

Abstract
Polycystic liver diseases are genetic disorders characterized by progressive bile duct dilatation and/or cyst development. The large volume of hepatic cysts causes different symptoms and complications such as abdominal distension, local pressure with back pain, hypertension, gastro-oesophageal reflux and dyspnea as well as bleeding, infection and rupture of the cysts. Current therapeutic strategies are based on surgical procedures and pharmacological management, which partially prevent or ameliorate the disease. However, as these treatments only show short-term and/or modest beneficial effects, liver transplantation is the only definitive therapy. Therefore, interest in understanding the molecular mechanisms involved in disease pathogenesis is increasing so that new targets for therapy can be identified. In this Review, the genetic mechanisms underlying polycystic liver diseases and the most relevant molecular pathways of hepatic cystogenesis are discussed. Moreover, the main clinical and preclinical studies are highlighted and future directions in basic as well as clinical research are indicated.
Introduction
Polycystic liver diseases (PCLDs) are genetic disorders characterized by bile duct dilatation and/or cyst development derived from the bile duct epithelial cells, cholangiocytes. PCLDs are inherited in a dominant or recessive form and can develop alone or in association with polycystic kidney diseases (PKDs).1 The most common symptoms and complications of PCLDs are hypertension, back pain, bloating and abdominal discomfort, dyspnea, gastrooesophageal reflux, bleeding, infection and cyst rupture. Patients progressively worsen and surgical procedures such as aspiration, sclerotherapy, fenestration and/or segmental hepatic resection are commonly used in the management of patients with PCLDs, but have short-term beneficial effects. High rates of recurrence and complications make liver transplantation the only curative treatment. As the possibilities to prevent and cure PCLDs are limited, this Review appraises the most up-to-date research into the underlying molecular mechanisms of PCLDs and the identification of potential therapeutic targets.
Genetic mechanisms of PCLDs
The formation of multiple cysts scattered throughout the liver alone (autosomal dominant polycystic liver disease [ADPLD]), or in association with similar kidney lesions (autosomal dominant polycystic kidney disease [ADPKD]) and autosomal recessive polycystic kidney disease (ARPKD; Caroli disease and congenial hepatic fibrosis [CHF] in infants), is the result of germ line and/or somatic mutations (listed in Table 1, along with disease frequency). Three genes, PRKCSH,2,3 SEC634 and LRP55 are associated with the aetiology of ADPLD, whereas PKD16 and PKD27 have been identified as causative genes for ADPKD. Mutations in PKHD1 are responsible for ARPKD as well as Caroli disease and CHF.8,9 The genetic diagnosis of ADPKD have revealed that mutations in PKD1 are more frequent (~85%) than PKD2 (~15%) and are also associated with a more severe phenotype.10 On the other hand, 20% of patients with ADPLD show mutations in PRKCSH, SEC63 or LRP5, with PRKCSH being the most frequent (occurring in ~15% of all ADPLD patients);11,12 however, the type of mutation (if any) present in the remaining 80% of patients with ADPLD remains unknown.
Table 1.
Genes, proteins and animal models of polycystic liver diseases
| Mutated genes | Protein | Localization | Function | Animal models |
|---|---|---|---|---|
| ADPLD (~1:100,000) | ||||
| PRKCSH | Glucosidase 2 subunit β, protein kinase C substrate 80K-H or hepatocystin | ER | N-linked glycan-processing enzyme in the endoplasmic reticulum | Prkcshflox/flox:pCXCreER mice and zebrafish |
| SEC63 | Translocation protein SEC63 homologue | ER | Translocation of proteins in the endoplasmic reticulum | Sec63flox/flox:pCXCreER mice and zebrafish |
| LRP5 | Low density lipoprotein receptor-related protein 5 | Plasma membrane | Canonical Wnt signalling | Lrp5KO mouse |
| ADPKD (~1:500–1:1,000) | ||||
| PKD1 | Polycystin-1 | Primary cilium, plasma membrane and cell junctions | Mechanoreceptor involved in calcium signalling | Pkd1flox/:pCxCreER™ (Pkd1cKO) and zebrafish |
| PKD2 | Polycystin-2 | Primary cilium and endoplasmic reticulum | Nonselective calcium channel | Pkd2flox/:pCxCreER™ (Pkd2cKO) and Pkd2WS25/− |
| ARPKD, CHF or CD (~1:20,000) | ||||
| PKHD1 | Fibrocystin or polyductin | Primary cilium | Tubulogenesis and/or maintenance of bile duct architecture | PCK rat, and Pkhd1del2/del2 mouse |
Abbreviations: ADPKD, autosomal dominant polycystic kidney disease; ADPLD, autosomal dominant polycystic liver disease; ARPKD, autosomal recessive polycystic kidney disease; CD, Caroli disease; CHF, congenital hepatic fibrosis; ER, endoplasmic reticulum.
PKD1 encodes the mechanoreceptor polycystin-1 and PKD2 encodes the nonselective calcium channel polycystin-2, which are coupled in the ciliary membrane to form a functional complex. Activation of polycystin-1 facilitates calcium uptake through polycystin-2,13 a large pool of which is also present in the endoplasmic reticulum.14 PRKCSH and SEC63 encode two proteins resident in the endoplasmic reticulum. PRKCSH (also known as hepatocystin) is the β subunit of glucosidase 2, a heterodimer complex with N-linked glycan-processing activity that is involved in the maturation and/or folding of glyco proteins.2,3 SEC63 is a component of the protein translocation machinery required for transport of glycoproteins into and out of the endoplasmic reticulum (Table 1).4 LRP5 encodes a transmembrane protein that acts as a co-receptor with Frizzled protein members to transduce Wnt signalling.5 PKHD1 encodes fibrocystin (also known as polyductin), a plasma membrane protein localized in the primary cilium that has a role in tubulogenesis and maintaining the architecture of the epithelial duct lumen (Table 1).15
To date, the Online Mendelian Inheritance in Man (OMIM) database includes 46 mutations that affect genes involved in the development of PCLDs (Table 2), which are functionally and clinically important. Moreover, four new mutations in the LRP5 gene have been described, but are not yet included in the OMIM database. Additional genetic variations that result in changes to protein functions have been identified but their pathogenic consequences have yet to be elucidated.
Table 2.
Mutations in genes causing polycystic liver diseases resulting in change of function with clinical significance
| Gene | Locus | Nucleotide change | Amino acid change | OMIM number | Phenotype MIM number |
|---|---|---|---|---|---|
| PRKCSH | 19p13.2 | IVS16, A-G, −2 | – | 177060.0001 | 174050 |
| IVS4, G-C, +1 | – | 177060.0002 | |||
| 2-bp del, IVS16GT, +1 | – | 177060.0003 | |||
| c.1240C>T | p.Gln414Ter | 177060.0004 | |||
| c.1269C>G | p.Tyr423Ter | 177060.0005 | |||
| 1-bp ins, 216A | – | 177060.0006 | |||
|
| |||||
| SEC63 | 6q21 | c.173G>A | p.Trp58Ter | 608648.0001 | 174050 |
| 1-bp ins, 442A | – | 608648.0002 | |||
| IVS8ds, G-A, +1 | – | 608648.0003 | |||
| c.1702_1704del | p.Glu568del | 608648.0004 | |||
|
| |||||
| LRP5 | 11q13.2 | c.1360G>A | p.Val454Met | Not assigned | Not assigned |
| c.3562C>T | p.Arg1188Trp | Not assigned | |||
| c.4587G>C | p.Arg1529Ser | Not assigned | |||
| c.4651G>A | p.Asp1551Asn | Not assigned | |||
|
| |||||
| PKD1 | 16p13.3 | IVSds, G-C, +1 | – | 601313.0001 | 173900 |
| c.12124C>T | p.Gln4042Ter | 601313.0002 | |||
| 15-bp del | – | 601313.0003 | |||
| c.12682C>T | p.Arg4228Ter | 601313.0004 | |||
| c.11512C>T | p.Gln3838Ter | 601313.0005 | |||
| c.12261T>A | p.Cys4087Ter | 601313.0006 | |||
| c.11457C>A | p.Tyr3819Ter | 601313.0007 | |||
| 12036G-A | – | 601313.0008 | |||
| 28-bp del, nt6434 | – | 601313.0009 | |||
| IVS14, G-A, −1 | – | 601313.00010 | |||
| c.971G>T | p.Arg324Leu | 601313.00011 | |||
| c.2534T>C | p.Leu845Ser | 601313.00012 | |||
| c.5764C>T | p.Gln1922Ter | 601313.00013 | |||
| 2-bp del, 5224AG | – | 601313.00014 | |||
| c.12420G>A | p.Trp4140Ter | 601313.00015 | |||
| Gly2579del, 8-bp del | – | 601313.00016 | |||
|
| |||||
| PKD2 | 4q22.1 | c.1139G>A | p.Trp380Ter | 173910.0001 | 613095 |
| c.2224C>T | p.Arg742Ter | 173910.0002 | |||
| c.1213C>T | p.Gln405Ter | 173910.0003 | |||
| 1-bp ins, 693C | – | 173910.0004 | |||
| c.1390C>T | p.Arg464Ter | 173910.0005 | |||
| 1-bp ins, 2160A | – | 173910.0006 | |||
| 1-bp ins, 197-203C | – | 173910.0007 | |||
| c.1532A>T | p.Asp511Val | 173910.0008 | |||
| 2-bp del/1-bp ins, nt1934 | – | 173910.0009 | |||
| Ex3dup | – | 173910.00010 | |||
| c.305_306insGAG | p.Glu102_Val103insArg | 173910.00011 | |||
|
| |||||
| PKHD1 | 6p12.2 | c.107C>T | p.Thr36Met | 606702.0001 | 263200 |
| c.4991C>T | p.Ser1664Phe | 606702.0002 | |||
| c.9053C>T | p.Ser3018Phe | 606702.0003 | |||
| c.5221G>A | p.Val1741Met | 606702.0004 | |||
| c.8011C>T | p.Arg2671Ter | 606702.0005 | |||
| c.10658T>C | p.Ile3553Thr | 606702.0006 | |||
| c.1486C>T | p.Arg496Ter | 606702.0007 | |||
| c.10412T>G | p.Val3471Gly | 606702.0008 | |||
| IVS46ds, A-G, +653 | – | 606702.0009 | |||
Data were obtained in June 2014 from the Online Mendelian Inheritance in Man (OMIM) of the National Center for Biotechnology Information (NCBI). Four new mutations in LRP5 have been described but the OMIM number and the phenotype (MIM number) have not yet been assigned. The mutations listed consist of single nucleotide polymorphisms resulting in an amino acid change, exon duplications, small deletions and insertions, and splice site mutations. Abbreviations: bp, base pair; del, small deletion; ds, donor splice site; ER, endoplasmic reticulum; Ex3dup, exon 3 duplication; ins, insertion; IVS, intervening sequence. Permission obtained from the Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, John Hopkins University (Baltimore, MD), September 2014. World Wide Web URL: http://omim.org/.
Although ADPLD and ADPKD are inherited in a dominant fashion, how heterozygous mutations lead to disease is unclear. Previous reports have indicated that mutations in a single allele do not have severe consequences for cholangiocyte function and that PCLD will only develop after loss of heterozygosity.16 Indeed, PRKCSH loss of heterozygosity has been found in a high proportion of the cysts in patients carrying a germ line mutation in this gene,17 whereas only a small proportion of SEC63-mutated cysts acquire SEC63 loss of heterozygosity.16 Second-hit mechanisms, such as loss of heterozygosity, have also been reported in cysts of patients with ADPKD who have PKD1 germ line mutations18 and in people with PKD2 germline mutations.19 Additionally, transheterozygous mutations in other genes associated with PCLDs have been suggested.20 The importance of somatic second-hit mutations in the pathogenesis of PCLDs has been reviewed elsewhere.21
Hepatic cystogenesis
Accumulating evidence suggests that pathophysiological alterations in ductal plate remodelling, the primary cilium and in many intracellular signalling pathways and cellular functions (proliferation, angiogenesis, secretion, cell–matrix interaction) account for hepatic cystogenesis. Given the multifactorial nature of these defects, their relative involvement in cyst growth is discussed below.
Embryology and ductal plate malformation
Hepatic cystogenesis in PCLDs is associated with ductal plate malformation, which is a defect of ductal plate remodelling.1,22–26 Development of the human liver starts on the eighteenth day of gestation when the hepatic diverticulum is formed.27 At the sixth week of gestation, hepatoblasts immediately adjacent to the portal tract mesenchyme flatten and establish the ductal plate, a layer of biliary-type cuboidal cells expressing the biliary markers CK19 and SOX9, as well as high levels of cadherin-1.23,28 Ductal plate malformation is defined as embryological arrest of ductal plate development29 and falls into three categories: the inability of biliary precursor cells to differentiate; defects in maturation of primitive bile ducts; and abnormal bile duct enlargement.24,30
ARPKD and CHF are both characterized by the presence of ductal plate remnants along the portal tract margins. In patients with ARPKD, liver histology depicts incompletely developed ductal plates,31 whereas in CHF bile ducts are embedded in fibrous stroma and form cystic lesions (called von Meyenburg complexes).29 Both ADPKD and ADPLD are associated with ductal plate malformation; however, is not clear to which category of malformation they belong.24
Hepatoblast commitment to biliary lineage and ductal plate remodelling is controlled by a network of the following: signalling molecules including Notch,32–34 transforming growth factor β,35,36 Wnt37–39 and fibroblast growth factor;40–42 transcription factors;43–50 and microRNAs51 (Box 1). Many components of ductal plate remodelling have also been implicated in the regulation of ciliary function and are aberrantly expressed in cystic cholangiocytes, thus linking ductal plate malformation and hepatic cystogenesis (Box 1).
Box 1. Regulators of ductal plate remodelling.
Signaling pathways
-
■
Notch receptors and Notch-processing enzymes are expressed in primary cilia and regulate their length.32–34
-
■
TGF-β signalling also regulates biliary commitment of hepatoblasts.23 TGF-β and its receptor are overexpressed in cystic cholangiocytes and renal epithelial cells.35,36
-
■
Wnt signalling is implicated in both ciliary sensation and cystogenesis.37–39
-
■
FGF signalling regulates cilia length and hepatoblast differentiation towards the biliary lineage. Patients with ADPKD have raised FGF23 levels.40–42
Transcription factors23,24
-
■
HNF1β, 4 and 6
-
■
HHEX
-
■
C/EBPα
-
■
Smad2 and Smad3, Onecut 2
-
■
FOXM1b
-
■
SALL4
-
■
TBX3
Examples include deletion of Hnf6 and Hhex in mice results in accumulation of ductal plate remnants, cyst development and cilia absence in cystic cholangiocytes.23,43,44 In addition, in the pancreas, Hnf6 stimulates the expression of Pkhd1 and Cys1 genes, mutations of which cause hepatic cystogenesis and affect ciliary length.45 In a further example, mice deficient in Cys1,46,47Hnf-1β48,49 or C/EBPα50 exhibit ductal plate malformation.
miRNAs
miRNA-30 family. miR-30a depletion in zebrafish affects bile duct morphogenesis.51
Abbreviations: ADPKD, autosomal dominant polycystic kidney disease; C/EBPα, CCAAT/enhancer binding protein α; FGF, fibroblast growth factor; FOXM1b, forkhead box factor M1b; HHEX, hematopoietically expressed homeobox; HNF, hepatocyte nuclear factor; miRNA, microRNA; SALL4, spalt-like transcription factor 4; TBX3, T-box transcription factor 3; TGF, transforming growth factor.
Cholangiocyte abnormalities
Primary cilium and centrosomes
Cholangiocytes, the epithelial cells that line the bile ducts, are key liver cells involved in the regulation of the flow and composition of bile. Cholangiocytes contain a single primary cilium that extends from the apical membrane into the bile duct lumen, which is formed by an axoneme and a centriole-derived basal body.52 The primary cilium, an antenna-like bulge, functions as a sensory organelle detecting changes in bile flow, composition and osmolarity and has an important role in cholangiocyte physiology and pathophysiology (Figure 1a).52,53 Hepatic cystogenesis is thought to be associated with disturbances in ciliary sensation that result from structural and functional changes owing to aberrant expression of PCLD-related and ciliary-associated proteins. Indeed, shortened, unusually long or entirely absent cilia are present in the cystic cholangiocytes of animal models with PCLD47,54–56 and patients with ADPKD (Figure 1b).57 Abnormalities in the primary cilium are accompanied by atypical centrosome positioning, supernumerary centrosomes and multipolar spindles.48,49,58 Notably, cholangiocytes with hyperamplified centrosomes represent a small but notably abnormal portion of cells lining liver cysts. In this regard, a link between centrosome amplification and renal cystogenesis was reported, emphasizing the importance of centrosome abnormality in hepatorenal cystogenesis.59
Figure 1.
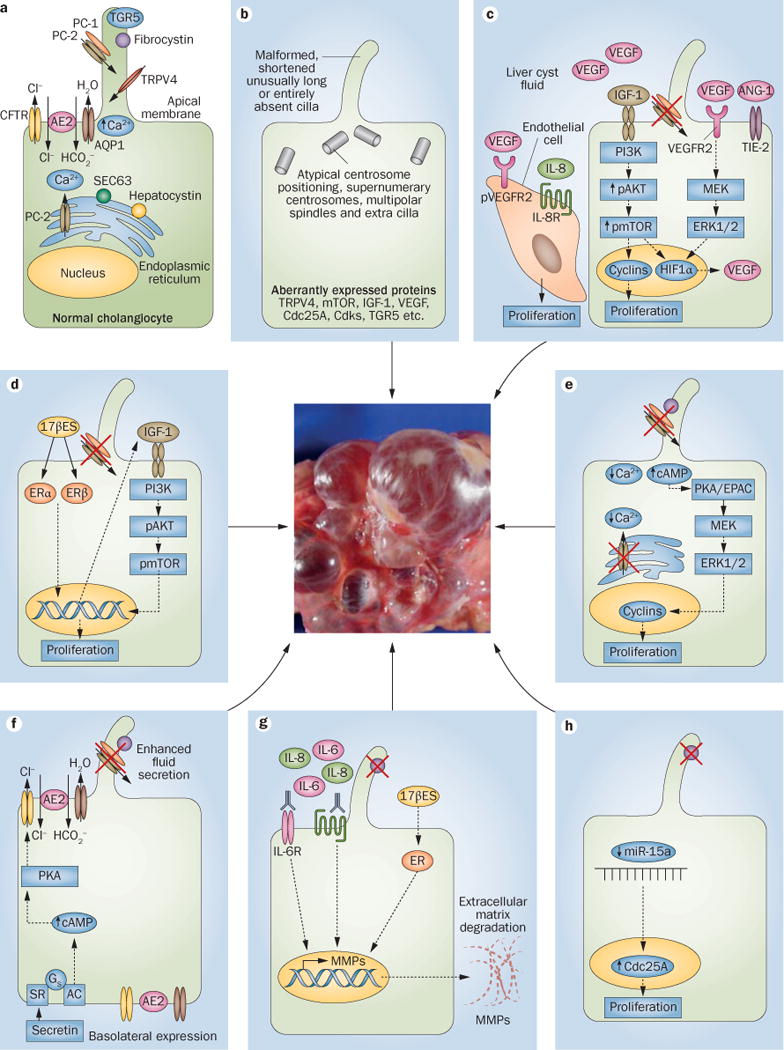
Cellular alterations and molecular mechanisms involved in hepatic cystogenesis. a | The normal structure of both a cholangiocyte and a primary cilium. b | The primary cilium and gene expression levels of key intracellular mediators can be altered in polycystic liver diseases. c | Different growth factors and cytokines stimulate the proliferation of cystic cholangiocytes and endothelial cells in an autocrine and/or paracrine fashion. Moreover, d | oestrogens and e | changes in intracellular calcium and cAMP levels might also induce the proliferation of cystic cholangiocytes. Hepatic cystogenesis is associated with f | alterations in fluid secretion and g | extracellular matrix remodelling. h | Global downregulation of microRNAs occurs in cystic cholangiocytes, which facilitates the proliferation of cystic cholangiocytes. The relative involvement of each pathway in different forms of hepatic cystogenesis has been highlighted in the main body text. The central image is of human liver tissue with cysts. Abbreviations: 17βES, 17β oestradiol; AC, adenylate cyclase; AE2, anion exchanger 2; ANG-1, angiopoetin-1; AQP1, aquaporin 1; cAMP, cyclic adenosine monophosphate; Cdc25A, cell division cycle 25A; Cdks, cyclin dependent kinase; CFTR, cystic fibrosis transmembrane conductance regulator; EPAC, rap guanine nucleotide exchange factor 3; ER, oestrogen receptor; ERα, oestrogen receptor α; ERβ, oestrogen receptor β; ERK1/2, extracellular signal regulated kinase 1/2; Gs, Gs protein; HIF1α, hypoxia inducible factor 1α; IGF1, insulin-like growth factor 1; MEK, mitogen-activated protein kinase kinase 1; miR-15a, microRNA 15a; MMP, matrix metalloproteinase; mTOR, mammalian target of rapamycin; pAKT, phosphorylated v-akt murine thymoma viral oncogene homolog 1; PC-1, polycystin-1; PC-2, polycystin-2; PI3K, phosphatidylinositol 4,5-bisphosphate 3-kinase; PKA, protein kinase A; pmTOR, phosphorylated mTOR; pVEGFR2, phosphorylated vascular endothelial growth factor receptor 2; SEC63, SEC63 homolog; SR, serotonin receptor; TGR5, g-protein coupled bile acid receptor-1; TIE-2, TEK tyrosine kinase; TRPV4, transient receptor potential cation subfamily V member 4; VEGF, vascular endothelial growth factor.
The absence of PCLD-related proteins (fibrocystin,54,55,60 polycystin-1 and polycystin-253,61,62) and cAMP-associated G-protein-coupled receptor TGR5,63 or the overexpression of the calcium channel TRPV4,64,65 causes functional abnormalities in cholangiocyte cilia and enhances proliferation and fluid secretion, which contributes to progressive cyst growth.1,25,66–68 Interestingly, a strong piece of evidence that might implicate cilia in the pathogenesis of cyst growth comes from outstanding research carried out in animal models of PCLDs. Polycystin-1, polycystin-2, fibrocystin, hepatocystin and SEC63 seem to interact together in a complex network; hepatocystin and SEC63 are necessary for the adequate expression of polycystin-1 and polycystin-2 functional complex, and importantly, polycystin-1 was defined as the rate-limiting component that determines cyst formation.69 Thus, polycystin-1 expression levels seem to be involved in determining the severity of ADPKD, ARPKD and ADPLD phenotypes, providing a direct link between cyst growth and the primary cilium.70 In addition, the use of proteasome inhibitors has been suggested as a therapy, which might inhibit cystogenesis in patients with ADPLD by increasing the steady-state levels of polycystin-1 and also by inducing apoptosis due to increased toxic levels of unfolded proteins in cyst-lining cells.70 On the other hand, studies carried out in experimental animal models of ADPKD suggest that, in the absence of polycystin-1 or polycystin-2, signalling mechanism present in the remaining cilium are required to promote cyst growth, whereas total absence of the cilium results in inhibition of cystogenesis.71 Evidence supporting the concept that ciliary dysfunctions underlie hepatic cystogenesis is summarized in Box 2.
Box 2. Ciliary dysfunction underlies hepatic cystogenesis.
-
■
Disappearance of fibrocystin (PKHD1) from cholangiocyte cilia leads to ciliary malformations and accelerated cyst expansion.54,55,60
-
■
Somatic inactivation of Pkd2 in mice results in hepato-cystic phenotype.56
-
■
Polycystin-1 and polycystin-2 function as ciliary sensors of cell injury activating the cholangiocyte proliferation as a reparative mechanism.62
-
■
Ciliary structural malformations (shortened, unusually long or entirely absent cilia) are present in cystic cholangiocytes of PCLD animal models47,54–56 and patients with ADPKD.57
-
■
Ciliary defects are linked to basal body abnormalities (that is atypical centrosome positioning, supernumerary centrosomes, multipolar spindles and extra cilia).58
-
■
Hnf1β deficient mice lack cholangiocyte cilia due to mispositioning of the basal body resulting in ductal plate malformation and biliary dysgenesis that resembles the lesions observed in patients with ARPKD.30,49,50
-
■
Abnormal ciliary structure is associated with enhanced fluid secretion and ion transport.62,66–68
-
■
Absence of polycystin-1 in cholangiocyte cilia decreases levels of [Ca2+]i and increases cAMP production via a Ca2+-inhibitable adenylyl cyclase 6 (which is also localized within the cilium) increasing cell proliferation and accelerating fluid secretion.53,61
-
■
TGR5, the G-protein coupled receptor linked to cAMP signalling, is overexpressed in cystic cholangiocytes and is mislocalized from cilia.63
-
■
Decreased levels of [Ca2+]i in hepatic cysts are linked to aberrantly expressed calcium channel TRPV4 in cholangiocyte cilia.65
-
■
Glucosidase 2 subunit β and Sec63 are required in mice for adequate expression of a functional complex of polycystin-1 and polycystin-2. Polycystin-1 is the rate-limiting component of this complex.69,70
-
■
In the absence of polycystin-1 or polycystin-2, the resulting cilium is required to promote cyst growth, whereas the total absence of the cilium results in inhibition of cystogenesis.71
Abbreviations: ADPKD, autosomal dominant polycystic kidney disease; ARPKD, autosomal recessive polycystic kidney disease; cAMP, cyclic adenosine monophosphate; Hnf1β, hepatocyte nuclear factor 1β; PCLD, polycystic liver disease; PKDH1, polycystic kidney and hepatic disease 1; Sec63, Sec63 homolog; TGR5, g-protein coupled bile acid receptor-1; TRPV4, transient receptor potential cation subfamily V member 4.
Proliferation and angiogenesis
Normal cholangiocytes are quiescent with almost no expression of proliferating cell nuclear antigen (PCNA) detectable by immunohistochemistry,72 whereas cystic cholangiocytes show intense PCNA staining.72–75 However, proliferation of cystic cholangiocytes in patients with ADPLD is insignificant, with few Ki67 positive cells,76 which suggests that other events such as secretion, cell–matrix interactions and ductal plate malformations could have a major role in ADPLD pathogenesis. The hyperproliferative phenotype of cystic cholangiocytes is regulated by growth factors and hormones present in the cystic fluid and/or secreted by cystic cholangiocytes (Figure 1c).72,77–79
The growth factors epidermal growth factor (EGF), vascular endothelial growth factor (VEGF) and insulin-like growth factor 1 (IGF1) all participate in autocrine and/or paracrine loops that stimulate cellular proliferation. Cholangiocytes from the PCK rat, an animal model of ARPKD, show more pronounced hyperproliferative features in response to Egf than normal rat cholangiocytes.80 Egf-stimulated hyperproliferation was linked to an overexpression of mitogen-activated protein kinase kinase 5 (Map2k5 also known as Mek5) and subsequent phosphorylation of Erk5, which was abolished by molecular or pharmacological targeting of either Mek5 or the Egf receptor with gefitinib.80 However, the role of EGF in hepatic cystogenesis is uncertain because a different study demonstrated that chronic administration of EGF receptor inhibitors (EKI-785 or EKB-569) had no effect on hepatic cystogenesis in PCK rats.81
VEGF and angiopoietin 1 are pleiotropic growth factors that have key roles in hepatic cytogenesis promoting cyst growth and their vascular supply. The expression of VEGF and angiopoietin 1 and their respective receptors (that is, VEGFR1, VEGFR2 and TIE2) are upregulated in cholangio cytes of patients with ADPKD.78 In addition, VEGF is found in liver cyst fluids of patients with ADPKD.82 VEGF stimulates proliferation of cystic cholangiocytes in patients with ADPKD, in the Pkd2WS25/− and Pkd2flox/:pCxCreER (Pkd2cKO) mouse models of ADPKD74,78,82 and promotes liver cyst growth in Pkd2cKO mice but not in Pkd1flox/:pCxCreER (Pkd1cKO) mice.74 The effects of VEGF secretion and VEGFR2 signalling are dependent on protein kinase A (PKA) and/or extracellular signal regulated kinase 1/2 (ERK1/2).74 In addition, the effect of VEGF is synergized by angiopoietin 1.78 Biliary cysts are surrounded by vascular networks that are critical for hepatic cystogenesis. Different intracellular signalling pathways and growth factors present in the cystic fluid, such as VEGF and IL-8, promote proliferation of endothelial cells (Figure 1c),83 which suggests that autocrine and paracrine mechanisms could be involved in neovascularization.78 The potential therapeutic value of targeting VEGF in patients with PCLDs is supported by the fact that the VEGFR2 inhibitor, SU5416, blunts liver cyst growth in animal models.74,82
Another potential therapeutic target is IGF1, a promitotic factor present in the cystic fluid of patients with ADPKD.72 IGF1, its main receptor IGF1R and their downstream effectors phosphorylated AKT and phosphorylated mammalian target of rapamycin (mTOR) are all overexpressed in the cystic epithelium.72 Moreover, the proliferation of cholangio cytes stimulated by cystic fluid, 17β-estradiol or IGF1 was inhibited by an IGF1R antagonist.72
As well as being downstream of IGF1 signalling, AKT is an activator of mTOR, through which it regulates the expression of hypoxia inducible factor 1 α (encoded by HIF1α), a major transcriptional activator of VEGF. As mTOR is involved in both VEGF and IGF1 signalling pathways it was thought that mTOR could be a useful therapeutic target for treating patients with PCLD. The expression of phosphorylated mTOR is increased in the liver cystic epithelium of Pkd2cKO mice;73 inhibiting mTOR in vivo with sirolimus decreased IGF1-stimulated HIF1α accumulation, VEGF secretion in cystic cholangiocytes and cyst growth.73 Accordingly, sirolimus and SU5416 (VEGFR2 inhibitor) inhibited the proliferation of cholangiocytes stimulated by IGF1 in Pkd2cKO mice.73 However, the inhib ition of mTOR by sirolimus did not attenuate hepatic and renal cystogenesis in PCK rats,84 highlighting the necessity to clarify in clinical trials the potential therapeutic role of mTOR inhibitors in the different forms of PCLDs. Interestingly, a reduction in liver volume was observed in a clinical trial using sirolimus as an immunosuppressant after renal transplantation in 16 patients with ADPKD (Table 3).85 However, in two independent clinical trials, chronic everolimus (an mTOR inhibitor derived from sirolimus) treatment in patients with ADPKD did not slow the progression of renal impairment and kidney growth.86,87 Unfortunately, effects of everolimus on liver volume were not examined in these trials. In a 2013 clinical trial, the efficacy of combining everolimus and octreotide (a somatostatin analogue) was compared with octreotide monotherapy. Everolimus did not enhance the beneficial effect of octreotide in reducing liver volume in patients with PCLDs (Table 3).88 Overall, the role of mTOR inhibitors in the treatment of patients with PCLDs has not fulfilled expectations and is not clinically recommended.
Table 3.
Clinical trials in polycystic liver diseases
| Study | Patient randomization | Treatment | Liver volume reduction | ||
|---|---|---|---|---|---|
| Dose | Duration | Route of administration | |||
| Octreotide (targets cAMP) | |||||
| Caroli et al. (2010)100 | ADPKD, n = 12 (5 treated, 7 placebo) | 40 mg every 28 days | 6 months | Intramuscular | 4.5% (vs 0.9% increase in placebo) |
| Hogan et al. (2010)101 | ADPKD and ADPLD, n = 42 (28 treated, 14 placebo) | 40 mg every 28 (± 5) days | 12 months | Intramuscular | 4.95% (vs 0.92% increase in placebo) |
| Hogan et al. (2012)102 | Extension study: ADPKD and ADPLD, n = 41/42 (all received treatment) | 40 mg every 28 (± 5) days | Additional 12 months (24 months total) | Intramuscular | No significant changes (−0.77%) after an additional year of therapy |
| Lanreotide (targets cAMP) | |||||
| Temmerman et al. (2013)105 | ADPKD and ADPLD, n = 132 (106 treated, 26 placebo) | 90 mg (n = 55) Or 120 mg (n = 51) every 28 days | 6 months | Subcutaneous | 1.4% (LAN 90 mg) 2.8% (LAN 120 mg) (vs 1.1% increase in placebo) LAN 90 mg had fewer adverse effects |
| van Keimpema et al. (2009)103 | ADPKD and ADPLD, n = 54 (28 treated, 27 placebo) | 120 mg every 28 days | 24 weeks | Subcutaneous | 2.9% (vs 1.6% increase in placebo) |
| Chrispijn et al. (2012)104 | Extension study: ADPKD and ADPLD, n = 41/54 (all received treatment) | 120 mg every 28 days | 12 months | Subcutaneous | 4% |
| Sirolimus vs tacrolimus (targets mTOR) | |||||
| Qian et al. (2008)85 | ADPKD (sirolimus n = 7, tacrolimus n = 9) | 5–10 mg daily (sirolimus) 3 mg twice day (tacrolimus) |
Retrospective analysis 19.4 months | Oral | 11.9% decrease with sirolimus vs 14.1% increase with tacrolimus |
| Everolimus alone or in combination with octreotide (targets cAMP and mTOR) | |||||
| Chrispijn et al. (2013)88 | ADPKD and ADPLD, n = 44 (23 received octreotide and 21 received octreotide + everolimus) | 40 mg octreotide every 4 weeks, 2.5 mg everolimus daily | 48 weeks | Intramuscular (octreotide) and oral (everolimus) | 3.5 ± 5.2% octreotide monotherapy vs 3.8 ± 4.7% in the octreotide/everolimus group (in response to octreotide, everolimus does not further reduce liver volume) |
Clinical trial NCT01670110 testing pasireotide in severe polycystic liver disease is underway (Mayo Clinic).107 Abbreviations: ADPKD, autosomal dominant polycystic kidney disease; ADPLD, autosomal dominant polycystic liver disease; cAMP, cyclic adenosine monophosphate; mTOR, mammalian target of rapamycin.
The potential therapeutic value of sorafenib, a tyrosine kinase inhibitor that might block the action of all EGF, VEGF and IGF1 receptors, has been tested for its ability to attenuate the proliferation of cystic cholangiocytes. However, evidence indicates that, paradoxically, sorafenib transactivates Raf1 thereby promoting cystogenesis in Pkd2cKO mice.89
Peroxisome proliferator-activated receptor γ (PPAR-γ) is also involved in hepatic cystogenesis by inhibiting genes involved in proliferation, inflammation and fibrosis. The PPAR-γ agonist, pioglitazone, inhibited kidney and liver disease in PCK rats,90 but its use in clinical practice is discouraged because of the increased risk of bladder cancer associated with this drug.90,91 However, telmisartan—an angiotensin II type 1 receptor antagonist that has off-target PPAR-γ agonist properties but fewer associated risks—reduced liver cystogenesis in PCK rats,92 which suggests that PPAR-γ might be a potential therapeutic target in PCLDs.
Although PCLDs affect both men and women, the latter usually exhibit a more severe phenotype. A number of clinical observations suggest that oestrogens are key regulators of hepatic cystogenesis.93–95 Cholangiocytes that line bile ducts of healthy individuals and unaffected bile ducts of patients with ADPKD do not express the oestrogen receptors α or β.72 By contrast, cystic cholangiocytes in patients with ADPKD are positive for oestrogen receptors α and β. Oestrogens exert promitotic effects on cystic cholangiocytes either directly or by inducing IGF1 secretion (Figure 1d);96 these effects can be partially inhibited by oestrogen receptor antagonists.72 All these data support the rationale for studying the potential therapeutic value of antioestrogen therapies (such as tamoxifen) for the t reatment of female patients with PCLDs.
cAMP and calcium levels
Intracellular signalling abnormalities associated with increased cAMP levels and decreased [Ca2+]i levels underlie the hyperproliferative phenotype of cystic cholangiocytes and represent potential therapeutic targets (Figure 1e). Raised cAMP levels in PCK rat cholangiocytes97 stimulates proliferation via two intracellular effectors involved in the Mek/Erk pathway—rap guanine nucleotide exchange factor 3 (Rapgef3, also known as Epac) and Pka.98 Octreotide treatment decreased these raised cAMP levels in the cholangiocytes and serum of PCK rats, reducing liver weight, cyst volume, hepatic fibrosis and mitotic indices.97 A study has shown that pasireotide, a more potent somatostatin analogue than octreotide with broader receptor specificity and a longer half-life, is more effective than octreotide in reducing hepatorenal cystogenesis in PCLD rodent models.99 Several clinical trials have evaluated the effects of octreotide100–102 and lanreotide103–105 in patients with PCLDs (Table 3). Both drugs moderately decreased liver volume (by ~5%) in patients with ADPLD and ADPKD, and improved quality of life.61,101,106 In addition, a clinical trial (NCT01670110107) to evaluate the therapeutic potential of pasireotide in patients with ADPKD and ADPLD is now underway at the Mayo Clinic. For these reasons, current pharmacological strategies are based on the chronic administration of somatostatin analogues, which are clinically recommended but might result in gastrointestinal adverse effects such as diarrhoea, abdominal cramps, flatulence, bloating, gas and injection site granulomas.101,105
Cholangiocytes from both PCK rats and Pkd2cKO mice are also characterized by diminished levels of [Ca2+]i.98,108 Restoration of intracellular calcium by a calcium ionophore inhibited both the basal and cAMP/Pka-stimulated proliferation of PCK rat cholangiocytes via the Pi3k/Akt pathway, whereas cAMP/Epac-stimulated proliferation was not affected.98 Thus, cAMP might regulate the proliferation of cystic cholangiocytes through calcium-dependent (Pka) and calcium-independent (Epac) mechanisms.98 Restoration of intracellular calcium could be a useful therapeutic approach in treating patients with PCLD. Pharmacological activation of the calcium-entry channel Trpv4 (which is overexpressed in PCLDs) inhibited the proliferation of PCK rat cholangiocytes in vitro, but did not affect hepatic cystogenesis in vivo.65 The in vivo study was limited by the low sublethal dose of the Trvp4 activator used (GSK1016790A), as it induces acute circulatory collapse when administered at higher doses. Therefore, we believe that new pharmacological strategies are needed to evaluate in vivo the potential therapeutic role of intracellular calcium restoration in PCLDs.
Secretion
Cholangioctes have a key role in the fluidization and alkalinization of the primary bile generated by hepatocytes, which are controlled by nucleotides, bile acids and hormones, such as secretin.109–110 Secretin interacts with its receptor (localized to the basolateral membrane of cholangiocytes), which leads to increased cAMP levels and further activation of PKA. PKA then mediates the exocitosis of intracellular vesicles containing the chloride channel, cystic fibrosis transmembrane conductance regulator (CFTR), the chloride/bicarbonate exchanger, anion exchanger 2 (AE2) and the water channel, aquaporin 1 (AQ1), which results in bicarbonate-rich choleresis.109–111 Increased fluid secretion is one of the contributing mechanisms of bile duct dilatation and cyst expansion in PCLDs (Figure 1f). PCK rat cholangiocytes cultured in a 3D collagen matrix showed enhanced expansion under basal conditions in response to secretin and hypotonicity. Observed alterations were associated with abnormal expression and location of Cftr, anion exchanger 2 and aquaporin 1.68 These carriers are preferentially localized to the apical membrane of normal rat cholangiocytes, but they were mainly found overexpressed and mislozalized at the basolateral membrane of PCK rat cholangiocytes. The basolateral presence of Cftr or anion exchanger 2 inhibitors blocked secretin-stimulated hypersecretion in cysts of PCK rats but not in normal cystic structures,68 which suggests that targeting these aberrant mechanisms could have potential therapeutic value in treating patients with PCLDs.
However, the role of secretin and flux proteins in cyst progression is more complex than expected. In contrast to the aforementioned data, human cholangiocytes from patients with ADPKD exhibit decreased anion exchanger activity due to diminished expression of mature glycosylated anion exchanger 2 polypeptide and decreased membrane-localized anion exchanger 2.112 The secretory differences between PCK rat cholangiocytes and cholangiocytes from patients with ADPKD could be linked to differences related to the particular form of PCLD. Thus, as cysts from PCK rats flow into the bile ducts, cysts in patients with ADPKD might be disconnected from the biliary tree,55 which could result in different pathological or adaptive secretory processes. Of note, chronic administration of secretin had negligible effects on hepatic cystogenesis of PCK rats and Pkd2WS25/− mice and the absence of the secretin receptor in Pkd2WS25/−:SCTR−/− double mutant mice did not alter the severity of PCLD.113 Together, these data suggest that secretin could have a modest role in the pathogenesis of PCLDs.
Cell–matrix interactions
Interactions between cells and the extracellular matrix are involved in normal ductal plate formation but also in the development and progression of PCLDs. The extracellular matrix is a complex structure of glycoproteins and is remodelled by matrix metalloproteases, tissue inhibitors of metalloproteases and hormones. Free space for cyst growth is generated by overexpression and hyper secretion of matrix metalloproteases by cholangiocytes, which is regulated by oestrogens and cytokines in an autocrine and paracrine fashion (Figure 1g).114 IL-6 and IL-8 present in the cystic fluid interact with their plasma membrane receptors (IL-6R and CXCR1, respectively) localized in cholangiocytes, which enhances the expression and secretion of matrix metalloproteases. Oestrogens, present in the cystic fluid of female patients, have the same effect.114 By contrast, other factors present in the cystic fluid such as VEGF, EGF, hepatocyte growth factor, epithelial neutrophil attractant 78 and growth-related oncogene α did not alter the matrix metalloprotease activity of normal cholangiocytes or cholangiocytes from patients with ADPKD.114 Altered cell–extracellular matrix inter actions detailed above are in agreement with the observation that the expression of basement membrane proteins such as laminin and collagen type IV around bile ducts is degraded in CHF, Caroli disease and in PCK rats.115 Finally, the importance of matrix metalloprotease hyperactivity in PCLDs is highlighted by the fact that chronic pharmacological inhibition of matrix metalloproteases with marimastat halts hepatic cystogenesis in PCK rats, decreasing fibrosis and inflammation.114 Importantly, the dose of marimastat used was reported as nontoxic in clinical trials for cancer treatment.116,117 Thus, inhibition of matrix metalloprotease hyperactivity in cystic cholangio cytes has emerged as a potential therapeutic tool for the treatment of patients with PCLDs and needs to be validated in future clinical trials.
Epigenetic abnormalities
In addition to genetic mutations, epigenetic alterations such as abnormal expression of microRNAs (miRNAs) and histone deacetylases have been associated with the hyperproliferative phenotype of cystic cholangiocytes (Figure 1h). Global changes in miRNA levels were reported in cholangiocytes isolated from PCK rats compared with control animals. Out of 109 differentially expressed miRNAs analysed by microarray, 97 (~89%) were downregulated and 12 (~11%) were overexpressed in PCK rats.118 Downregulation of the majority of miRNAs in PCK rat cholangiocytes was associated with overexpression of their predicted target proteins involved in proliferation, secretion and cell–extracellular matrix interactions.118 In particular, miRNA-15a was highly underexpressed in cultured PCK rat cholangiocytes, as well as in cystic tissue from PCK rats and patients with PCLDs. Downregulation of miRNA-15a led to overexpression of the cell division cycle 25A (Cdc25A) protein.118 Experimental upregulation of miRNA-15a in PCK rat cholangiocytes decreased levels of Cdc25A, which halted cholangiocyte hyperproliferation and cyst growth. By contrast, inhibition of miRNA-15a in normal rat cholangiocytes promoted cell proliferation, increased Cdc25A levels and accelerated cyst growth.118 To determine the potential therapeutic value of targeting Cdc25A in patients with PCLDs, Cdc25A+/− mice (with decreased Cdc25A expression but normal liver morphology) were cross-bred with Pkhd1del2/del2 mice (which overexpress Cdc25A and develop hepatic cysts). As expected, liver weights, hepatic cystogenesis and fibrosis were reduced in double mutants compared with Pkhd1del2/del2 mice.75 In addition, pharmacological inhibition of Cdc25A with vitamin K3 or PM-20 decreased hepatorenal cystogenesis and fibrosis in PCK rats and Pkd2WS25/− mice by affecting cell cycle progression and proliferation.75
Histone deacetylase 6, which is involved in the regulation of the cell cycle and ciliary disassembly, has also been associated with the pathogenesis of PCLDs. Histone deacetylase 6 is overexpressed in cystic cholangiocytes from both PCK rats and patients with PCLDs,119 and pharmacological inhibition of this complex with tubastatin-A, tubacin or ACY-1215 all decreased proliferation of PCK rat cholangiocytes in vitro in a dose-dependent and time-dependent manner. Moreover, treatment of PCK rats with ACY-1215 attenuated hepatic cystogenesis and fibrosis, which indicates that targeting histone deacetylase 6 might be a useful therapeutic approach.119
Conclusions
Despite advances in our understanding of the mechanisms of hepatic cystogenesis and the discovery of potential therapeutic targets, the available treatment options (conservative management, surgery and medical therapies) are limited.1,22,25,120,121 Novel approaches are mainly focused on the cAMP signalling pathway1,61,120,122 and evaluating the effect of somatostatin analogues on hepatic cystogenesis. However, given that the benefits of using somatostatin analogues are modest, identification of new targets for therapeutic intervention is urgently needed. Several new targets (for example, histone deacetylase 6, Cdc25A phosphatase, PPAR-γ and matrix metalloproteases)75,90,92,114,119 have already been evaluated in preclinical studies and need to be tested clinically (Table 4). Moreover, downregulation of the intracellular calcium levels in cholangiocytes seems to be a central event in hepatic cystogenesis. Therefore, restoration of these levels in cystic cholangiocytes seems to be a promising therapeutic strategy. Advances in the field of PCLD research and the discovery of new mutations in genes involved in disease susceptibility, such as LRP5 in ADPLD, has highlighted new signalling pathways, such as Wnt signalling, that could be pharmacologically important to target. Finally, the possibility exists that combined therapeutic strategies might have an additive or synergistic effect.
Table 4.
Preclinical studies in animal models of polycystic liver diseases
| Study | Drug | Animal model | Treatment | Results |
|---|---|---|---|---|
| cAMP | ||||
| Masyuk et al. (2007)97 | Octreotide | PCK rat | Intraperitoneally (10μg/kg daily) for 4, 8, 12, and 16 weeks or 100μg/kg daily for 4 weeks | Reduction of liver weight, cyst volume, hepatic fibrosis, and mitotic indices |
| Masyuk et al. (2013)99 | Pasireotide | PCK rat and Pkd2WS25/− mouse | Osmotic mini-pumps (20 μg/kg daily) for 6 weeks | Reduction of liver weight, cyst volume, hepatic fibrosis and mitotic indices |
| Raf kinase | ||||
| Spirli et al. (2012)89 | Sorafenib | Pkd2fox/−:pCxCreER™ (Pkd2cKO mouse) | Orally (20–60 mg/kg daily) for 8 weeks | Increase in liver cyst area, cell proliferation and phosphorylated Erk |
| mTOR | ||||
| Spirli et al. (2010)73 | Sirolimus | Pkd2fox/−:pCxCreER™ (Pkd2cKO mouse) | Intraperitoneally (1.5 mg/kg daily) for 8 weeks | Reduction of liver cyst area, liver weight and Pcna expression |
| Renken et al. (2011)84 | Sirolimus | PCK rat | In drinking water (2 mg/kg daily) for 4, 8 or 12 weeks | Failed to attenuate hepatorenal cystogenesis |
| Cdc25A | ||||
| Masyuk et al. (2012)75 | Vitamin K3 | Pkd2WS25/− mouse and PCK rat | In drinking water (0.15 g/l) for 4 and 8 weeks | Reduction of both liver and kidney weights and hepatorenal cystic and fibrotic areas |
| Masyuk et al. (2012)75 | PM-20 | Pkd2WS25/− mouse | Intraperitoneally (1 mg/kg) for 4 weeks | Reduction of hepatorenal cystogenesis |
| MMPs | ||||
| Urribarri et al. (2014)114 | Marimastat | PCK rat | Orally twice a day (0.29 mg/kg daily) for 8 weeks | Reduction of hepatic cystogenesis, fibrosis and inflammation |
| Hdac6 | ||||
| Gradilone et al. (2014)119 | ACY-1215 | PCK rat | Intraperitoneally (30 mg/kg daily) for 4 weeks | Reduction of hepatic cystogenesis and fibrosis |
| Ppar-γ | ||||
| Yoshihara et al. (2011)90 | Pioglitazone (full agonist) | PCK rat | Orally (10 mg/kg daily) for 16 weeks | Reduction of liver weight, liver cystic area and fibrotic index |
| Yoshihara et al. (2013)92 | Telmisartan (partial agonist) | PCK rat | Orally (3 mg/kg daily) for 16 weeks | Reduction of liver weight, cystic and fibrotic areas |
| Vegfr2 | ||||
| Spirli et al. (2010)74 | SU5416 | Pkd1KO and Pkd2KO mice | Intraperitoneally (12.5 mg/kg) twice a week for 8 weeks | Reduction of hepatic cystogenesis, liver weight and levels of phosphorylated Erk and Pcna in Pkd2KO but not Pkd1KO mice |
| Amura et al. (2007)82 | SU5416 | Pkd2WS25/ mouse | Subcutaneously (0.75 mg) every 2 weeks for 4 and 8 months | Reduction of liver cystic area |
| Egfr | ||||
| Torres et al. (2004)81 | EKI-785 and EKB-569 | PCK rat | Intraperitoneally (EKI-785: 90 mg/kg) every 3 days Intraperitoneally (EKB-569: 20 mg/kg) every 3 days Orally (EKB-569: 5, 10 or 20 mg/kg) every 3 days All administered for 7 weeks |
No effects on fibrocystic liver disease |
| Trpv4 | ||||
| Gradilone et al. (2010)65 | Trpv4 activator GSK1016790A | PCK rat | Intraperitoneally (0.01 mg/kg daily) for 8 weeks | Trpv4 activation induced a significant decrease in renal cystic area and a nonsignificant decrease in liver cysts |
Abbreviations: cAMP, cyclic adenosine monophosphate; Cdc25A, cyclin division cycle 25A; Egfr, epidermal growth factor receptor; Erk, extracellular-regulated kinase; Hdac6, histone deacetylase 6; MMP, matrix metalloprotease; mTOR, mammalian target of rapamycin; Pcna, proliferating cell nuclear antigen; PPAR-y, peroxisome proliferator-activated receptor gamma; Raf, rapidly accelerated fibrosarcoma; Trpv4, transient receptor potential cation channel, subfamily V, member 4; Vegfr2, vascular endothelial growth factor receptor 2.
Key points.
-
■
Proteins encoded by the genes that cause polycystic liver diseases are predominantly localized in the primary cilium, plasma membrane and/or the endoplasmic reticulum of cholangiocytes
-
■
Current treatments are based on surgical procedures and/or pharmacological management; however, their beneficial effects are modest, leaving liver transplantation as the only definitive remedy
-
■
Elucidating the molecular mechanisms involved in the pathogenesis of these disorders is crucial in order to identify new potential targets for therapy
-
■
Hepatic cystogenesis is characterized by ductal plate malformation, abnormalities of the cholangiocyte primary cilium, centrosome amplification, hyperproliferation, hypersecretion, matrix-metalloprotease hyperactivity, angiogenesis, epigenetic alterations and atypical levels of key intracellular mediators
-
■
Preclinical studies have revealed new potential therapeutic targets that need to be validated in future clinical trials
Footnotes
Review criteria
We searched PubMed (June 2014) using the following terms: “polycystic liver diseases”, “cystogenesis”, “ADPKD”, “ADPLD”, “ARPKD”, “congenital hepatic fibrosis”, “Caroli disease”, “primary cilium”, “ductal plate malformation”, “cystic fluid”, “cystogenesis”, “biliary dilatation”, “cystic cholangiocytes”, “molecular pathways”, “proliferation”, “secretion”, “angiogenesis”, “cell–extracellular matrix”, “microRNAs”, “epigenetics”, “oestrogens”, “cAMP”, “calcium”, “treatment” and “clinical trials”. All selected papers were full-text in English. We searched the reference lists of identified papers for further relevant papers.
Competing interests
The authors declare no competing interests.
Author contributions
All authors contributed equally to all aspects of this manuscript.
References
- 1.Gevers TJ, Drenth JP. Diagnosis and management of polycystic liver disease. Nat Rev Gastroenterol Hepatol. 2013;10:101–108. doi: 10.1038/nrgastro.2012.254. [DOI] [PubMed] [Google Scholar]
- 2.Drenth JP, te Morsche RH, Smink R, Bonifacino JS, Jansen JB. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat Genet. 2003;33:345–347. doi: 10.1038/ng1104. [DOI] [PubMed] [Google Scholar]
- 3.Li A, et al. Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am J Hum Genet. 2003;72:691–703. doi: 10.1086/368295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davila S, et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet. 2004;36:575–577. doi: 10.1038/ng1357. [DOI] [PubMed] [Google Scholar]
- 5.Cnossen WR, et al. Whole-exome sequencing reveals LRP5 mutations and canonical Wnt signaling associated with hepatic cystogenesis. Proc Natl Acad Sci USA. 2014;111:5343–5348. doi: 10.1073/pnas.1309438111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. The European Polycystic Kidney Disease Consortium. Cell. 1994;77:881–894. doi: 10.1016/0092-8674(94)90137-6. [No authors listed] [DOI] [PubMed] [Google Scholar]
- 7.Mochizuki T, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–1342. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 8.Ward CJ, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet. 2002;30:259–269. doi: 10.1038/ng833. [DOI] [PubMed] [Google Scholar]
- 9.Strazzabosco M, Somlo S. Polycystic liver diseases: congenital disorders of cholangiocyte signaling. Gastroenterology. 2011;140:1855–1859. doi: 10.1053/j.gastro.2011.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rossetti S, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007;18:2143–2160. doi: 10.1681/ASN.2006121387. [DOI] [PubMed] [Google Scholar]
- 11.Waanders E, te Morsche RH, de Man RA, Jansen JB, Drenth JP. Extensive mutational analysis of PRKCSH and SEC63 broadens the spectrum of polycystic liver disease. Hum Mutat. 2006;27:830. doi: 10.1002/humu.9441. [DOI] [PubMed] [Google Scholar]
- 12.Waanders E, et al. Secondary and tertiary structure modeling reveals effects of novel mutations in polycystic liver disease genes PRKCSH and SEC63. Clin Genet. 2010;78:47–56. doi: 10.1111/j.1399-0004.2009.01353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ong AC, Harris PC. Molecular pathogenesis of ADPKD: the polycystin complex gets complex. Kidney Int. 2005;67:1234–1247. doi: 10.1111/j.1523-1755.2005.00201.x. [DOI] [PubMed] [Google Scholar]
- 14.Geng L, et al. Syntaxin 5 regulates the endoplasmic reticulum channel-release properties of polycystin-2. Proc Natl Acad Sci USA. 2008;105:15920–15925. doi: 10.1073/pnas.0805062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Al-Bhalal L, Akhtar M. Molecular basis of autosomal recessive polycystic kidney disease (ARPKD) Adv Anat Pathol. 2008;15:54–58. doi: 10.1097/PAP.0b013e31815e5295. [DOI] [PubMed] [Google Scholar]
- 16.Janssen MJ, Salomon J, te Morsche RH, Drenth JP. Loss of heterozygosity is present in SEC63 germline carriers with polycystic liver disease. PLoS ONE. 2012;7:e50324. doi: 10.1371/journal.pone.0050324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janssen MJ, et al. Secondary, somatic mutations might promote cyst formation in patients with autosomal dominant polycystic liver disease. Gastroenterology. 2011;141:2056–2063. doi: 10.1053/j.gastro.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 18.Watnick TJ, et al. Somatic mutation in individual liver cysts supports a two-hit model of cystogenesis in autosomal dominant polycystic kidney disease. Mol Cell. 1998;2:247–251. doi: 10.1016/s1097-2765(00)80135-5. [DOI] [PubMed] [Google Scholar]
- 19.Pei Y, et al. Somatic PKD2 mutations in individual kidney and liver cysts support a “two-hit” model of cystogenesis in type 2 autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1999;10:1524–1529. doi: 10.1681/ASN.V1071524. [DOI] [PubMed] [Google Scholar]
- 20.Watnick T, et al. Mutations of PKD1 in ADPKD2 cysts suggest a pathogenic effect of trans-heterozygous mutations. Nat Genet. 2000;25:143–144. doi: 10.1038/75981. [DOI] [PubMed] [Google Scholar]
- 21.Banales JM, Munoz-Garrido P, Bujanda L. Somatic second-hit mutations leads to polycystic liver diseases. World J Gastroenterol. 2013;19:141–143. doi: 10.3748/wjg.v19.i1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chandok N. Polycystic liver disease: a clinical review. Ann Hepatol. 2012;11:819–826. [PubMed] [Google Scholar]
- 23.Raynaud P, Carpentier R, Antoniou A, Lemaigre FP. Biliary differentiation and bile duct morphogenesis in development and disease. Int J Biochem Cell Biol. 2011;43:245–256. doi: 10.1016/j.biocel.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 24.Wills ES, Roepman R, Drenth JP. Polycystic liver disease: ductal plate malformation and the primary cilium. Trends Mol Med. 2014;20:261–270. doi: 10.1016/j.molmed.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 25.Temmerman F, et al. Systematic review: the pathophysiology and management of polycystic liver disease. Aliment Pharmacol Ther. 2011;34:702–713. doi: 10.1111/j.1365-2036.2011.04783.x. [DOI] [PubMed] [Google Scholar]
- 26.Gunay-Aygun M. Liver and kidney disease in ciliopathies. Am J Med Genet C Semin Med Genet. 2009;151C:296–306. doi: 10.1002/ajmg.c.30225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Godlewski G, Gaubert-Cristol R, Rouy S, Prudhomme M. Liver development in the rat and in man during the embryonic period (Carnegie stages 11–23) Microsc Res Tech. 1997;39:314–327. doi: 10.1002/(SICI)1097-0029(19971115)39:4<314::AID-JEMT2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 28.Carpentier R, et al. Embryonic ductal plate cells give rise to cholangiocytes, periportal hepatocytes, and adult liver progenitor cells. Gastroenterology. 2011;141:1432–1438. doi: 10.1053/j.gastro.2011.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Desmet VJ. Ludwig symposium on biliary disorders—part I. Pathogenesis of ductal plate abnormalities. Mayo Clin Proc. 1998;73:80–89. doi: 10.4065/73.1.80. [DOI] [PubMed] [Google Scholar]
- 30.Raynaud P, et al. A classification of ductal plate malformations based on distinct pathogenic mechanisms of biliary dysmorphogenesis. Hepatology. 2011;53:1959–1966. doi: 10.1002/hep.24292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crawford JM. Development of the intrahepatic biliary tree. Semin Liver Dis. 2002;22:213–226. doi: 10.1055/s-2002-34508. [DOI] [PubMed] [Google Scholar]
- 32.Ezratty EJ, et al. A role for the primary cilium in Notch signaling and epidermal differentiation during skin development. Cell. 2011;145:1129–1141. doi: 10.1016/j.cell.2011.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lopes SS, et al. Notch signalling regulates left-right asymmetry through ciliary length control. Development. 2010;137:3625–3632. doi: 10.1242/dev.054452. [DOI] [PubMed] [Google Scholar]
- 34.Shih HP, et al. A Notch-dependent molecular circuitry initiates pancreatic endocrine and ductal cell differentiation. Development. 2012;139:2488–2499. doi: 10.1242/dev.078634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sato Y, et al. Cholangiocytes with mesenchymal features contribute to progressive hepatic fibrosis of the polycystic kidney rat. Am J Pathol. 2007;171:1859–1871. doi: 10.2353/ajpath.2007.070337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hassane S, et al. Elevated TGFβ-Smad signalling in experimental Pkd1 models and human patients with polycystic kidney disease. J Pathol. 2010;222:21–31. doi: 10.1002/path.2734. [DOI] [PubMed] [Google Scholar]
- 37.Decaens T, et al. Stabilization of β-catenin affects mouse embryonic liver growth and hepatoblast fate. Hepatology. 2008;47:247–258. doi: 10.1002/hep.21952. [DOI] [PubMed] [Google Scholar]
- 38.May-Simera HL, Kelley MW. Cilia, Wnt signaling, and the cytoskeleton. Cilia. 2012;1:7. doi: 10.1186/2046-2530-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benzing T, Simons M, Walz G. Wnt signaling in polycystic kidney disease. J Am Soc Nephrol. 2007;18:1389–1398. doi: 10.1681/ASN.2006121355. [DOI] [PubMed] [Google Scholar]
- 40.Yanai M, et al. FGF signaling segregates biliary cell-lineage from chick hepatoblasts cooperatively with BMP4 and ECM components in vitro. Dev Dyn. 2008;237:1268–1283. doi: 10.1002/dvdy.21520. [DOI] [PubMed] [Google Scholar]
- 41.Neugebauer JM, Amack JD, Peterson AG, Bisgrove BW, Yost HJ. FGF signalling during embryo development regulates cilia length in diverse epithelia. Nature. 2009;458:651–654. doi: 10.1038/nature07753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pavik I, et al. Patients with autosomal dominant polycystic kidney disease have elevated fibroblast growth factor 23 levels and a renal leak of phosphate. Kidney Int. 2011;79:234–240. doi: 10.1038/ki.2010.375. [DOI] [PubMed] [Google Scholar]
- 43.Clotman F, et al. The onecut transcription factor HNF6 is required for normal development of the biliary tract. Development. 2002;129:1819–1828. doi: 10.1242/dev.129.8.1819. [DOI] [PubMed] [Google Scholar]
- 44.Hunter MP, et al. The homeobox gene Hhex is essential for proper hepatoblast differentiation and bile duct morphogenesis. Dev Biol. 2007;308:355–367. doi: 10.1016/j.ydbio.2007.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pierreux CE, et al. The transcription factor hepatocyte nuclear factor-6 controls the development of pancreatic ducts in the mouse. Gastroenterology. 2006;130:532–541. doi: 10.1053/j.gastro.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 46.Guay-Woodford LM, Green WJ, Lindsey JR, Beier DR. Germline and somatic loss of function of the mouse cpk gene causes biliary ductal pathology that is genetically modulated. Hum Mol Genet. 2000;9:769–778. doi: 10.1093/hmg/9.5.769. [DOI] [PubMed] [Google Scholar]
- 47.Hou X, et al. Cystin, a novel cilia-associated protein, is disrupted in the cpk mouse model of polycystic kidney disease. J Clin Invest. 2002;109:533–540. doi: 10.1172/JCI14099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hiesberger T, et al. Mutation of hepatocyte nuclear factor-1β inhibits Pkhd1 gene expression and produces renal cysts in mice. J Clin Invest. 2004;113:814–825. doi: 10.1172/JCI20083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gresh L, et al. A transcriptional network in polycystic kidney disease. EMBO J. 2004;23:1657–1668. doi: 10.1038/sj.emboj.7600160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamasaki H, et al. Suppression of C/EBPα expression in periportal hepatoblasts may stimulate biliary cell differentiation through increased Hnf6 and Hnf1b expression. Development. 2006;133:4233–4243. doi: 10.1242/dev.02591. [DOI] [PubMed] [Google Scholar]
- 51.Hand NJ, et al. The microRNA-30 family is required for vertebrate hepatobiliary development. Gastroenterology. 2009;136:1081–1090. doi: 10.1053/j.gastro.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Masyuk AI, Masyuk TV, LaRusso NF. Cholangiocyte primary cilia in liver health and disease. Dev Dyn. 2008;237:2007–2012. doi: 10.1002/dvdy.21530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Masyuk AI, et al. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling. Gastroenterology. 2006;131:911–920. doi: 10.1053/j.gastro.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Masyuk TV, et al. Defects in cholangiocyte fibrocystin expression and ciliary structure in the PCK rat. Gastroenterology. 2003;125:1303–1310. doi: 10.1016/j.gastro.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 55.Masyuk TV, et al. Biliary dysgenesis in the PCK rat, an orthologous model of autosomal recessive polycystic kidney disease. Am J Pathol. 2004;165:1719–1730. doi: 10.1016/S0002-9440(10)63427-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stroope A, et al. Hepato-renal pathology in Pkd2ws25/– mice, an animal model of autosomal dominant polycystic kidney disease. Am J Pathol. 2010;176:1282–1291. doi: 10.2353/ajpath.2010.090658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alvaro D, Mancino MG. New insights on the molecular and cell biology of human cholangiopathies. Mol Aspects Med. 2008;29:50–57. doi: 10.1016/j.mam.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 58.Masyuk TV, et al. Centrosomal abnormalities characterize human and rodent cystic cholangiocytes and are associated with Cdc25A overexpression. Am J Pathol. 2014;184:110–121. doi: 10.1016/j.ajpath.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Battini L, et al. Loss of polycystin-1 causes centrosome amplification and genomic instability. Hum Mol Genet. 2008;17:2819–2833. doi: 10.1093/hmg/ddn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Woollard JR, et al. A mouse model of autosomal recessive polycystic kidney disease with biliary duct and proximal tubule dilatation. Kidney Int. 2007;72:328–336. doi: 10.1038/sj.ki.5002294. [DOI] [PubMed] [Google Scholar]
- 61.Torres VE, Harris PC. Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol. 2014;25:18–32. doi: 10.1681/ASN.2013040398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Torrice A, et al. Polycystins play a key role in the modulation of cholangiocyte proliferation. Dig Liver Dis. 2010;42:377–385. doi: 10.1016/j.dld.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 63.Masyuk AI, et al. Ciliary subcellular localization of TGR5 determines the cholangiocyte functional response to bile acid signaling. Am J Physiol Gastrointest Liver Physiol. 2013;304:G1013–G1024. doi: 10.1152/ajpgi.00383.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gradilone SA, et al. Cholangiocyte cilia express TRPV4 and detect changes in luminal tonicity inducing bicarbonate secretion. Proc Natl Acad Sci USA. 2007;104:19138–19143. doi: 10.1073/pnas.0705964104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gradilone SA, et al. Activation of Trpv4 reduces the hyperproliferative phenotype of cystic cholangiocytes from an animal model of ARPKD. Gastroenterology. 2010;139:304–314. doi: 10.1053/j.gastro.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Onori P, et al. Polycystic liver diseases. Dig Liver Dis. 2010;42:261–271. doi: 10.1016/j.dld.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Muchatuta MN, Gattone VH, 2nd, Witzmann FA, Blazer-Yost BL. Structural and functional analyses of liver cysts from the BALB/c-cpk mouse model of polycystic kidney disease. Exp Biol Med (Maywood) 2009;234:17–27. doi: 10.3181/0807-RM-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Banales JM, et al. Hepatic cystogenesis is associated with abnormal expression and location of ion transporters and water channels in an animal model of autosomal recessive polycystic kidney disease. Am J Pathol. 2008;173:1637–1646. doi: 10.2353/ajpath.2008.080125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fedeles SV, et al. A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nat Genet. 2011;43:639–647. doi: 10.1038/ng.860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fedeles SV, Gallagher AR, Somlo S. Polycystin-1: a master regulator of intersecting cystic pathways. Trends Mol Med. 2014;20:251–260. doi: 10.1016/j.molmed.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet. 2013;45:1004–1012. doi: 10.1038/ng.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Alvaro D, et al. Morphological and functional features of hepatic cyst epithelium in autosomal dominant polycystic kidney disease. Am J Pathol. 2008;172:321–332. doi: 10.2353/ajpath.2008.070293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Spirli C, et al. Mammalian target of rapamycin regulates vascular endothelial growth factor-dependent liver cyst growth in polycystin 2 defective mice. Hepatology. 2010;51:1778–1788. doi: 10.1002/hep.23511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Spirli C, et al. ERK1/2-dependent vascular endothelial growth factor signaling sustains cyst growth in polycystin-2 defective mice. Gastroenterology. 2010;138:360–371. doi: 10.1053/j.gastro.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Masyuk TV, et al. Inhibition of Cdc25A suppresses hepato-renal cystogenesis in rodent models of polycystic kidney and liver disease. Gastroenterology. 2012;142:622–633. doi: 10.1053/j.gastro.2011.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Waanders E, Van Krieken JH, Lameris AL, Drenth JP. Disrupted cell adhesion but not proliferation mediates cyst formation in polycystic liver disease. Mod Pathol. 2008;21:1293–1302. doi: 10.1038/modpathol.2008.115. [DOI] [PubMed] [Google Scholar]
- 77.Nichols MT, et al. Secretion of cytokines and growth factors into autosomal dominant polycystic kidney disease liver cyst fluid. Hepatology. 2004;40:836–846. doi: 10.1002/hep.20401. [DOI] [PubMed] [Google Scholar]
- 78.Fabris L, et al. Effects of angiogenic factor overexpression by human and rodent cholangiocytes in polycystic liver diseases. Hepatology. 2006;43:1001–1012. doi: 10.1002/hep.21143. [DOI] [PubMed] [Google Scholar]
- 79.Amura CR, et al. CXCR2 agonists in ADPKD liver cyst fluids promote cell proliferation. Am J Physiol Cell Physiol. 2008;294:C786–C796. doi: 10.1152/ajpcell.00457.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sato Y, et al. Activation of the MEK5/ERK5 cascade is responsible for biliary dysgenesis in a rat model of Caroli’s disease. Am J Pathol. 2005;166:49–60. doi: 10.1016/S0002-9440(10)62231-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Torres VE, et al. Epidermal growth factor receptor tyrosine kinase inhibition is not protective in PCK rats. Kidney Int. 2004;66:1766–1773. doi: 10.1111/j.1523-1755.2004.00952.x. [DOI] [PubMed] [Google Scholar]
- 82.Amura CR, et al. VEGF receptor inhibition blocks liver cyst growth in pkd2WS25/− mice. Am J Physiol Cell Physiol. 2007;293:C419–C428. doi: 10.1152/ajpcell.00038.2007. [DOI] [PubMed] [Google Scholar]
- 83.Brodsky KS, McWilliams RR, Amura CR, Barry NP, Doctor RB. Liver cyst cytokines promote endothelial cell proliferation and development. Exp Biol Med (Maywood) 2009;234:1155–1165. doi: 10.3181/0903-RM-112. [DOI] [PubMed] [Google Scholar]
- 84.Renken C, Fischer DC, Kundt G, Gretz N, Haffner D. Inhibition of mTOR with sirolimus does not attenuate progression of liver and kidney disease in PCK rats. Nephrol Dial Transplant. 2011;26:92–100. doi: 10.1093/ndt/gfq384. [DOI] [PubMed] [Google Scholar]
- 85.Qian Q, et al. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol. 2008;19:631–638. doi: 10.1681/ASN.2007050626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Walz G, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:830–840. doi: 10.1056/NEJMoa1003491. [DOI] [PubMed] [Google Scholar]
- 87.Serra AL, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:820–829. doi: 10.1056/NEJMoa0907419. [DOI] [PubMed] [Google Scholar]
- 88.Chrispijn M, et al. Everolimus does not further reduce polycystic liver volume when added to long acting octreotide: results from a randomized controlled trial. J Hepatol. 2013;59:153–159. doi: 10.1016/j.jhep.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 89.Spirli C, et al. Cyclic AMP/PKA-dependent paradoxical activation of Raf/MEK/ERK signaling in polycystin-2 defective mice treated with sorafenib. Hepatology. 2012;56:2363–2374. doi: 10.1002/hep.25872. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 90.Yoshihara D, et al. PPAR-γ agonist ameliorates kidney and liver disease in an orthologous rat model of human autosomal recessive polycystic kidney disease. Am J Physiol Renal Physiol. 2011;300:F465–F474. doi: 10.1152/ajprenal.00460.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yoshihara D, et al. Global gene expression profiling in PPAR-γ agonist-treated kidneys in an orthologous rat model of human autosomal recessive polycystic kidney disease. PPAR Res. 2012;2012:695898. doi: 10.1155/2012/695898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yoshihara D, et al. Telmisartan ameliorates fibrocystic liver disease in an orthologous rat model of human autosomal recessive polycystic kidney disease. PLoS ONE. 2013;8:e81480. doi: 10.1371/journal.pone.0081480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chapman AB. Cystic disease in women: clinical characteristics and medical management. Adv Ren Replace Ther. 2003;10:24–30. doi: 10.1053/jarr.2003.50005. [DOI] [PubMed] [Google Scholar]
- 94.Gabow PA, et al. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology. 1990;11:1033–1037. doi: 10.1002/hep.1840110619. [DOI] [PubMed] [Google Scholar]
- 95.Sherstha R, et al. Postmenopausal estrogen therapy selectively stimulates hepatic enlargement in women with autosomal dominant polycystic kidney disease. Hepatology. 1997;26:1282–1286. doi: 10.1002/hep.510260528. [DOI] [PubMed] [Google Scholar]
- 96.Alvaro D, et al. Estrogens and the pathophysiology of the biliary tree. World J Gastroenterol. 2006;12:3537–3545. doi: 10.3748/wjg.v12.i22.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3′, 5′-cyclic monophosphate. Gastroenterology. 2007;132:1104–1116. doi: 10.1053/j.gastro.2006.12.039. [DOI] [PubMed] [Google Scholar]
- 98.Banales JM, et al. The cAMP effectors Epac and protein kinase A (PKA) are involved in the hepatic cystogenesis of an animal model of autosomal recessive polycystic kidney disease (ARPKD) Hepatology. 2009;49:160–174. doi: 10.1002/hep.22636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Masyuk TV, et al. Pasireotide is more effective than octreotide in reducing hepatorenal cystogenesis in rodents with polycystic kidney and liver diseases. Hepatology. 2013;58:409–421. doi: 10.1002/hep.26140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Caroli A, et al. Reducing polycystic liver volume in ADPKD: effects of somatostatin analogue octreotide. Clin J Am Soc Nephrol. 2010;5:783–789. doi: 10.2215/CJN.05380709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hogan MC, et al. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol. 2010;21:1052–1061. doi: 10.1681/ASN.2009121291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hogan MC, et al. Somatostatin analog therapy for severe polycystic liver disease: results after 2 years. Nephrol Dial Transplant. 2012;27:3532–3539. doi: 10.1093/ndt/gfs152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.van Keimpema L, et al. Lanreotide reduces the volume of polycystic liver: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2009;137:1661–1668. doi: 10.1053/j.gastro.2009.07.052. [DOI] [PubMed] [Google Scholar]
- 104.Chrispijn M, et al. The long-term outcome of patients with polycystic liver disease treated with lanreotide. Aliment Pharmacol Ther. 2012;35:266–274. doi: 10.1111/j.1365-2036.2011.04923.x. [DOI] [PubMed] [Google Scholar]
- 105.Temmerman F, et al. Safety and efficacy of different lanreotide doses in the treatment of polycystic liver disease: pooled analysis of individual patient data. Aliment Pharmacol Ther. 2013;38:397–406. doi: 10.1111/apt.12384. [DOI] [PubMed] [Google Scholar]
- 106.Gevers TJ, et al. Young women with polycystic liver disease respond best to somatostatin analogues: a pooled analysis of individual patient data. Gastroenterology. 2013;145:357–365. doi: 10.1053/j.gastro.2013.04.055. [DOI] [PubMed] [Google Scholar]
- 107.US National Library of Medicine. ClincalTrials.gov [online] 2012 http://www.clinicaltrials.gov/ct2/show/NCT01670110.
- 108.Spirli C, et al. Altered store operated calcium entry increases cyclic 3′, 5′-adenosine monophosphate production and extracellular signal-regulated kinases 1 and 2 phosphorylation in polycystin 2 defective cholangiocytes. Hepatology. 2012;55:856–868. doi: 10.1002/hep.24723. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 109.Banales JM, Prieto J, Medina JF. Cholangiocyte anion exchange and biliary bicarbonate excretion. World J Gastroenterol. 2006;12:3496–3511. doi: 10.3748/wjg.v12.i22.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Banales JM, et al. Bicarbonate-rich choleresis induced by secretin in normal rat is taurocholate-dependent and involves AE2 anion exchanger. Hepatology. 2006;43:266–275. doi: 10.1002/hep.21042. [DOI] [PubMed] [Google Scholar]
- 111.Tietz PS, et al. Agonist-induced coordinated trafficking of functionally related transport proteins for water and ions in cholangiocytes. J Biol Chem. 2003;278:20413–20419. doi: 10.1074/jbc.M302108200. [DOI] [PubMed] [Google Scholar]
- 112.Perrone RD, et al. Autosomal dominant polycystic kidney disease decreases anion exchanger activity. Am J Physiol. 1997;272:C1748–C1756. doi: 10.1152/ajpcell.1997.272.5.C1748. [DOI] [PubMed] [Google Scholar]
- 113.Wang X, et al. Insignificant effect of secretin in rodent models of polycystic kidney and liver disease. Am J Physiol Renal Physiol. 2012;303:F1089–F1098. doi: 10.1152/ajprenal.00242.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Urribarri AD, et al. Inhibition of metalloprotease hyperactivity in cystic cholangiocytes halts the development of polycystic liver diseases. Gut. doi: 10.1136/gutjnl-2013-305281. http://dx.doi.org/10.1136/gutjnl-2013-305281. [DOI] [PMC free article] [PubMed]
- 115.Yasoshima M, et al. Matrix proteins of basement membrane of intrahepatic bile ducts are degraded in congenital hepatic fibrosis and Caroli’s disease. J Pathol. 2009;217:442–451. doi: 10.1002/path.2472. [DOI] [PubMed] [Google Scholar]
- 116.Nemunaitis J, et al. Combined analysis of studies of the effects of the matrix metalloproteinase inhibitor marimastat on serum tumor markers in advanced cancer: selection of a biologically active and tolerable dose for longer-term studies. Clin Cancer Res. 1998;4:1101–1109. [PubMed] [Google Scholar]
- 117.Rosenbaum E, et al. Marimastat in the treatment of patients with biochemically relapsed prostate cancer: a prospective randomized, double-blind, phase I/II trial. Clin Cancer Res. 2005;11:4437–4443. doi: 10.1158/1078-0432.CCR-04-2252. [DOI] [PubMed] [Google Scholar]
- 118.Lee SO, et al. MicroRNA15a modulates expression of the cell-cycle regulator Cdc25A and affects hepatic cystogenesis in a rat model of polycystic kidney disease. J Clin Invest. 2008;118:3714–3724. doi: 10.1172/JCI34922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gradilone SA, et al. HDAC6 is overexpressed in cystic cholangiocytes and its inhibition reduces cystogenesis. Am J Pathol. 2014;184:600–608. doi: 10.1016/j.ajpath.2013.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gevers TJ, Drenth JP. Somatostatin analogues for treatment of polycystic liver disease. Curr Opin Gastroenterol. 2011;27:294–300. doi: 10.1097/MOG.0b013e328343433f. [DOI] [PubMed] [Google Scholar]
- 121.Takiar V, Caplan MJ. Polycystic kidney disease: pathogenesis and potential therapies. Biochim Biophys Acta. 2011;1812:1337–1343. doi: 10.1016/j.bbadis.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Abu-Wasel B, Walsh C, Keough V, Molinari M. Pathophysiology, epidemiology, classification and treatment options for polycystic liver diseases. World J Gastroenterol. 2013;19:5775–5786. doi: 10.3748/wjg.v19.i35.5775. [DOI] [PMC free article] [PubMed] [Google Scholar]