related to discrete events persist over time and context. Thus, treatments of both PTSD and substance abuse require an understanding of the mechanisms that make memories long-lasting and protective against disruption.
One potential mechanism through which stress may induce relapse in individuals with PTSD is through the interaction of glucocorticoids and mesolimbic dopamine neurons. The mesolimbic dopamine system constitutes the brains primary reward circuitry, and nearly all drugs of abuse cause increases in dopamine in the nucleus accumbens (NAc), a primary component of this circuit. Glucocorticoids can activate dopamine neurons by binding directly to them and inducing excitation patterns similar to those observed with drugs of abuse (Saal et al. 2003). Therefore, stress is able to induce a state of drug seeking even when drugs of abuse are not present by stimulating similar neural pathways. Corticotropin-releasing factor antagonists are able to prevent reinstatement to footshock in self-administration studies, suggesting that CRF is a critical mediator of stress-induced reinstatement (Shaham et al. 1998).
As previously discussed, cortisol levels do not always increase in individuals with PTSD, and in fact may stay the same or decrease. One explanation for this is the glucocorticoid insensitivity hypothesis, which defines insensitivity as any state in which the normal negative feedback mechanisms fail to restrain the stress response (Raison & Miller 2003). This may be through hypocortisolism, where levels of cortisol are decreased, or reduced GR sensitivity. When levels of cortisol remain unrestrained, individuals experience high levels of stress, and this may trigger relapse to drugs of abuse. For example, Sinha et al. (1999) found that psychological stress induced craving for cocaine in human addicts. In animals, a wide range of stressful stimuli have been shown to induce reinstatement of responding for drugs of abuse (Shaham et al. 2000; Shalev et al. 2000).
Thus, a major goal of treatment interventions for PTSD and substance abuse is to weaken the ability of external and internal cues to evoke stress and/or drug seeking. A growing literature suggests that behavioral intervention paired with a pharmacological approach that targets the molecular mechanisms involved in stressful and drug-related memory formation may lead to a weakening of the behavior evoked by those memories (e.g. Davis et al. 2006). It is abundantly clear that a key aspect of these treatments is a behavioral component that relies on principles of extinction.
Behavioral extinction as a therapeutic
At the level of behavior, treatment interventions for PTSD and substance abuse have focused on exposure therapy – presenting patients with cues associated with trauma or drug seeking, and allowing the responses evoked by those cues (anxiety or craving) to occur and extinguish as the expected outcome does not occur. This basic principle of exposure therapy is similar regardless of whether the treatment is directed toward behaviors that produce a positive outcome (attenuation of substance abuse) or remove a negative outcome (anxiety reduction), and there are numerous demonstrations of the power of extinction to change maladaptive behavior that occurs in a variety of disorders (e.g. Campbell 2003).
Despite some success of exposure therapy based on extinction processes (e.g. Wachen et al. 2014), there are several challenges that can lead to poor clinical outcomes in the treatment of PTSD or substance abuse. First, extinction is known to suppress behavior (fear, avoidance, drug seeking) without necessarily altering the content of the original memory. This means that that memory can still influence future behavior given the right circumstances (see Delamater & Westbrook 2014). Second, the changes that occur during extinction may not be long lasting; extinguished behavior returns with changes in context, with reminders of the original association, and after the simple passage of time. This means that enhancements in extinction that may occur within a therapeutic setting may not persist outside of that treatment context. This is problematic, of course, because patients are likely to encounter cues associated with trauma or drug taking within their daily lives outside of the therapist’s office. This is especially true in the case of addiction, in which, for example, an alcoholic may experience intense cravings when walking by the neighborhood bar or a nicotine addict may crave a cigarette with a morning cup of coffee. Third, exposure therapy involves the mental re-experiencing of anxiety or cravings and this type of experience may be aversive, which may decrease the likelihood that a patient will return to treatment sessions. Thus, there is a need for interventions that promote extinction, making it occur more rapidly and persist outside of the treatment context.
A fourth challenge that is beginning to emerge in thinking about models for PTSD is that extremely stressful experiences have deleterious effects on the long-term efficacy of extinction (e.g. Knox et al. 2012; Rau & Fanselow 2009). This means that much of what we have come to understand about extinction after simple conditioning may not reflect what is actually happening in PTSD, further underscoring the need for different models and approaches to promoting extinction.
Common extinction circuits in fear and substance abuse
As noted above, a survey of the literature reveals that similar extinction processes appear to operate independent of the type of outcome that is involved in conditioning the original behavior. Unmasking phenomena associated with extinction (such as spontaneous recovery, contextual renewal and outcome-induced reinstatement) are ubiquitous, having been demonstrated in procedures with outcomes such as nausea, psychostimulants, alcohol, food, and shock, to name a few. It is therefore not surprising that there appears to be some common neurobiological circuits that underlie the development and maintenance of extinction.
Early work with lesions and temporary inactivation demonstrated a role for the medial prefrontal cortex (mPFC) in the expression and extinction of fear (e.g. Morgan et al. 1993). Since then, many studies have extended these findings to other preparations, including extinction of drug seeking in conditioned place preference (CPP; e.g. Groblewski et al. 2012) and self-administration paradigms with drugs of abuse (LaLumiere et al. 2010). There is also increasing evidence that the mPFC may become dysregulated with prolonged stress (Knox et al. 2010, 2012). The specific circuits regulating extinction within the mPFC and between the mPFC and other structures are still being identified and may differ as a function of strain and species (e.g. Camp et al. 2012; Chang & Maren 2010; MacPherson et al. 2013). Nonetheless, some general properties are emerging.
Subregions of the mPFC are involved in regulating both the expression and extinction of fear. Although there is clear overlap in function, it is generally thought that the infralimbic (IL) region is involved in the development and consolidation of extinction, whereas the prelimbic (PL) region is involved in the expression and contextual modulation of fear (Laurent & Westbrook, 2009; Sharpe & Killcross, 2015). Effects on fear modulation are due to excitatory glutamatergic projections from the mPFC to the amygdala (Krettek & Price 1977; Paré & Smith 1994). The more dorsally located PL mPFC sends projections to the basal amygdala (BA), which then synapses onto the central amygdala (CeA), which promotes fear behavior through projections to the periaqueductal gray area (e.g. McNally et al. 2011). Projections from the IL target GABAergic cells found in the CeA and intercalated cells between the basolateral and central amygdala (reviewed in Pape & Pare 2010). Many studies using a variety of manipulations have demonstrated a role for these structures in expression and consolidation of fear and fear extinction (e.g. Moustafa et al. 2013). Overlapping circuits also appear to regulate extinction of drug-seeking behavior (see McNally 2014).
Divergent projections from the mPFC regulate the increase in glutamate that leads to locomotor sensitization, as well as drug seeking and the cessation of drug seeking in a similar manner to that of fear expression. The IL, which attenuates fear expression by inhibiting the CeA through the GABAergic-intercalated cell projections, sends an excitatory projection to the medial portion of the NAc shell, which in turn sends an inhibitory GABAergic projection to the medial ventral pallidum (VP) which inhibits the motor output necessary for drug-seeking behavior (McFarland et al. 2004; Peters et al. 2009). The PL, which promotes the expression of fear through a glutamatergic projection to CeA, sends an excitatory projection to the dorsal NAc core, which putatively sends enkephalins to VP (Peters et al. 2009). These enkephalins may bind to mu opioid receptors on GABAergic inhibitory interneurons in the VP, thus disinhibiting the VP and promoting the locomotor behavior necessary for drug seeking (Peters et al. 2009). Therefore, the IL promotes the attenuation of drug-seeking behavior, just as it attenuates the expression of fear, and the PL promotes drug-seeking behavior, just as it promotes the expression of fear.
Additional similarities may underlie the converging circuitry that promotes both fear expression and drug seeking. For example, the basolateral amygdala (BLA), an integral structure for the expression of fear, is critically involved in the initial consolidation and subsequent modulation of memory associated with different rewards, such as food, ethanol and cocaine (Fuchs et al. 2006). Similarly, a large literature implicates the hippocampus in the contextual modulation of extinction in both fear and substance abuse paradigms (reviewed in Maren et al. 2013).
The question that remains is how acute activity in these circuits leads to changes in the brain that persist over time and that produce long-term changes in behavior. This is a particularly important question consider in the context of stress, PTSD and addiction, because many different experiences can disinhibit the circuit, resulting in relapse. One strategy that is emerging is to pair behavioral extinction treatment with drugs that target receptors that are involved in modulating stress and extinction.
Targeting receptor-level processes to promote extinction
One way to promote extinction is to pharmacologically promote the function of cellular and molecular pathways involved in memory. The thinking behind this approach is that potentiating these signals at the time of extinction will strengthen the memory that forms during extinction, resulting in a persistent form of extinction that transfers to situations outside of the treatment context (see Bukalo et al. 2014; Fitzgerald et al. 2014; Lattal & Wood 2013).
Several principles are beginning to emerge from studies that use pharmacological tools to promote extinction. First, extinction can be promoted behaviorally by drugs that both promote memory – behavioral enhancements in extinction are likely due to enhanced memory consolidation processes – and inhibit memory – behavioral enhancements in extinction are likely due to changes in the emotional content of the memory. Thus, simply knowing how a compound works at the cellular level does not necessarily speak to the behavioral mechanism of action of that compound. For example, one target that has received a great deal of attention both at the basic science level and at the translational level is the N-methyl-D-aspartate (NMDA) receptor. This receptor has been the focus of years of behavioral and electrophysiological work because of its involvement in the development of memory and long-term potentiation. Many studies have demonstrated that extinction can be impaired by drugs that antagonize this receptor and enhanced by drugs that agonize the receptor (e.g. Falls et al. 1992; Smits et al. 2013; Thompson & Disterhoft 1997; Walker et al. 2002). These enhancements in extinction are consistent with how the field regards the role of NMDA receptors in memory – they allow new memories to be encoded and consolidated, so facilitating their action should promote that process and lead to a strong extinction memory.
Other compounds promote extinction, but do so by antagonizing receptors that are involved in stress and memory. For example, the noradrenergic system is critically involved in modulating the emotional aspects of memory, is altered during periods of stress and is involved in extinction (e.g. Bernardi & Lattal 2010; Holmes & Quirk 2010; but see Font & Cunningham 2012, Janak & Corbit 2011). Antagonizing the β-adrenergic class of receptors is a widely used strategy for treating stress and anxiety. When paired with behavioral extinction, drugs such as propranolol can facilitate extinction (Bernardi et al. 2006, 2009; Rodriguez-Romaguera et al. 2009). These findings are much less theoretically straightforward compared with the results of d-cycloserine (DCS) studies because noradrenergic antagonists block the function of receptors that are involved in memory. There have been multiple ways offered to think about this and it seems that the most parsimonious interpretation is that the drugs alter the emotional content of the memory without necessarily altering the subjects’ memory of the original contingencies (e.g. Bernardi & Lattal 2012; Bernardi et al. 2009; Kindt et al. 2009; Rodriguez-Romaguera et al. 2009). Some of these interpretations of pharmacological experiments are entirely consistent with theoretical accounts that have been developed for extinction at the behavioral level. These accounts have long recognized that extinction operates through a variety of mechanisms, including alterations in processing of the conditioned and unconditioned stimuli, new inhibitory and/or excitatory associations that may develop during extinction and changes in affective state during extinction. Thus, extinction can be promoted by enhancing memory for the extinction contingencies or by attenuating some aspect of the original memory.
A second principle that has emerged from pharmacological studies of extinction is that drugs that promote extinction when paired with a behavioral experience are often completely ineffective on their own. This means that the drug works by targeting the memory that develops during a behavioral experience, suggesting that drugs targeting extinction need to be administered in controlled settings. There are, however, demonstrations that drugs on their own may promote extinction, even in the absence of behavioral experience (e.g. Peters et al. 2010), meaning that if the extinction circuit can be selectively targeted, there may be opportunity for extinction to develop by changing the function of that circuit. This is an exciting possibility that is just beginning to be explored.
A third principle is that if the behavioral experience is not enough to cause behavioral extinction, drugs may actually have deleterious effects on extinction. This appears to be true particularly if the extinction session is short enough to allow the animal to retrieve the original memory, but not long enough to allow extinction mechanisms to suppress the response. The same drugs that promote extinction, such as DCS, may impair extinction or even potentiate memory with very brief retrieval trials (e.g. Flavell et al. 2011; Lee et al. 2009). When DCS is targeted to circuits that mediate fear (such as the basolateral amygdala), impairments in extinction may occur even with longer durations that allow within-session extinction (Bolkan & Lattal 2014). Further, there are many documented effects of drugs failing to alter extinction, even under conditions in which there should be effects (e.g. Vurbic et al. 2011).
Finally, pharmacological approaches to extinction are complicated by the demonstration that enhancements in extinction are not always persistent. Some drugs may promote extinction, but may not weaken spontaneous recovery or contextual renewal, which may question the utility of these compounds in the clinic (e.g. Bouton et al. 2008; Stafford & Lattal 2009; Woods & Bouton 2006). It is this inconsistent persistence that has been the thorn in the side of the potential for using pharmacology to potentiate extinction. Recent studies have moved from targeting cellular receptor-level processes to processes that may lead to a longer lasting molecular signature, potentially resulting in a longer lasting signature at the level of memory. This includes some of the final steps of transcriptional regulation necessary for memory formation – the expression of genes and the proteins they code during the formation of long-term memories.
Understanding transcriptional regulation and gene expression may ultimately be the best path toward developing treatments for PTSD and addiction because both of these disorders are associated with long-term epigenetic marks that contribute to the persistence of the problems at the level of behavior. Drugs that target some of these processes, when paired with behavioral extinction, may be especially useful as treatment interventions.
The need for a molecular approach: epigenetic mechanisms in PTSD and addiction
Given that one of the key challenges in using extinction as a therapeutic is that it may not always create lasting effects, there is a need to look for molecular mechanisms that may provide a long-term signature for extinction. As we understand more about persistent epigenetic changes that are associated with stress, PTSD and addiction, it is becoming clear that targeting epigenetic processes may provide a long-term solution to the problem of creating persistent extinction (Fig. 1; reviewed in Stafford & Lattal 2011; Zovkic & Sweatt 2012).
Figure 1. Potential interactions between epigenetic mechanisms and cues associated with stress and drug-seeking behavior.
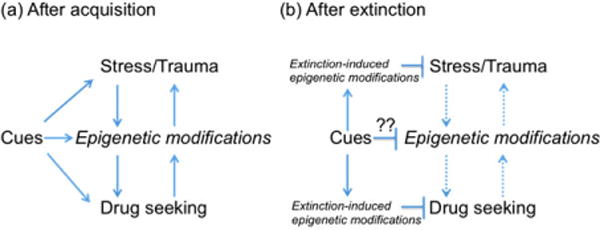
(a) Both stress and drug intake lead to epigenetic changes that result in persistent behaviors. These changes create a vicious cycle, in which deleterious effects may occur either directly through these epigenetic changes or through exposure to cues associated with stress or drug- seeking. An issue of continuing research is to understand the relation between epigenetic marks induced by early life experiences, stress, and drug seeking. (b) After extinction, new epigenetic modifications occur that inhibit conditioned fear responses and drug seeking. This inhibitory process can be strengthened by drugs that target epigenetic mechanisms, such as histone deacetylation or DNA methylation. A major unresolved question is whether extinction can reverse or at least inhibit the actions of the persistent marks induced by stress or drug -seeking and how those marks may continue to cause relapse even after successful extinction treatment.
One of the reasons that epigenetic mechanisms are receiving so much attention in the fields of fear and drug seeking is because these mechanisms provide experience-dependent positive and negative regulation of gene expression in the circuits that mediate memory. Broadly speaking, epigenetic processes provide molecular mechanisms through which environmental experience can have lasting effects on behavior. Because these mechanisms lead to lasting cellular changes, there is great focus on how these mechanisms translate into lasting memories involving trauma and drugs of abuse. By enhancing the formation of memories where stressful stimuli no longer produce fear or drug-related stimuli no longer predict drug availability using agents that influence the epigenetic machinery, the extinction memory may persist outside the treatment context, preventing relapse in vulnerable populations.
Broadly, epigenetics refers to changes in chromatin structure that influence gene expression without affecting gene sequence (Kwapis & Wood 2014; Levenson & Sweatt 2005). Two primary mechanisms of epigenetic regulation are through direct DNA methylation and histone modification, and these mechanisms allow for the integration and storage of information in species ranging from yeast and plants up to humans (reviewed in Hitchcock & Lattal 2014; Vanyushin 2006). Chromatin refers to the complex of DNA and proteins that reside in the nucleus of a cell, and the primary proteins that comprise chromatin are histones. Chromatin and DNA interact as an octamer of histones known as a nucleosome (Luger et al. 1997). The default structure of chromatin is largely in a repressed state, with changes in either DNA methylation or histone modification on lysine residues along the N-terminal tail affecting the activity of a number of transcription factors and the transcriptional machinery itself, which in turns affects gene expression. There are many types of modifications that can occur on DNA or histones (see Strahl & Allis 2000), but the majority of memory and addiction work has focused on DNA methylation and histone acetylation, with a particular emphasis on effects on methylation and acetylation levels on histones 3 and 4.
Histone acetylation involves two key molecules, histone acetyl transferase (HAT) and histone deacetylase (HDAC). The addition of an acetyl group by HAT causes chromatin to relax, thus making the DNA available to the transcription machinery, which subsequently leads to the enhanced production of proteins that contribute to the formation of memories. Acetyl groups are removed by HDACs, which results in the tightening of chromatin that renders the DNA unavailable to transcription machinery. One mechanism through which memories may be enhanced is through the use of HDAC inhibitors, which relax chromatin, causing it to remain open and increasing transcription. DNA methylation is often associated with transcriptional silencing, and the class of molecules responsible for methylating DNA are known as DNA methyltransferases (DNMTs; Zovkic et al. 2013). DNMT inhibitors have also been shown to induce deficits in learning related to different forms of fear and substance abuse in a circuit-specific fashion (e.g. Bali et al. 2011; Levenson et al. 2006; Miller et al. 2010; Miller 2008; Monsey et al. 2011).
One of the early demonstrations of the importance of acute epigenetic mechanisms in learning and memory came from Levenson et al. (2004), who found that contextual fear conditioning induced acetylation of histone H3 in the CA1 region of the hippocampus and that injection of an HDAC inhibitor before a conditioning session enhanced the long-term fear memory. Many subsequent studies have demonstrated that HDAC inhibitors promote initial memories (e.g. Raybuck et al. 2013; Vecsey et al. 2007) and memories that form during extinction (Bredy & Barad 2008; Gräff et al. 2014; Lattal et al. 2007; Marek et al. 2011; Stafford et al. 2012; Wei et al. 2012). This is true in multiple behavioral procedures that examine learning processes involved in everything from fear to object recognition to substance abuse.
Enhancements in extinction are not always the consequence of promoting epigenetic mechanisms after retrieval. Some studies have found that acetylation of histone H3, but not histone H4, is regulated following fear conditioning in rodents in the lateral amygdala (LA), and infusion of an HDAC inhibitor in the LA impairs extinction, while DNMT inhibitor infusions to the LA promote extinction (Maddox & Schafe 2011; Monsey et al. 2011). Similarly, although HDAC inhibitors are associated with promoting memory, this depends ultimately on which genes are expressed; hyperacetylation of regions coding memory repressor genes, for example, may have deleterious effects on memory and extinction. Findings like these again raise the issue of how to interpret behavioral data as evidence for enhancements in extinction or impairments in reconsolidation. Others have shown that histone acetylation and DNA methylation work in concert to regulate the formation of long-term memories. The DNMT inhibitor 5-Aza-2′-deoxycytidine (5-AZA) prevents fear conditioning-related increases in H3 acetylation, but pretreatment with the HDAC inhibitor trichostatin A (TSA) prevents the memory deficit following 5-AZA (Monsey et al. 2011).
Epigenetic mechanisms have similarly been implicated in drug addiction. Both acute and chronic cocaine administration increase histone acetylation on H3 and H4 in the NAc, a critical brain region associated with reward (Kumar et al. 2005). Mice that lack class 1 HDAC1, but not HDAC2 or HDAC3, in the NAc specifically show deficits in cocaine-induced locomotor sensitization (Kennedy et al. 2013). Knockout of HDAC1 in the striatum attenuates amphetamine-induced desensitization of the c-fos gene, an immediate early response gene that is rapidly induced in the striatum following acute exposure to psychostimulants (Renthal et al. 2008). Epigenetic changes have been implicated in the transition from drug use to drug addiction, as well as in chronic stress, through the activity of HDAC5 (Renthal et al. 2007). Mice that received site-specific injections of the HDAC class I and II inhibitor suberoylanilide hydroxamic acid (SAHA) to the NAc during place conditioning show enhancements in cocaine-induced CPP (Renthal et al. 2007). This group also found that a single injection of cocaine in mice chronically exposed to cocaine induced HDAC5 expression, an effect not present following a single dose of cocaine in naïve mice. Conversely, overexpression of HDAC5 using HSV-mediated transgene expression attenuates cocaine-induced CPP (Renthal et al. 2007). Similarly, chronic, but not acute, social defeat downregulates Hdac5 mRNA in the NAc (Renthal et al. 2007). Taken together, these findings strongly suggest that HDAC5 regulates behavioral responses to cocaine.
DNA methylation is also involved in modulating responses to drugs of abuse. DNMT3a has been found to be upregulated 4 h following both acute and chronic cocaine administration, but is downregulated 24 h later (LaPlant et al. 2010). Injection of the DNA methylation inhibitor RG108 to the NAc enhanced cocaine-induced CPP and cocaine-induced locomotor sensitivity at a dose that decreased methylation globally (LaPlant et al. 2010). Conversely, viral-mediated overexpression of DNMT3a in the NAc attenuated cocaine-induced CPP (LaPlant et al. 2010). Epigenetic regulation has also been observed in experiments looking at non-psychostimulants, including heroin. Heroin induces phosphoacetylation of H3 in the NAc, and intra-accumbal injection of TSA increases heroin-induced CPP, suggesting increased reward salience (Sheng et al. 2011).
Both stress and exposure to drugs of abuse induce epigenetic changes that result in persistent behavioral changes, some of which may contribute to the formation of a drug addiction or a stress-related psychiatric disorder. It is therefore of great interest to find new therapeutic options to treat these disorders, and the use of agents that promote extinction as a promising new class of drugs.
Theoretical considerations in interpreting effects on memory and extinction
Emerging evidence is consistent with the idea that drugs that target epigenetic processes may create a form of extinction that is persistent, potentially reducing relapse. As noted above, there are always multiple theoretical interpretations of extinction enhancements. The enhancements induced by HDAC inhibitors are particularly challenging to interpret at the theoretical level because they function to turn a weak extinction experience that does not change behavior on its own into a lasting extinction memory that persistently eliminates behavior. This type of effect is usually interpreted as an effect on reconsolidation, with the idea being that the memory is retrieved and the drug prevents the reconsolidation of that memory. However, this type of interpretation is particularly problematic with HDAC inhibitors because so many studies have shown their positive effect on memory regulation. Instead, it seems that the most parsimonious interpretation of HDAC inhibitor effects after retrieval is that these drugs promote the development of extinction, resulting in a persistent form of extinction that may be resistant to some of the behavioral properties associated with extinction, such as spontaneous recovery, contextual renewal and reinstatement. Indeed, one of the key findings from HDAC inhibitor studies in extinction is that these drugs can transform a learning experience that on its own does not result in extinction into an experience that results in rapid and persistent attenuation of behavior.
In a particularly convincing demonstration of the power of HDAC inhibitors, Malvaez et al. (2013) examined whether the same drug injection that promotes extinction could also promote initial memory formation in a different task. Mice were first trained in a cocaine-induced CPP procedure. During a post-conditioning test, mice expressed a high level of preference for the side of the apparatus previously associated with cocaine. Immediately after this test, mice received an injection of the HDAC3 inhibitor RGFP966 or vehicle, followed 1 h later by training in an object recognition task. The next day, mice treated with the specific inhibitor of HDAC3 showed a reduced CPP and an enhanced object location memory, consistent with the general idea that having an HDAC inhibitor on board resulted in a general memory enhancement. This demonstrates that pairing the HDAC inhibitor with retrieval promoted extinction, even in a case in which very little extinction occurred in vehicle-control animals. Malvaez et al. (2013) found that the HDAC3 inhibitor induced a specific pattern of acetylation in the hippocampus, NAc and infralimbic cortex, all of which are brain regions that are heavily involved in the processing of drug-related information and memory formation. What makes this discovery especially exciting are the implications for treatment in humans; HDAC inhibition may be able to simultaneously promote the extinction learning associated with attenuating drug use, while simultaneously promoting the extinction learning associated with exposure therapy in individuals with PTSD, thus conferring a twofold benefit in individuals who are especially prone to relapse to drugs of abuse when confronted with stressful stimuli.
From bench to bedside: testing treatments in better models of PTSD
PTSD is a difficult disorder to treat. It results from a variety of mechanisms and individuals with PTSD may show lifetime changes in stress responses. This means that treating PTSD is not as simple as exposing a PTSD patient to cues associated with a traumatic memory and attempting to enhance extinction processes pharmacologically. Although PTSD may be related to a specific event, it often occurs as a function of exposure to chronic stress at key developmental time points. Laboratory studies of fear conditioning, substance abuse and extinction may capture important components of development and treatment of PTSD, but PTSD often does not develop in humans the way that we often model it in the animal laboratory.
Indeed, one of the challenges in studying PTSD in animals is that it is a complicated disorder that often involves more than a traumatic memory. Basic researchers often discuss fear conditioning effects as modeling PTSD and it is true that a single aversive experience can result in lasting psychological impairments associated with PTSD. But PTSD often develops after exposure to chronic stressors, such as long-term abuse or sustained military combat. This type of exposure results in a hyper-responsivity to very mild stressors, an effect that is often not captured in animal models using conventional forms of fear conditioning. Rodent models of PTSD have included exposure to predators (Adamec et al. 1997) or predator urine (Adamec & Shallow 1993; Wallace & Rosen 2000), single prolonged stress (SPS), which consists of animals being restrained for 2 h, run through a forced swim procedure, then exposed to ether until loss of consciousness (Khan & Liberzon 2004), exposure to unsignaled footshock (Mikics et al. 2008; Rau et al. 2005) and social defeat (Buwalda et al. 2005). Some of these models show that fear conditioning and extinction are different after exposure to these stressors, compared to when conditioning and extinction occur in the absence of previous stress.
Many studies have documented the effects of stress on memory (e.g. Conrad et al. 1996; Luine et al. 1994; Sandi et al. 2003; Venero et al. 2002) and the effects of glucocorticoid receptor manipulations on memory (e.g. Diamond et al. 2004; Oitzl et al. 2001). Strikingly, although stress leads to deficits in memory associated with spatial tasks, it has been shown to lead to enhancements in fear learning (Conrad et al. 1999; Rau et al. 2009). Rau et al. (2005) found that exposure to a battery of shocks causes animals to develop robust and persistent fear conditioning after relatively mild conditioning procedures in a different context. This stress-enhanced fear learning persists across time and context, increases voluntary alcohol intake and is resistant to both extinction and NMDA receptor antagonism, which generally impairs memory formation (e.g. Meyer et al. 2013; Ponomarev et al. 2010; Rau & Fanselow 2009). Others have found that even if fear conditioning and extinction occur normally, long-term retention of extinction is impaired following various stress protocols (e.g. Bentz et al. 2010; Garcia et al. 2008; Knox et al. 2012; Merz et al. 2014; Miracle et al. 2006; Raio et al. 2014) or following exposure to corticosterone during critical developmental periods (Den et al. 2014). This underscores the need to understand the long-term biochemical changes that occur with PTSD and how those changes may influence subsequent response to new fearful situations and subsequent extinction.
Epigenetics as the path forward in treating PTSD and addiction
Epigenetic changes that result from stressful situations, experienced either early in life or during adulthood, may predispose individuals to aberrant stress responses, as well as SUDs (Fig. 1). For example, changes in methylation of both the genes for glucocorticoid receptors and BDNF have been associated with early-life trauma in human populations (McGowan et al. 2009; Roth et al. 2009). McGowan et al. (2009) found that individuals who suffered early-life trauma have decreased levels of GR mRNA and increased methylation on the promoter region of the neuron-specific exon l7 glucocorticoid receptor (NR3C1) gene, both resulting in decreased GR levels. Mice that were chronically exposed to CORT via drinking water demonstrated epigenetic changes in genes associated with the HPA axis (Lee et al. 2010). Specifically, this group investigated changes in the following genes: Fkbp5, Nr3c1, Hsp90, Crh and Crhr1. Following chronic CORT Fkbp5 levels were elevated, and differences in methylation were observed in hippocampus and hypothalamus. The protein Fkbp5 codes for FKBP5 regulates the sensitivity of GRs to glucocorticoids by binding and tagging the receptor for translocation to the nucleus of the cell. Enhanced levels of FKBP5 are associated with decrements in HPA sensitivity and decreased negative feedback (Binder 2009). These changes may lay the groundwork for long-term dysregulation of the HPA axis, which may account for increased rates of relapse to drugs of abuse in response to stressful stimuli.
Meaney and coworkers have investigated the influence of maternal care on epigenetic changes in rodent offspring. Mice display natural individual variability in the amount and quality of maternal care, and these differences result in changes to HPA responsiveness in offspring that persist into adulthood (Liu et al. 1997; Weaver et al. 2004). Adult offspring of high-licking mothers have increased GR density in the hippocampus, which results in enhanced negative feedback sensitivity, and reduced production of CRF (Liu et al. 1997; van Hasselt et al. 2012). Adult offspring of mothers displaying low-licking behavior have increased methylation of the GR promoter relative to offspring from high-licking mothers, and this leads to a decrease in HPA-negative feedback and increased sensitivity to stress (Liu et al. 1997). Together, these findings may account for decreased negative feedback in individuals with a history of trauma.
What is particularly promising for an epigenetic approach to PTSD is that these long-term changes can be reversed through environmental or pharmacological means. Cross fostering or injection with the HDAC inhibitor TSA can reverse changes in DNA methylation and HPA responsiveness to restraint stress (Weaver et al. 2004). This finding suggests that the changes that lead to sensitivity to PTSD can be reversed using epigenetic regulators in adulthood, and that this reversal can normalize HPA activity and behavioral responses to stress.
Of course, these long-term epigenetic changes may result in developmental programming effects that set in motion a chain of events that lead to endpoints that are far removed from their initial triggers. This means that even if, for example, methylation of certain genes can be reversed long after a stressful experience, the developmental program induced by that methylation pattern may have already resulted in long-term cellular changes that now function independently of that initial pattern.
Nonetheless, the long-term changes that can be induced in adulthood by drugs that target epigenetic mechanisms have exciting implications for potential treatment options for PTSD. Given the vicious circle that exists between stress, PTSD and substance abuse, as well as many common underlying cellular and molecular mechanisms, drugs that can successfully treat one aspect of these disorders may simultaneously treat other aspects of those disorders. The power of an epigenetic approach that is informed by basic science on the neurobiology of stress and extinction is the potential to ultimately change a signal within a specific circuit that mediates memory.
Acknowledgments
This article was supported by NIH grant R01 DA025922 (K.M.L.), US Army/DOD-TATRC grant W81XWH-12-2-0048 (K.M.L.) and NIH grant T32 DA 007262-23 (C.L.P.).
Footnotes
The authors declare no conflicts of interest.
References
- Adamec RE, Shallow T. Lasting effects on rodent anxiety of a single exposure to a cat. Physiol Behav. 1993;54:101–109. doi: 10.1016/0031-9384(93)90050-p. [DOI] [PubMed] [Google Scholar]
- Adamec RE, Shallow T, Budgell J. Blockade of CCK(B) but not CCK(A) receptors before and after the stress of predator exposure prevents lasting increases in anxiety-like behavior: implications for anxiety associated with posttraumatic stress disorder. Behav Neurosci. 1997;111:435–449. doi: 10.1037//0735-7044.111.2.435. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th. American Psychiatric Publishing; Arlington, VA: 2013. [Google Scholar]
- Baker DG, West SA, Nicholson WxE, Ekhator NN, Kasckow JW, Hill KK, Bruce AB, Orth DN, Geracioti TD. Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am J Psychiatry. 1999;156:585–588. doi: 10.1176/ajp.156.4.585. [DOI] [PubMed] [Google Scholar]
- Bali P, Im HI, Kenny PJ. Methylation, memory and addiction. Epigenetics. 2011;6:671–674. doi: 10.4161/epi.6.6.15905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentz D, Michael T, de Quervain DJF, Wilhelm FH. Enhancing exposure therapy for anxiety disorders with glucocorticoids: from basic mechanisms of emotional learning to clinical applications. J Anxiety Disord. 2010;24:223–230. doi: 10.1016/j.janxdis.2009.10.011. [DOI] [PubMed] [Google Scholar]
- Bernardi RE, Lattal KM. A role for α1-adrenergic receptors in extinction of conditioned fear and cocaine conditioned preference. Behav Neurosci. 2010;124:204–210. doi: 10.1037/a0018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi RE, Lattal KM. Prazosin differentially affects extinction of cocaine CPP based on dose and initial preference. Neuroreport. 2012;23:1048–1051. doi: 10.1097/WNR.0b013e32835ad246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi RE, Lattal KM, Berger SP. Postretrieval propranolol disrupts a cocaine conditioned place preference. Neuroreport. 2006;17:1443–1447. doi: 10.1097/01.wnr.0000233098.20655.26. [DOI] [PubMed] [Google Scholar]
- Bernardi RE, Ryabinin A, Berger SP, Lattal KM. Post-retrieval disruption of a cocaine conditioned place preference by systemic and intra-basolateral amygdala β2 and α1-adrenergic antagonists. Learn Mem. 2009;16:777–789. doi: 10.1101/lm.1648509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. 2009;34(Suppl. 1):S186–S195. doi: 10.1016/j.psyneuen.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Bolkan SS, Lattal KM. Opposing effects of D-cycloserine on fear despite a common extinction duration: interactions between brain regions and behavior. Neurobiol Learn Mem. 2014;113:25–34. doi: 10.1016/j.nlm.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouton ME, Vurbic D, Woods AM. D-cycloserine facilitates context-specific fear extinction learning. Neurobiol Learn Mem. 2008;90:504–510. doi: 10.1016/j.nlm.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradizza CM, Stasiewicz PR, Paas ND. Relapse to alcohol and drug use among individuals diagnosed with co-occurring mental health and substance use disorders: a review. Clin Psychol Rev. 2006;26:162–178. doi: 10.1016/j.cpr.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Bredy TW, Barad M. The histone deacetylase inhibitor valproic acid enhances acquisition, extinction, and reconsolidation of conditioned fear. Learn Mem. 2008;15:39–45. doi: 10.1101/lm.801108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremner JD, Vermetten E. Stress and development: behavioral and biological consequences. Dev Psychopathol. 2001;13:473–489. doi: 10.1017/s0954579401003042. [DOI] [PubMed] [Google Scholar]
- Bremner JD, Licinio J, Darnell A. Elevated CSF corticotropin-releasing factor concentrations in posttraumatic stress disorder. Am J Psychiatry. 1997;154:624–629. doi: 10.1176/ajp.154.5.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PJ, Stout RL, Mueller T. Posttraumatic stress disorder and substance abuse relapse among women: a pilot study. Psychol Addict Behav. 1996;10:124–128. [Google Scholar]
- Bukalo O, Pinard C, Holmes A. Mechanisms to medicines: elucidating neural and molecular substrates of fear extinction to identify novel treatments for anxiety disorders. Br J Pharmacol. 2014;171:4690–4718. doi: 10.1111/bph.12779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buwalda B, Kole MHP, Veenema AH, Huininga M, de Boer SF, Korte SM, Koolhaas JM. Long-term effects of social stress on brain and behavior: a focus on hippocampal functioning. Neurosci Biobehav Rev. 2005;29:83–97. doi: 10.1016/j.neubiorev.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Camp MC, MacPherson KP, Lederle L, Graybeal C, Gaburro S, DeBrouse LM, Ihne JL, Bravo JA, O’Connor RM, Ciocchi S, Wellman CL, Luthi A, Cryan JF, Singewald N, Holmes A. Genetic strain differences in learned fear inhibition associated with variation in neuroendocrine, autonomic, and amygdala dendritic phenotypes. Neuropsychopharmacology. 2012;37:1534–1547. doi: 10.1038/npp.2011.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JM. Efficacy of behavioral interventions for reducing problem behavior in persons with autism: a quantitative synthesis of single-subject research. Res Dev Disabil. 2003;24:120–138. doi: 10.1016/s0891-4222(03)00014-3. [DOI] [PubMed] [Google Scholar]
- Chang C, Maren S. Strain difference in the effect of infralimbic cortex lesions on fear extinction in rats. Behav Neurosci. 2010;124:391–397. doi: 10.1037/a0019479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad CD, Galea LA, Kuroda Y, McEwen BS. Chronic stress impairs rat spatial memory on the Y maze, and this effect is blocked by tianeptine pretreatment. Behav Neurosci. 1996;110:1321–1334. doi: 10.1037//0735-7044.110.6.1321. [DOI] [PubMed] [Google Scholar]
- Conrad CD, LeDoux JE, Magarinos AM, McEwen BS. Repeated restraint stress facilitates fear conditioning independently of causing hippocampal CA3 dendritic atrophy. Behav Neurosci. 1999;113:902–913. doi: 10.1037//0735-7044.113.5.902. [DOI] [PubMed] [Google Scholar]
- Davis M, Myers KM, Chhatwal J, Ressler KJ. Pharmacological treatments that facilitate extinction of fear: relevance to psychotherapy. NeuroRx. 2006;3:82–96. doi: 10.1016/j.nurx.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delamater AR, Westbrook RF. Psychological and neural mechanisms of experimental extinction: a selective review. Neurobiol Learn Mem. 2014;108:38–51. doi: 10.1016/j.nlm.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Den ML, Altmann SR, Richardson R. A comparison of the short- and long-term effects of corticosterone exposure on extinction in adolescence versus adulthood. Behav Neurosci. 2014;128:722–735. doi: 10.1037/bne0000022. [DOI] [PubMed] [Google Scholar]
- Diamond DM, Park CR, Woodson JC. Stress generates emotional memories and retrograde amnesia by inducing an endogenous form of hippocampal LTP. Hippocampus. 2004;14:281–291. doi: 10.1002/hipo.10186. [DOI] [PubMed] [Google Scholar]
- Falls WA, Miserendino MJ, Davis M. Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci. 1992;12:854–863. doi: 10.1523/JNEUROSCI.12-03-00854.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald PJ, Seemann JR, Maren S. Can fear extinction be enhanced? A review of pharmacological and behavioral findings. Brain Res Bull. 2014;105:46–60. doi: 10.1016/j.brainresbull.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell CR, Barber DJ, Lee JL. Behavioural memory reconsolidation of food and fear memories. Nat Commun. 2011;2:504. doi: 10.1038/ncomms1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Font L, Cunningham CL. Post-retrieval propranolol treatment does not modulate reconsolidation or extinction of ethanol-induced conditioned place preference. Pharmacol Biochem Behav. 2012;101:222–230. doi: 10.1016/j.pbb.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs RA, Feltenstein MW, See RE. The role of the basolateral amygdala in stimulus–reward memory and extinction memory consolidation and in subsequent conditioned cued reinstatement of cocaine seeking. Eur J Neurosci. 2006;23:2809–2813. doi: 10.1111/j.1460-9568.2006.04806.x. [DOI] [PubMed] [Google Scholar]
- Garcia R, Spennato G, Nilsson-Todd L, Moreau JL, Deschaux O. Hippocampal low-frequency stimulation and chronic mild stress similarly disrupt fear extinction memory in rats. Neurobiol Learn Mem. 2008;89:560–566. doi: 10.1016/j.nlm.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Glover DA, Poland RE. Urinary cortisol and catecholamines in mothers of child cancer survivors with and without PTSD. Psychoneuroendocrinology. 2002;27:805–819. doi: 10.1016/s0306-4530(01)00081-6. [DOI] [PubMed] [Google Scholar]
- Gräff J, Joseph NF, Horn ME, Samiei A, Meng J, Seo J, Rei D, Bero AW, Phan TX, Wagner F, Holson E, Xu J, Sun J, Neve RL, Mach RH, Haggarty SJ, Tsai LH. Epigenetic priming of memory updating during reconsolidation to attenuate remote fear memories. Cell. 2014;156:261–276. doi: 10.1016/j.cell.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groblewski PA, Ryabinin AE, Cunningham CL. Activation and role of the medial prefrontal cortex (mPFC) in extinction of ethanol-induced associative learning in mice. Neurobiol Learn Mem. 2012;97:37–46. doi: 10.1016/j.nlm.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hasselt FN, Cornelisse S, Zhang TY, Meaney MJ, Velzing EH, Krugers HJ, Joëls M. Adult hippocampal glucocorticoid receptor expression and dentate synaptic plasticity correlate with maternal care received by individuals early in life. Hippocampus. 2012;22:255–266. doi: 10.1002/hipo.20892. [DOI] [PubMed] [Google Scholar]
- Hitchcock LN, Lattal KM. Histone-mediated epigenetics in addiction. In: Lubin FD, Akbarian S, editors. Progress in Molecular Biology and Translational Science. Vol. 128. Academic Press; Burlington, MA: 2014. pp. 51–87. (Epigenetics and Neuroplasticity: Evidence and Debate). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes A, Quirk GJ. Pharmacological facilitation of fear extinction and the search for adjunct treatments for anxiety disorders-the case of yohimbine. Trends Pharmacol Sci. 2010;31:2–7. doi: 10.1016/j.tips.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsboer F. Corticotropin-releasing hormone modulators and depression. Curr Opin Investig Drugs. 2003;4:46–50. [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Jacobsen LK, Southwick SM, Kosten TR. Substance use disorders in patients with posttraumatic stress disorder: a review of the literature. Am J Psychiatry. 2001;158:1184–1190. doi: 10.1176/appi.ajp.158.8.1184. [DOI] [PubMed] [Google Scholar]
- Janak PH, Corbit LH. Deepened extinction following compound stimulus presentation: noradrenergic modulation. Learn Mem. 2011;18:1–10. doi: 10.1101/lm.1923211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy PJ, Feng J, Robison AJ, Maze I, Badimon A, Mouzon E, Chaudhury D, Damez-Werno DM, Haggarty SJ, Han MH, Bassel-Duby R, Olson EN, Nestler EJ. Class I HDAC inhibition blocks cocaine-induced plasticity by targeted changes in histone methylation. Nat Neurosci. 2013;16:434–440. doi: 10.1038/nn.3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S, Liberzon I. Topiramate attenuates exaggerated acoustic startle in an animal model of PTSD. Psychopharmacology (Berl) 2004;172:225–229. doi: 10.1007/s00213-003-1634-4. [DOI] [PubMed] [Google Scholar]
- Kindt M, Soeter M, Vervliet B. Beyond extinction: erasing fear responses and preventing the return of fear. Nat Neurosci. 2009;12:256–258. doi: 10.1038/nn.2271. [DOI] [PubMed] [Google Scholar]
- Knox D, Perrine SA, George SA, Galloway MP, Liberzon I. Single prolonged stress decreases glutamate, glutamine, and creatine concentrations in the rat medial prefrontal cortex. Neurosci Lett. 2010;480:16–20. doi: 10.1016/j.neulet.2010.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox D, Nault T, Henderson C, Liberzon I. Glucocorticoid receptors and extinction retention deficits in the single prolonged stress model. Neuroscience. 2012;223:163–173. doi: 10.1016/j.neuroscience.2012.07.047. [DOI] [PubMed] [Google Scholar]
- Kofoed L, Friedman MJ, Peck R. Alcoholism and drug abuse in patients with PTSD. Psychiatr Q. 1993;64:151–171. doi: 10.1007/BF01065867. [DOI] [PubMed] [Google Scholar]
- Krettek JE, Price JL. Projections from the amygdaloid complex and adjacent olfactory structures to the entorhinal cortex and to the subiculum in the rat and cat. J Comp Neurol. 1977;172:723–752. doi: 10.1002/cne.901720409. [DOI] [PubMed] [Google Scholar]
- Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DEH, Truong HT, Russo SJ, Laplant Q, Sasaki TS, Whistler KN, Neve RL, Self DW, Nestler EJ. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in stria-tum. Neuron. 2005;48:303–314. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Kwapis JL, Wood MA. Epigenetic mechanisms in fear conditioning: implications for treating post-traumatic stress disorder. Trends Neurosci. 2014;37:706–720. doi: 10.1016/j.tins.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaLumiere RT, Niehoff KE, Kalivas PW. The infralimbic cortex regulates the consolidation of extinction after cocaine self-administration. Learn Mem. 2010;17:168–175. doi: 10.1101/lm.1576810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPlant Q, Vialou V, Covington L, et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat Neurosci. 2010;13:1137–1143. doi: 10.1038/nn.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattal KM, Wood MA. Epigenetics and persistent memory: implications for reconsolidation and silent extinction beyond the zero. Nat Neurosci. 2013;16:124–129. doi: 10.1038/nn.3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattal KM, Barrett R, Wood MA. Systemic or intrahippocampal delivery of histone deacetylase inhibitors facilitates fear extinction. Behav Neurosci. 2007;121:1125–1131. doi: 10.1037/0735-7044.121.5.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent V, Westbrook RF. Inactivation of the infralimbic but not the prelimbic cortex impairs consolidation and retrieval of fear extinction. Learn Mem. 2009;16:520–529. doi: 10.1101/lm.1474609. [DOI] [PubMed] [Google Scholar]
- Lee JL, Gardner RJ, Butler VJ, Everitt BJ. D-cycloserine potentiates the reconsolidation of cocaine-associated memories. Learn Mem. 2009;16:82–85. doi: 10.1101/lm.1186609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RS, Tamashiro KLK, Yang X, Purcell RH, Harvey A, Willour VL, Huo Y, Rongione M, Wand GS, Potash JB. Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology. 2010;151:4332–4343. doi: 10.1210/en.2010-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeMieux AM, Coe CL. Abuse-related posttraumatic stress disorder: evidence for chronic neuroendocrine activation in women. Psychosom Med. 1995;57:105–115. doi: 10.1097/00006842-199503000-00002. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Sweatt JD. Epigenetic mechanisms in memory formation. Nat Rev Neurosci. 2005;6:108–118. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- Levenson JM, O’Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, Malone LM, Sweatt JD. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem. 2006;281:15763–15773. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- Liu D, Diorio J, Tannenbaum B, Caldji C, Francis D, Freedman A, Sharma S, Pearson D, Plotsky PM, Meaney MJ. Maternal care, hippocampal glucocorticoid receptors, and hypothalamic-pituitary-adrenal responses to stress. Science. 1997;277:1659–1662. doi: 10.1126/science.277.5332.1659. [DOI] [PubMed] [Google Scholar]
- Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Luine V, Villegas M, Martinez C, McEwen BS. Repeated stress causes reversible impairments of spatial memory performance. Brain Res. 1994;639:167–170. doi: 10.1016/0006-8993(94)91778-7. [DOI] [PubMed] [Google Scholar]
- MacPherson K, Whittle N, Camp M, Gunduz-Cinar O, Singewald N, Holmes A. Temporal factors in the extinction of fear in inbred mouse strains differing in extinction efficacy. Biol Mood Anxiety Disord. 2013;3:13. doi: 10.1186/2045-5380-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox SA, Schafe GE. Epigenetic alterations in the lateral amygdala are required for reconsolidation of a Pavlovian fear memory. Learn Mem. 2011;18:579–593. doi: 10.1101/lm.2243411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvaez M, McQuown SC, Rogge GA, Astarabadi M, Jacques V, Carreiro S, Rusche JR, Wood MA. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc Natl Acad Sci U S A. 2013;110:2647–2652. doi: 10.1073/pnas.1213364110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek R, Coelho CM, Sullivan RKP, Baker-Andresen D, Li X, Ratnu V, Dudley KJ, Meyers D, Mukherjee C, Cole PA, Sah P, Bredy TW. Paradoxical enhancement of fear extinction memory and synaptic plasticity by inhibition of the histone acetyltransferase p300. J Neurosci. 2011;31:7486–7491. doi: 10.1523/JNEUROSCI.0133-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Phan KL, Liberzon I. The contextual brain: implications for fear conditioning, extinction and psychopathology. Nat Rev Neurosci. 2013;14:417–428. doi: 10.1038/nrn3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Davidge SB, Lapish CC, Kalivas PW. Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. J Neurosci. 2004;24:1551–1560. doi: 10.1523/JNEUROSCI.4177-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonté B, Szyf M, Turecki G, Meaney MJ. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12:342–348. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally GP. Extinction of drug seeking: neural circuits and approaches to augmentation. Neuropharmacology. 2014;76:528–532. doi: 10.1016/j.neuropharm.2013.06.007. [DOI] [PubMed] [Google Scholar]
- McNally GP, Johansen JP, Blair HT. Placing prediction into the fear circuit. Trends Neurosci. 2011;34:283–292. doi: 10.1016/j.tins.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz CJ, Hermann A, Stark R, Wolf OT. Cortisol modifies extinction learning of recently acquired fear in men. Soc Cogn Affect Neurosci. 2014;9:1426–1434. doi: 10.1093/scan/nst137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer EM, Long V, Fanselow MS, Spigelman I. Stress increases voluntary alcohol intake, but does not alter established drinking habits in a rat model of posttraumatic stress disorder. Alcohol Clin Exp Res. 2013;37:566–574. doi: 10.1111/acer.12012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikics E, Tóth M, Varjú P, Gereben B, Liposits Z, Ashaber M, Halasz J, Barna I, Farkas I, Haller J. Lasting changes in social behavior and amygdala function following traumatic experience induced by a single series of foot-shocks. Psychoneuroen-docrinology. 2008;33:1198–1210. doi: 10.1016/j.psyneuen.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Miller CA, Campbell SL, Sweatt JD. DNA methylation and histone acetylation work in concert to regulate memory formation and synaptic plasticity. Neurobiol Learn Mem. 2008;89:599–603. doi: 10.1016/j.nlm.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, Gavin CF, White JA, Parrish RR, Honasoge A, Yancey CR, Sweatt JD. Cortical DNA methylation maintains remote memory. Nat Neurosci. 2010;13:664–666. doi: 10.1038/nn.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miracle AD, Brace MF, Huyck KD, Singler SA, Wellman CL. Chronic stress impairs recall of extinction of conditioned fear. Neurobiol Learn Mem. 2006;85:213–218. doi: 10.1016/j.nlm.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Monsey MS, Ota KT, Akingbade IF, Hong ES, Schafe GE. Epigenetic alterations are critical for fear memory consolidation and synaptic plasticity in the lateral amygdala. PLoS One. 2011;6 doi: 10.1371/journal.pone.0019958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MA, Romanski LM, LeDoux JE. Extinction of emotional learning: contribution of medial prefrontal cortex. Neurosci Lett. 1993;163:109–113. doi: 10.1016/0304-3940(93)90241-c. [DOI] [PubMed] [Google Scholar]
- Moustafa AA, Gilbertson MW, Orr SP, Herzallah MM, Servatius RJ, Myers CE. A model of amygdala–hippocampal–prefrontal interaction in fear conditioning and extinction in animals. Brain Cogn. 2013;81:29–43. doi: 10.1016/j.bandc.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeroff CB, Widerlov E, Bissette G, Walleus H, Eklund K, Kilts CD, Loosen PT, Vale W. Elevated concentrations of CSF corticotropin release factor-like immunoreactivity in depressed patients. Science. 1984;226:1342–1344. doi: 10.1126/science.6334362. [DOI] [PubMed] [Google Scholar]
- Oitzl MS, Reichardt HM, Joëls M, de Kloet ER. Point mutation in the mouse glucocorticoid receptor preventing DNA binding impairs spatial memory. Proc Natl Acad Sci. 2001;98:12790–12795. doi: 10.1073/pnas.231313998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC, Pare D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev. 2010;90:419–463. doi: 10.1152/physrev.00037.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paré D, Smith Y. GABAergic projection from the intercalated cell masses of the amygdala to the basal forebrain in cats. J Comp Neurol. 1994;344:33–49. doi: 10.1002/cne.903440104. [DOI] [PubMed] [Google Scholar]
- Peters J, Kalivas PW, Quirk GJ. Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn Mem. 2009;16:279–288. doi: 10.1101/lm.1041309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, Dieppa-Perea LM, Melendez LM, Quirk GJ. Induction of fear extinction with hippocampal-infralimbic BDNF. Science. 2010;328:1288–1290. doi: 10.1126/science.1186909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman RK, Orr SP. Twenty-four hour urinary cortisol and catecholamine excretion in combat-related posttraumatic stress disorder. Biol Psychiatry. 1990;27:245–247. doi: 10.1016/0006-3223(90)90654-k. [DOI] [PubMed] [Google Scholar]
- Ponomarev I, Rau V, Eger EI, Harris RA, Fanselow MS. Amygdala transcriptome and cellular mechanisms underlying stress-enhanced fear learning in a rat model of posttraumatic stress disorder. Neuropsychopharmacology. 2010;35:1402–1411. doi: 10.1038/npp.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raio CM, Brignoni-Perez E, Goldman R, Phelps EA. Acute stress impairs the retrieval of extinction memory in humans. Neurobiol Learn Mem. 2014;112:212–221. doi: 10.1016/j.nlm.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry. 2003;160:1554–1565. doi: 10.1176/appi.ajp.160.9.1554. [DOI] [PubMed] [Google Scholar]
- Rau V, Fanselow MS. Exposure to a stressor produces a long lasting enhancement of fear learning in rats. Stress. 2009;12:125–133. doi: 10.1080/10253890802137320. [DOI] [PubMed] [Google Scholar]
- Rau V, DeCola JP, Fanselow MS. Stress-induced enhancement of fear learning: an animal model of posttraumatic stress disorder. Neurosci Biobehav Rev. 2005;29:1207–1223. doi: 10.1016/j.neubiorev.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Raybuck JD, McCleery EJ, Cunningham CL, Wood MA, Lattal KM. The histone deacetylase inhibitor sodium butyrate modulates acquisition and extinction of cocaine-induced conditioned place preference. Pharmacol Biochem Behav. 2013;106:109–116. doi: 10.1016/j.pbb.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, Covington HE, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, Kerstetter KA, Neve RL, Haggarty SJ, McKinsey TA, Bassel-Duby R, Olson EN, Nestler EJ. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Renthal W, Carle TL, Maze I, Covington HE, Truong HT, Alibhai I, Kumar A, Montgomery RL, Olson EN, Nestler EJ. Delta FosB mediates epigenetic desensitization of the c-fos gene after chronic amphetamine exposure. J Neurosci. 2008;28:7344–7349. doi: 10.1523/JNEUROSCI.1043-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinne T, de Kloet ER, Wouters L, Goekoop JG, DeRijk RH, van den Brink W. Hyperresponsiveness of hypothalamic-pituitary-adrenal axis to combined dexamethasone/corticotropin-releasing hormone challenge in female borderline personality disorder subjects with a history of sustained childhood abuse. Biol Psychiatry. 2002;52:1102–1112. doi: 10.1016/s0006-3223(02)01395-1. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Romaguera J, Sotres-Bayon F, Mueller D, Quirk GJ. Systemic propranolol acts centrally to reduce conditioned fear in rats without impairing extinction. Biol Psychiatry. 2009;65:887–892. doi: 10.1016/j.biopsych.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth TL, Lubin FD, Funk AJ, Sweatt JD. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol Psychiatry. 2009;65:760–769. doi: 10.1016/j.biopsych.2008.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Sandi C, Davies HA, Cordero MI, Rodriguez JJ, Popov VI, Stewart MG. Rapid reversal of stress induced loss of synapses in CA3 of rat hippocampus following water maze training. Eur J Neurosci. 2003;17:2447–2456. doi: 10.1046/j.1460-9568.2003.02675.x. [DOI] [PubMed] [Google Scholar]
- Shaham Y, Erb S, Leung S, Buczek Y, Stewart J. CP-154,526, a selective, non-peptide antagonist of the corticotropin-releasing factor. Psychopharmacology (Berl) 1998;137:1–7. doi: 10.1007/s002130050608. [DOI] [PubMed] [Google Scholar]
- Shaham Y, Erb S, Stewart J. Stress-induced relapse to heroin and cocaine seeking in rats: a review. Brain Res Brain Res Rev. 2000;33:13–33. doi: 10.1016/s0165-0173(00)00024-2. [DOI] [PubMed] [Google Scholar]
- Shalev U, Highfield D, Yap J, Shaham Y. Stress and relapse to drug seeking in rats: studies on the generality of the effect. Psychopharmacology (Berl) 2000;150:337–346. doi: 10.1007/s002130000441. [DOI] [PubMed] [Google Scholar]
- Sharpe MJ, Killcross S. The prelimbic cortex uses higher-order cues to modulate both the acquisition and expression of conditioned fear. Front Syst Neurosci. 2015;8:235. doi: 10.3389/fnsys.2014.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng J, Lv ZG, Wang L, Zhou Y, Hui B. Histone H3 phosphoacetylation is critical for heroin-induced place preference. Neuroreport. 2011;22:575–580. doi: 10.1097/WNR.0b013e328348e6aa. [DOI] [PubMed] [Google Scholar]
- Shvil E, Rusch HL, Sullivan GM, Neria Y. Neural, psychophysiological, and behavioral markers of fear processing in PTSD: a review of the literature. Curr Psychiatry Rep. 2013;15:1–10. doi: 10.1007/s11920-013-0358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha R, Catapano D, O’Malley S. Stress-induced craving and stress response in cocaine dependent individuals. Psychopharmacology (Berl) 1999;142:343–351. doi: 10.1007/s002130050898. [DOI] [PubMed] [Google Scholar]
- Smits JA, Rosenfield D, Otto MW, Powers MB, Hofmann SG, Telch MJ, Pollack MH, Tart CD. D-Cycloserine enhancement of fear extinction is specific to successful exposure sessions: evidence from the treatment of height phobia. Biol Psychiatry. 2013;73:1054–1058. doi: 10.1016/j.biopsych.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford JM, Lattal KM. Direct comparisons of the size and persistence of anisomycin-induced consolidation and reconsolidation deficits. Learn Mem. 2009;16:494–503. doi: 10.1101/lm.1452209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford JM, Lattal KM. Is an epigenetic switch the key to persistent extinction? Neurobiol Learn Mem. 2011;96:35–40. doi: 10.1016/j.nlm.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford JM, Raybuck JD, Ryabinin AE, Lattal KM. Increasing histone acetylation in the hippocampus-infralimbic network enhances fear extinction. Biol Psychiatry. 2012;72:25–33. doi: 10.1016/j.biopsych.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein MB, Yehuda R, Koverola C, Hanna C. Enhanced dexamethasone suppression of plasma cortisol in adult women traumatized by childhood sexual abuse. Biol Psychiatry. 1997;42:680–686. doi: 10.1016/s0006-3223(96)00489-1. [DOI] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Thompson LT, Disterhoft JF. Age-and dose-dependent facilitation of associative eyeblink conditioning by {d}-cycloserine in rabbits. Behav Neurosci. 1997;111:1303–1312. doi: 10.1037//0735-7044.111.6.1303. [DOI] [PubMed] [Google Scholar]
- Tipps ME, Raybuck JD, Lattal KM. Substance abuse, memory, and post-traumatic stress disorder. Neurobiol Learn Mem. 2014;112:87–100. doi: 10.1016/j.nlm.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanyushin BF. DNA methylation and epigenetics. Russ J Genet. 2006;42:985–997. [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venero C, Tilling T, Hermans-Borgmeyer I, Schmidt R, Schachner M, Sandi C. Chronic stress induces opposite changes in the mRNA expression of the cell adhesion molecules NCAM and L1. Neuroscience. 2002;115:1211–1219. doi: 10.1016/s0306-4522(02)00543-2. [DOI] [PubMed] [Google Scholar]
- Vurbic D, Gold B, Bouton ME. Effects of D-cycloserine on the extinction of appetitive operant learning. Behav Neurosci. 2011;125:551–559. doi: 10.1037/a0024403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachen JS, Jimenez S, Smith K, Resick PA. Long-term functional outcomes of women receiving cognitive processing therapy and prolonged exposure. Psychol Trauma: Theory Res Pract Policy. 2014;16:58–65. [Google Scholar]
- Walker DL, Ressler KJ, Lu KT, Davis M. Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of D-cycloserine as assessed with fear-potentiated startle in rats. J Neurosci. 2002;22:2343–2351. doi: 10.1523/JNEUROSCI.22-06-02343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace KJ, Rosen JB. Predator odor as an unconditioned fear stimulus in rats: elicitation of freezing by trimethylthiazoline, a component of fox feces. Behav Neurosci. 2000;114:912–922. doi: 10.1037//0735-7044.114.5.912. [DOI] [PubMed] [Google Scholar]
- Weaver ICG, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Wei W, Coelho CM, Li X, Marek R, Yan S, Anderson S, Meyers D, Mukherjee C, Sbardella G, Milite C, Rotili D, Mai A, Cole PA, Sah P, Kobor MS, Bredy TW. p300/CBP-associated factor selectively regulates the extinction of conditioned fear. J Neurosci. 2012;32:11930–11941. doi: 10.1523/JNEUROSCI.0178-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods AM, Bouton ME. D-cycloserine facilitates extinction but does not eliminate renewal of the conditioned emotional response. Behav Neurosci. 2006;120:1159–1162. doi: 10.1037/0735-7044.120.5.1159. [DOI] [PubMed] [Google Scholar]
- Yehuda R. Current status of cortisol findings in post-traumatic stress disorder. Psychiatr Clin North Am. 2002;25:341–368. doi: 10.1016/s0193-953x(02)00002-3. [DOI] [PubMed] [Google Scholar]
- Yehuda R. Advances in understanding neuroendocrine alterations in PTSD and their therapeutic implications. Ann N Y Acad Sci. 2006;1071:137–166. doi: 10.1196/annals.1364.012. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Antelman SM. Criteria for rationally evaluating animal models of postraumatic stress disorder. Biol Psychiatry. 1993;33:479–486. doi: 10.1016/0006-3223(93)90001-t. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Southwick SM, Nussbaum G, Wahby V, Giller EL, Mason JW. Low urinary cortisol excretion in patients with posttraumatic stress disorder. J Nerv Ment Dis. 1990;178:366–369. doi: 10.1097/00005053-199006000-00004. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Boisoneau D, Lowy MT, Giller EL. Dose-response changes in plasma cortisol and lymphocyte glucocorticoid receptors following dexamethasone administration in combat veterans with and without posttraumatic stress disorder. Arch Gen Psychiatry. 1995;52:583–593. doi: 10.1001/archpsyc.1995.03950190065010. [DOI] [PubMed] [Google Scholar]
- Zovkic IB, Sweatt JD. Epigenetic mechanisms in learned fear: implications for PTSD. Neuropsychopharmacology. 2012;38:77–93. doi: 10.1038/npp.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zovkic IB, Guzman-Karlsson MC, Sweatt JD. Epigenetic regulation of memory formation and maintenance. Learn Mem. 2013;20:61–74. doi: 10.1101/lm.026575.112. [DOI] [PMC free article] [PubMed] [Google Scholar]