

Abstract
RNA replicons represent potential vaccine delivery vehicles, but are considered too unstable for such use. This study examined the recovery, integrity and function of in vitro transcribed replicon RNA encoding hepatitis C virus (HCV) proteins. To remove residual template DNA, the RNA was digested with TURBO DNase followed by RNeasy DNase set and purified through an RNeasy column. The RNA was freeze-dried in distilled water or trehalose, stored under nitrogen gas for up to 10 months and analyzed at different time points. The recovery of RNA stored at ≤4°C that was freeze-dried in distilled water varied between 66% to zero of that recovered from RNA freeze-dried in 10% trehalose, a figure that depended on the duration of storage. In contrast, the recovery of the RNA stored in trehalose was consistently high for all time points. After recovery, both RNAs were translationally competent and expressed high levels of proteins after transfection, although the level of expression from the trehalose-stored RNA was consistently higher. Thus the addition of trehalose permitted stable storage of functional RNA at 4°C for up to 10 months and this permits the development of RNA vaccines, even in developing countries where only minimum storage conditions (e.g., 4°C) can be achieved.
INTRODUCTION
The use of nucleic acid vaccines is being investigated worldwide as a viable option for the prevention of infectious diseases (1–4; reviewed in 5,6). A major advantage of nucleic acid vaccines results from endogenous expression of the encoded protein(s) that may result in either direct processing of the protein in an antigen presenting cell or in cross presentation (7). Thus, expressed proteins are processed and presented through major histocompatibility complex (MHC) class I and class II pathways and stimulate CD4+ and CD8+ T-lymphocytes (1,8–10). DNA vaccines, first introduced in 1988 (11), are able to generate good immunity in small animals, particularly in common laboratory animals, but with a few exceptions have not been equally effective in non-human primates and humans (12). Furthermore, the risk of integration of the introduced DNA resulting in insertional mutagenesis is a recognized, although perhaps only theoretical, risk (13).
An alternative is to use RNA vaccines that retain the advantages of DNA vaccines without the disadvantages, including the aforementioned insertional mutagenesis and the possibility of inducing autoimmunity. RNA-based vaccines can be delivered in a number of different formulations (14). In particular, RNA replicons derived from positive-sense RNA viruses have considerable potential as vaccine delivery vehicles (15) and may be delivered as DNA, naked RNA, or RNA that is packaged into virus-like particles. We have previously used Kunjin virus replicon RNA to generate cytotoxic T cell (CTL) responses in mice (16), and this replicon shows promise as a vaccine delivery vehicle (17,18). RNA vaccines have the potential to induce strong cell mediated immunity (CMI) including CTL as well as antibody responses that would be advantageous in combating viral infections (19,20).
A general assumption regarding RNA vaccines, however, is that they are unstable and expensive. A recent review (14) presented a convincing argument that since it is possible to synthesize large amounts of RNA in vitro from a DNA template and because the bulk of the costs of a vaccine are related to regulatory affairs, this assumption is incorrect. Furthermore, it is axiomatic that RNA is only unstable in the presence of RNases and consequently it should be possible to synthesize and store RNA for a considerable period if contact with nucleases can be avoided.
Trehalose is a diglucose molecule that accumulates in certain organisms that naturally survive dehydration (21) and has demonstrated a protective effect when freezing mammalian cells (22). The addition of trehalose to reverse transcription PCR (RT-PCR) or ligase reactions can result in thermostabilization of enzymes and resolve secondary structures of RNA resulting in increased yield of long cDNA transcripts (23).
Consequently, the aim of this study was to develop a protocol for the synthesis and storage of the RNA replicon which was devoid of protein, DNA, and nucleases, and to examine whether the addition of trehalose increased RNA stability as a first step toward the more general introduction of self-replicating RNA vaccines.
MATERIALS AND METHODS
In Vitro Transcription of RNA
The Kunjin replicon vector plasmid encoding either the hepatitis C virus (HCV) NS3 or HCV core proteins (24) was linearized using the restriction enzyme XhoI according to the manufacturer’s recommendations (Promega, Sydney, Australia), and the DNA purified using High Pure PCR purification kit (Roche, West End, Australia). RNA was transcribed in vitro in a 20-μL reaction mix using the SP6 mMessage mMachine kit containing a 7-mG cap analog (Ambion, Thebarton, Australia) with the addition of 1 mM guanosine-5′-triphosphate (GTP), as recommended by the manufacturer. After different DNase treatments (described in the Results section), the RNA was purified using RNeasy kit spin purification columns (Qiagen, Doncaster, Australia), eluted from the column with nuclease-free water, and adjusted to a final concentration of 50 μg/mL in nuclease-free water (Promega). The quality and integrity of the RNA was assessed by gel electrophoresis in a 1% agarose TAE gel and visualized under UV light using 1 μg/mL ethidium bromide.
DNase Treatment
The efficiency of removal of the DNA template from the RNA preparation was compared using three sources of DNase: a) DNase I supplied with the mMessage mMachine in vitro transcription kit (Ambion), b) TURBO-DNase (Ambion) and c) the DNase-set for use with the spin purification columns (QIAGEN). These were each used according to the manufacturer’s instructions individually or in combination as described in Results. The integrity of the treated RNA was assessed on a 1% agarose Tris-acetate-EDTA (TAE) gel and visualized under UV light using 1 μg/ mL ethidium bromide.
PCR Detection of Residual DNA
Residual DNA in purified RNA preparations was examined by PCR by the addition of 1 μL RNA to a 50-μL reaction using HCV core gene-specific primers CORE S 578→5′-TCTATGGCAATGAGGGCATGG-3′ and CORE AS 741→5′-GAGCGGAATGTACCCCATGAG-3′.
The parameters of the reaction were 94°C for 5 min; 30 cycles of 94°C for 30 s; 55°C for 30 s; 72°C for 30 s; and a final extension of 72°C for 7 min. PCR products (163 bp) were detected by gel electrophoresis in a 3% agarose TAE gel and visualized under UV light using 1 μg/mL ethidium bromide. Distilled water and linearized plasmid DNA, or non-DNase-treated RNA represented negative and positive controls, respectively.
Freeze-Drying
The purified RNA was diluted 1:1 in water or 20% trehalose (in nuclease-free water) to a final concentration of 25 μg/mL RNA in water (RNA-W) or 10% trehalose (RNA-T). The RNA was dispensed in 200-μL aliquots (5 μg/vial) into sterile glass vials. The samples were frozen at −70°C, loaded into a freeze-drying apparatus, and a vacuum applied for 6 h at room temperature. The vacuum was broken with nitrogen gas and the vials sealed in situ. The samples were then stored at −70°, −20°, 4°, 37°C, or room temperature. Individual vials were removed for analysis at the following time points to examine if the freeze-drying process had any effect on RNA integrity, baseline (within 24 h of freeze-dry), 1 week, 1, 3, 6, and 10 months. At each time point, the vials were allowed to return to room temperature, and the RNA was re-dissolved in 200 μL nuclease-free water.
The yield and quality of the recovered RNA was examined by agarose gel electrophoresis analysis of a 40-μL aliquot. The intensity of the RNA bands at each time point was measured and analyzed using ImageGauge V4.2 software (Fujifilm, Berthold, Bundoora, Australia) by subtracting the background reading from that generated by the specific band and expressed as arbitrary units. The proportion of RNA recovered from the RNA-W samples was then estimated by comparison of the intensity of the RNA-W and RNA-T bands by ImageGauge.
Transfection
Huh7 cells were seeded into a 24-well plate (Corning, Clayton, Australia) and grown to 60% confluence. For each sample, 6 μL DMRIE (Invitrogen, Mt Waverley, Australia) and 80 μL of the reconstituted freeze-dried RNA (equivalent to a maximum of 2 μg) were added to 600 μL OptiMEM serum free medium (Invitrogen). The cells were washed once with OptiMEM and 300 μL transfection mixture were added to each well. Additional cultures were plated on glass coverslips for immunofluorescence studies and treated in the same way. The cells were incubated in a 37°C humidified 5% CO2 incubator for 4 h, the transfection medium then replaced with Dulbecco’s modified Eagle’s medium (DMEM)/10% fetal calf serum (FCS) and incubated for a further 48 h.
Immunofluorescence
The cells were washed in phosphate-buffered saline (PBS), air-dried for 10 min, then fixed in ice-cold acetone for 10 min. The coverslips were attached, cells facing up, to glass slides using DePeX (Biolab, Clayton, Australia) and stored at 4°C overnight. The cells were rehydrated in PBS, blocked using 2% FCS in PBS for 45 min, then stained with a 1:50 dilution of monoclonal anti-HCV-NS3 antibody (NovoCastra, Mt Waverley, Australia) for 1 h at 37°C, washed, then stained with 2 μg/mL Alexa 488 goat anti-mouse immunoglobulin G (IgG; Molecular Probes, Mt Waverley, Australia) at 37°C for 1 h. The cells were counterstained with propidium iodide (1 μg/mL), washed, and mounted in 80% glycerol, 100 mM Tris-HCl, pH 8.5, 100 mM NaCl then examined with a confocal microscope (Bio-Rad, Regents Park, Australia).
Immunoblot
The cells were removed by scraping into 400 μL 2 mM phenyl methyl sulfonyl fluoride (PMSF) in PBS. The cells were stored at −80°C until analysis, lysed with 5× Laemmli buffer, and the lysate heated to 90°C for 5 min before analysis on duplicate 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels. After transfer, one nitrocellulose membrane (Perkin Elmer, Waltham, MA, USA) was probed with anti-HCV-NS3 goat polyclonal antibody (ViroStat, Portland, OR, USA), and the other with anti-calreticulin rabbit polyclonal antibody (Stressgen, Stepney, Australia) to act as a control. This was followed by the relevant horseradish peroxidase (HRP)-conjugated antibody (HRP rabbit anti-goat, Dako, Botany, Australia; HRP donkey anti-rabbit, Amersham, Castle Hill, Australia), and positive bands were detected using the ECL kit (Amersham). The level of expression was determined by phosphorimager analysis and the relative expression of NS3 measured against a standardized level of calreticulin expression in the different samples. Data were analyzed using ImageGauge V4.2 software (Fujifilm).
RESULTS
DNase Treatment to Remove DNA from In Vitro Transcribed RNA
The potential for the use of in vitro transcribed RNA in vaccines may be compromised by contamination with template DNA, as regulatory authorities are likely to prefer DNA-free RNA. Consequently, we examined the RNA for any residual DNA. The conditions used in the PCR would only be expected to generate a product from DNA rather than RNA. In our hands, DNase-1, supplied with the mMessage mMachine kit, consistently failed to remove all traces of template, as determined by PCR analysis of the RNA preparation (Figure 1A, lane 2). Although this product was superseded by TURBO-DNase, which reduced the amount of contaminating DNA compared with the original method, there was still a detectable amount of DNA contamination of the RNA sample (Figure 1A, lane 3). The results using the DNase set supplied by Qiagen (Figure 1A, lane 4) also failed to digest all DNA. These results showed that no single DNase treatment, commonly used by workers in the field, was sufficient to eradicate all residual DNA in the RNA preparations (Figure 1A, lanes 2–4). We then compared the levels of residual DNA after combining DNase treatments (Figure 1A, lanes 5 and 6) and observed that TURBO DNase treatment followed by treatment with the Qiagen DNase set in the Qiagen column resulted in undetectable levels of DNA after 30 cycles of PCR (Figure 1A, lane 6). As the sensitivity of the PCR was determined to be 1 pg DNA (data not shown), this experiment determined that the RNA samples containing approximately 1 μg/mL RNA showed a residual DNA concentration of <1 pg/μL. Consequently, the most effective protocol to remove the DNA, using TURBO DNase and the DNase set followed by a Qiagen column, removed at least 99.99% of the DNA. Importantly, none of the DNase treatments had any effect on the yield and integrity (as determined by gel electrophoresis) of the RNA (Figure 1B). These results show that it is possible to remove DNA from in vitro transcribed RNA without compromising the quality of the RNA, thus making it safer for use as a vaccine delivery vehicle.
Figure 1. Analysis of residual DNA in preparations of in vitro transcribed RNA.
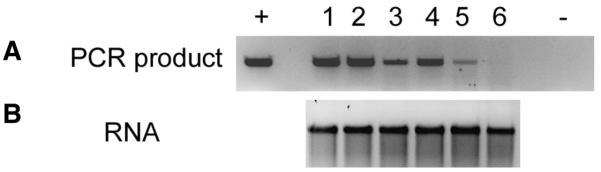
(A) The RNA samples were treated with DNase and then examined by PCR using primers from the hepatitis C virus (HCV) core gene region. Lanes + and − represent positive (linear vector DNA) and negative (distilled water) controls, respectively. Lane 1: no DNase treatment; lane 2, mMessage mMachine DNase 1; lane 3, TURBO DNase; lane 4, RNeasy DNase set; lane 5, mMessage mMachine DNase 1 + RNeasy DNase set; lane 6, TURBO DNase + RNeasy DNase-set. (B) Gel electrophoresis analysis of the RNA contained in each of the above samples.
Reconstitution of Freeze-Dried RNA
To determine if the freeze-dry process itself was detrimental to the integrity of the RNA, a vial of each sample was reconstituted and analyzed 24 h after freeze-drying. Initially the quantity of RNA which was recovered was determined by UV adsorption spectroscopy, but it became clear that this was inaccurate due to the high adsorption by degraded RNA and nucleotides. Thus, the efficiency and quality of RNA recovered was analyzed by agarose gel electrophoresis alongside freshly transcribed RNA. The results show that there was a 42% decrease in the intensity of the RNA-W band and 20% decrease in the RNA-T band relative to the fresh RNA (RNA-F) (Figure 2A). The intensity of the RNA-W band relative to the RNA-T band was 73%, suggesting that the RNA-W samples were either degraded during the freeze-dry process or that the recovery of these samples was reduced due to a decrease in the solubility of RNA dried in water as compared with that dried in trehalose.
Figure 2. Analysis of the integrity of stored, reconstituted RNA by gel electrophoresis.

The stored RNA was recovered in distilled water and a 40-μL aliquot, equivalent to 1 μg fresh RNA, analyzed by agarose gel electrophoresis. (A) Baseline RNA. RNA-F, freshly transcribed RNA; RNA-W, freeze-dried from water and stored for 24 h prior to recovery; RNA-T, freeze-dried from 10% trehalose and stored for 24 h prior to recovery. The numbers above each lane indicate the relative efficiency of recovery. The lower panels show the integrity of the RNA at different times after storage. (B) 1 week, (C) 1 month, (D) 3 months, (E) 6 months, (F) 10 months. The left and right panels show the results of the RNA-W and RNA-T, respectively. Note that in panel B, the exposure period was approximately half of the remaining panels, and in panel F the RNA-W gel was exposed for three times longer than the RNA-T panel to permit detection of the bands. Lanes marked with “F” in panel F represents freshly transcribed RNA.
The efficiency of recovery of RNA from the samples stored at different temperatures for increasing times was then determined. The efficiency of recovery of the RNA-T samples was determined in the 10-month samples by comparison of the intensity of the band against a fresh RNA sample using ImageGauge analysis (Figure 2F and Table 1). This showed that the efficiency of recovery of the RNA stored for 10 months at −70°, −20° or 4°C was approximately 80%, similar to the efficiency of recovery 24 h after freeze-drying (compare Figure 2, A and F). We assume that this level of efficiency was similar for recovery of the earlier RNA-T samples stored at these temperatures. The absolute recovery of the RNA-W was then estimated by comparison of the intensity of the RNA-W and RNA-T bands at each time point. The results (Figure 2, B–F and Table 1) show a reduced recovery of RNA-W (Table 1) with increased time in storage in contrast to the RNA-T samples. In contrast, the RNA in the RNA-T samples was observed as a distinct band, particularly at temperatures of 4°C or lower. In addition, as might be expected, there was minimal recovery of RNA-W stored at RT and 37°C, and little survived beyond week 1. The overall data are highly reproducible, and the lack of any RNA in the RNA-T sample in track 4, 1-month sample (Figure 2C) is clearly an outlier due to a technical anomaly. This result is inconsistent with the other RNA-T data which show strong bands in samples from all time points at lower temperatures but weaker bands from samples stored at room temperature and 37°C. Somewhat unexpectedly, RNA stored at 4°C in both water and trehalose appeared to be equally stable to RNA stored at −20° and −70°C. The data set for the 10 months samples was incomplete as no sample was stored at room temperature.
Table 1.
Quantity of RNA Recovered After Reconstitution as Visualized by Gel Electrophoresis
| RNA-W (μg) |
RNA-T (μg) |
|||||
|---|---|---|---|---|---|---|
| Temp (°C) | −70 | −20 | 4 | −70 | −20 | 4 |
| 1 week | 0.53 | 0.44 | 0.49 | 0.8 | 0.8 | 0.8 |
| 1 month | 0.55 | 0.43 | 0.39 | 0.8 | 0.8 | 0.8 |
| 3 months | 0.32 | 0.11 | 0.32 | 0.8 | 0.8 | 0.8 |
| 6 months | 0.15 | 0.15 | 0.20 | 0.8 | 0.8 | 0.8 |
| 10 months | 0.06 | 0.05 | 0 | 0.8 | 0.8 | 0.8 |
Forty-microliter samples from each time point were analyzed after recovery of the RNA. Complete recovery of the RNA would represent 1 μg. The method to calculate the recovery of each RNA sample is described in the Materials and Methods section.
In summary, the integrity of the RNA as determined by the appearance of a discrete band, and image analysis clearly demonstrated that RNA-T was superior to RNA-W and that storage at 4°C appeared superior to storage at −20° or −70°C. This improvement in RNA quality from the RNA-T samples is not likely to be due to increased solubility due to the presence of the sugar, as Figure 2A shows this to have a minimal effect on RNA recovery when the freeze-dried samples were reconstituted after 24 h.
Replication Competence of Reconstituted Replicon RNA
The described results showed distinct differences in the quality of the RNA stored at different temperatures and suggested that RNA-T was more stable than RNA-W. To confirm that structural integrity correlated with translational and replication competency, we transfected Huh7 cells and compared the level of expression of the HCV NS3 protein from the RNA samples stored for 6 months. The experiment was designed to ensure that the same proportion of the stored sample was used; although the data described previously showed that each sample contained unequal amounts of RNA, it was thought that this experimental design best reflected the aims of the study. The results of this experiment (Figure 3A) showed that transfection with the RNA-T resulted in a higher proportion of cells expressing the HCV NS3 protein compared with the RNA-W samples, although the latter produced a surprisingly high level of expression.
Figure 3. Stored RNA is translationally and replication competent after recovery.

RNA stored for 6 months was recovered, redissolved in water, and used to transfect Huh7 cells. The expression of the hepatitis C virus (HCV) NS3 protein was determined by (A) immunofluorescence and (B) immunoblot. (C) The immunoblot was quantified using ImageGauge software. (A) The upper panels show the immunofluorescence results using RNA-W, and the lower panels the results using RNA-T. The cells were counterstained with propidium iodide to identify nuclei (red) and the percentage of NS3-positive cells is noted in each panel. (B) Lysates of Huh7 cells, transfected with reconstituted RNA-W (upper left panel) or RNA-T (upper right panel), were analyzed by probing blots with anti-NS3 polyclonal antibody. The expression of NS3 in the RNA-W and RNA-T samples was compared against calreticulin immunoblotted in the same assay as a loading control. The −70°C RNA-W sample was unavailable due to technical problems. (C) Relative expression of NS3 normalized against calreticulin expression, as determined by ImageGauge analysis.
Approximately 45% of cells expressed the viral protein after transfection with RNA-T or RNA-W that was stored at −70°C (Figure 3A). RNA-T that was stored at −20° or 4°C also showed a high transfection efficiency (49% and 43%, respectively) and only the samples which were stored at RT or 37°C showed decreased levels of transfection of 22% and 12%, respectively (Figure 3A), consistent with the data in Figure 2E. RNA-W showed considerably reduced levels of transfection. Samples stored at temperatures higher than −70°C showed a temperature-related loss of transfection efficiency so that RNA-W stored at −20° and 4°C produced transfection efficiencies of 25% and 15%, respectively; at room temperature and 37°C storage temperatures, less than 3% of cells were positive (Figure 3A).
The level of HCV NS3 expression in the Huh7 cells was then examined by immunoblot analysis. The results of this experiment showed that the level of NS3 expression was considerably higher in the RNA-T transfected cells, as determined by macroscopic analysis (Figure 3B) and by phosphorimager analysis (Figure 3C) using ImageGauge 4.2 software (Fujifilm) for analysis. Only the 6-month time point data are shown. These data show that the protein expression was higher in the Huh7 cells transfected with RNA-T than in cells transfected with RNA-W, irrespective of the RNA storage temperature. The immunoblot to detect NS3 in the sample stored at −70°C was negative despite an intense immunofluorescent signal and we assume this was due to a technical problem during processing. The maximum relative expression after transfection was noted in the RNA-T samples stored at 4°C that showed a 3.6-fold expression over the corresponding RNA-W sample. Thus the ideal temperature for storage of RNA-T to achieve highest levels of protein expression in transfected cells is 4°C.
DISCUSSION
Vaccines are urgently required for many infectious diseases, including human immunodeficiency virus (HIV) and HCV, which are considered to require CMI for optimum efficiency. Although such responses can be generated by a number of strategies, it is thought that endogenous expression of the immunogen is likely to be most effective. CMI is readily generated with nucleic acid vaccines, and in this study we have outlined a method to store purified RNA for long periods without significant loss of activity. A major finding from the study is that replicon RNA, stored as a freeze-dried preparation for up to 10 months in trehalose at 4°, −20°, or −70°C, was translati onally and replication competent. Thus, suitably prepared RNase-free RNA vaccines are no less stable than other conventional vaccines which require a cold chain to retain efficacy.
It was considered that the increased stability of RNA freeze-dried in the presence of trehalose was an effect of increased solubility due to the presence of the sugar. However, this is an unlikely explanation as Figure 2A shows that although there was a decrease in the band intensity of reconstituted RNA-W relative to RNA-T and a freshly prepared sample, this was not sufficient to explain the great decrease in band intensity at the other time points and the decreased level of protein expression after transfection with RNA-W.
A second finding from the study describes a method for the removal of DNA template from in vitro transcribed RNA. Although not of direct scientific relevance, this is an important point that will be considered by regulatory bodies which assess future vaccine candidates. The level of residual DNA was much lower than the acceptable level (suggested to be 10 ng/dose) and this will increase the safety profile of RNA vaccines. Although we used the HCV core replicon in the DNase digestion study and the NS3 replicon in the study due to the availability of antibodies and primer pairs, the data are reproducible and will be equally applicable to any RNA molecule. Since the study was completed, Ambion has included the TURBO-DNase as standard in the mMessage mMachine in vitro transcription kit. Nevertheless, the additional purification step using the Qiagen DNase set followed by the RNeasy column is essential for effective removal of DNA. Generally, the method for DNA removal involves a single DNase step, which we have shown to be ineffective at removing all contaminating DNA. The method described herein is novel in that it uses a two-step method of DNA removal and uses PCR to confirm the absence of DNA. This is a highly sensitive way of determining the absence of any contaminating DNA. However, the significance of our finding has been reduced due to the recent release of a product by EPICENTRE Biotechnologies that also removes all DNA from RNA preparations, as determined by PCR.
A feature of the study is the remarkable stability of the RNA-T stored at or below 4°C (Figure 2), whereas the RNA-W showed increasing degradation with increased time in storage. NS3 expression was detected at high levels in cells transfected with RNA-T whether the samples were stored at or below 4°C, and even RNA-T stored at RT and 37°C retained some translational activity. However, it appears that 4°C is the optimum temperature for storage of RNA-T (Figure 3C). This is an important feature of trehalose stabilization. In contrast, the RNA-W was completely degraded with increasing temperature of storage.
A number of studies have used RNA immunization, either by direct injection (4,16,17,19,25) or by pulsing dendritic cells with RNA for immunotherapy of infectious disease (reviewed in 26) or cancer (27–29). However, very few studies have examined methods of long-term storage of RNA for vaccine studies. One group (30) which used gene gun technology for vaccination were able to store gold particles laden with mRNA at room temperature for up to 8 months without evidence of RNA degradation or loss of function, while another group (1) showed no loss in the activity of RNA-coated gold particles after storage for 5 weeks at 4°C. These are specific applications of RNA storage whereas freeze-drying RNA in the presence of trehalose, as described in our study, will have more widespread applications.
The results of this study provide support for the future potential of RNA vaccines. Using the disaccharide sugar, trehalose, as a stabilizing agent, DNA-free and protein-free RNA, stored for 10 months at 4°C, retained its function. As many of the viruses for which new vaccines are urgently required are transmitted by arthropod vectors in tropical countries (31), our novel findings will allow RNA vaccines to be considered as a viable option, even in developing countries where the cold chain may be restricted to 4°C.
ACKNOWLEDGMENTS
This work was supported by grant no. RO1 A1054459-01 from the U.S. National Institutes of Health. We thank Preethi Mayura Guru for technical help and Damon Warren for help in formatting the figures.
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare no competing interests.
REFERENCES
- 1.Mandl CW, Aberle JH, Aberl SW, Holzman H, Allison SL, Heinz FX. In vitro synthesized infectious RNA as an attenuated live vaccine in a flavivirus model. Nat. Med. 1998;4:1438–1440. doi: 10.1038/4031. [DOI] [PubMed] [Google Scholar]
- 2.Kofler RM, Aberle JH, Aberle SW, Allison SL, Heinz FX, Mandl CW. Mimicking live flavivirus immunization with a non-infectious RNA vaccine. Proc. Natl. Acad. Sci. USA. 2004;101:1951–1956. doi: 10.1073/pnas.0307145101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pijlman GP, Suhrbier A, Khromykh A. Kunjin virus replicons: and RNA based, non-cytopathic viral vector system for protein production, vaccine and gene therapy applications. Expert Opin. Biol. Ther. 2006;6:135–145. doi: 10.1517/14712598.6.2.135. [DOI] [PubMed] [Google Scholar]
- 4.Aberle JH, Aberle SW, Kofler RM, Mandl CW. Humoral and cellular immune response to RNA immunization with flavivirus replicons derived from tick-borne encephalitis virus. J. Virol. 2005;79:15107–15113. doi: 10.1128/JVI.79.24.15107-15113.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Restifo NP, Ying H, Hwang L, Leitner WW. The promise of nucleic acid vaccines. Gene Ther. 2000;7:89–92. doi: 10.1038/sj.gt.3301117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clark KR, Johnson PR. Gene delivery of vaccines for infectious disease. Curr. Opin. Mol. Ther. 2001;3:375–384. [PubMed] [Google Scholar]
- 7.Howarth M, Elliott T. The processing of antigens delivered as DNA vaccines. Immunol. Rev. 2004;199:27–39. doi: 10.1111/j.0105-2896.2004.00141.x. [DOI] [PubMed] [Google Scholar]
- 8.Carralot JP, Probst J, Hoerr I, Scheel B, Teufel R, Jung G, Rammensee HG, Pascolo S. Polarization of immunity induced by direct injection of naked sequence-stabilized mRNA vaccines. Cell. Mol. Life Sci. 2004;61:2418–2424. doi: 10.1007/s00018-004-4255-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoerr I, Obst R, Rammensee HG, Jung G. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur. J. Immunol. 2000;30:1–7. doi: 10.1002/1521-4141(200001)30:1<1::AID-IMMU1>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 10.Vignuzzi M, Gerbaud S, van der Werf S, Escriou N. Naked RNA immunization with replicons derived from poliovirus and Samliki Forest virus genomes for the generation of a cytotoxic T cell response against the influenza virus nucleoprotein. J. Gen. Virol. 2001;82:1737–1747. doi: 10.1099/0022-1317-82-7-1737. [DOI] [PubMed] [Google Scholar]
- 11.Kieny MP, Blancou J, Lathe R, Pastoret PP, Soulebot JP, Desmettre P, Lecocq JP. Development of animal recombinant DNA vaccine and its efficacy in foxes. Rev. Infect. Dis. 1988;10(Suppl4):S799–S802. doi: 10.1093/clinids/10.supplement_4.s799. [DOI] [PubMed] [Google Scholar]
- 12.O’Hagan DT, Singh M, Ulmer JB. Microparticles for the delivery of DNA vaccines. Immunol. Rev. 2004;199:191–200. doi: 10.1111/j.0105-2896.2004.00153.x. [DOI] [PubMed] [Google Scholar]
- 13.Estcourt MJ, McMichael AJ, Hanke T. DNA vaccines against human immunodeficiency virus. Immunol. Rev. 2004;199:144–155. doi: 10.1111/j.0105-2896.2004.00151.x. [DOI] [PubMed] [Google Scholar]
- 14.Pascolo S. Messenger RNA-based vaccines. Expert Opin. Biol. Ther. 2004;4:1285–1294. doi: 10.1517/14712598.4.8.1285. [DOI] [PubMed] [Google Scholar]
- 15.Khromykh AA. Replicon-based vectors of positive strand RNA viruses. Curr. Opin. Mol. Ther. 2000;2:555–569. [PubMed] [Google Scholar]
- 16.Ward SM, Tindle RW, Khromykh AA, Gowans EJ. Generation of CTL responses using Kunjin replicon RNA. Immunol. Cell Biol. 2003;81:73–78. doi: 10.1046/j.0818-9641.2002.01137.x. [DOI] [PubMed] [Google Scholar]
- 17.Anraku I, Harvey TJ, Linedale R, Gardner J, Harrich D, Suhrbier A, Khromykh AA. Kunjin virus replicon vaccine vectors induce protective CD8+ T-cell immunity. J. Virol. 2002;76:3791–3799. doi: 10.1128/JVI.76.8.3791-3799.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harvey TJ, Anraku I, Linedale R, Harrich D, Mackenzie J, Suhrbier A, Khromykh AA. Kunjin virus replicon vectors for human immunodeficiency virus vaccine development. J. Virol. 2003;77:7796–7803. doi: 10.1128/JVI.77.14.7796-7803.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoerr I, Obst R, Rammensee H-G, Jung G. In vivo application of RNA leads to induction of specific cytotoxic T-lymphocytes and antibodies. Eur. J. Immunol. 2000;30:1–7. doi: 10.1002/1521-4141(200001)30:1<1::AID-IMMU1>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 20.Teufel R, Carralot JP, Scheel B, Probst J, Walter S, Jung G, Hoerr I, Rammensee H-G, Pascolo S. Human peripheral blood mononuclear cells transfected with messenger RNA stimulate antigen-specific cytotoxic T-lymphocytes in vitro. Cell. Mol. Life Sci. 2005;62:1755–1762. doi: 10.1007/s00018-005-5067-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crowe JH, Hoekstra FA, Crowe LM. Anhydrobiosis. Annu. Rev. Physiol. 1992;54:579–599. doi: 10.1146/annurev.ph.54.030192.003051. [DOI] [PubMed] [Google Scholar]
- 22.Eroglu A, Russo MJ, Bieganski R, Fowler A, Cheley S, Bayley H, Toner M. Intracellular trehalose improves the survival of cryopreserved mammalian cells. Nat. Biotechnol. 2000;18:163–167. doi: 10.1038/72608. [DOI] [PubMed] [Google Scholar]
- 23.Carninci P, Nisjiyama Y, Westove A, Itoh M, Nagaoka S, Sasaki N, Okazaki Y, Muramatsu M, Hayashizaki Y. Thermostabilization and thermoactivation of thermolabile enzymes by trehalose and its application for the synthesis of full length cDNA. Proc. Natl. Acad. Sci. USA. 1998;95:520–524. doi: 10.1073/pnas.95.2.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Varnavski AN, Khromykh AA. Noncytopathic Flavivirus replicon RNA-based system for expression and delivery of heterologous genes. Virology. 1999;255:366–375. doi: 10.1006/viro.1998.9564. [DOI] [PubMed] [Google Scholar]
- 25.Scheel B, Aulwurm S, Probst J, Stitz L, Hoerr I, Rammensee HG, Weller M, Pascolo S. Therapeutic anti-tumour immunity triggered by injections of immunostimulating single-stranded RNA. Eur. J. Immunol. 2006;36:2807–2816. doi: 10.1002/eji.200635910. [DOI] [PubMed] [Google Scholar]
- 26.Gowans EJ, Jones KL, Bharadwaj M, Jackson DC. Prospects for dendritic cell vaccination in persistent infection with hepatitis C virus. J. Clin. Virol. 2004;30:283–290. doi: 10.1016/j.jcv.2004.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kyte JA, Kvalheim G, Aamdal S, Saeboe-Larsson S, Gaudernack G. Preclinical full-scale evaluation of dendritic cells transfected with autologous tumor-mRNA for melanoma vaccination. Cancer Gene Ther. 2005;12:579–591. doi: 10.1038/sj.cgt.7700837. [DOI] [PubMed] [Google Scholar]
- 28.Mu LJ, Gaudernack G, Saeboe-Larsson S, Hammerstad H, Tierens A, Kvalheim G. A protocol for generation of clinical grade mRNA-transfected monocyte-derived dendritic cells for cancer vaccines. Scand. J. Immunol. 2003;58:578–586. doi: 10.1046/j.1365-3083.2003.01333.x. [DOI] [PubMed] [Google Scholar]
- 29.Kalady MF, Onaitis MW, Padilla KM, Emani S, Tyler DS, Pruitt SK. Enhanced dendritic cell antigen presentation in RNA-based immunotherapy. J. Surg. Res. 2002;105:17–24. doi: 10.1006/jsre.2002.6435. [DOI] [PubMed] [Google Scholar]
- 30.Vassilev VB, Gil LH, Donis RO. Microparticle-mediated RNA immunization against bovine viral diarrhea virus. Vaccine. 2001;19:2012–2019. doi: 10.1016/s0264-410x(00)00438-2. [DOI] [PubMed] [Google Scholar]
- 31.Chang G-J, Kuno G, Purdy DE, Davis BS. Recent advancement in Flavivirus vaccine development. Expert Rev. Vaccines. 2004;3:199–220. doi: 10.1586/14760584.3.2.199. [DOI] [PubMed] [Google Scholar]