

Abstract
Although hepatitis C virus (HCV) is classified in the Hepacivirus genus in the family Flaviviridae, it is unlike most of the other members of this family due to its propensity to cause persistent infections. This persistent infection eventually results in chronic liver disease, cirrhosis and hepatocellular carcinoma in a proportion of infected individuals. It has been difficult to examine correlates of clearance or persistence because most acute phase HCV infections are subclinical or result in symptoms which are non-specific; consequently, acute infections are not generally recognised and patients often present many years later with persistent infection and accompanying chronic liver disease. Nevertheless, seminal studies, performed during the acute phase, have identified a number of factors which are likely to influence the outcome of infection, although it is possible that the mechanism is multifactorial. One of these factors is impairment of dendritic cell function by a mechanism resulting from expression of an HCV protein(s) in these cells. This may be a major factor in the failure of the immune response to expand after HCV infection, leading to persistence. Nevertheless, it may be possible to overcome this defect by autologous transfusion of HCV antigen-loaded, mature dendritic cells and the purpose of this review is to highlight the need and general approaches for developing dendritic cell-based immunotherapy for HCV infection.
Keywords: Hepatitis C virus, Dendritic cell, Intravenous drug use, Therapeutic
1. Introduction
It has been estimated that 80% of HCV-infected individuals fail to clear the infection during the acute phase and develop persistent infection. As a result, there are approximately 200 million HCV carriers in the world. Estimates suggest that 20% of these individuals will develop cirrhosis and 1–5% of these will eventually develop hepatocellular carcinoma (Lavanchy and McMahon, 2001). These HCV carriers also represent an infectious pool for transmission of the virus to uninfected individuals. Transmission is by the parenteral route; traditionally, in developed countries, transfusion with contaminated blood and blood products, prior to testing blood donations, represented a common mechanism for transmission, but the explosion in intravenous drug use (IDU) in the 1960s and 1970s is now recognized as a major mechanism. A large proportion of HCV carriers in the world probably resulted from iatrogenic infection e.g. shared use of needles during mass vaccination campaigns, poor medical practice, and this continues to be a major contributor to the ever-increasing numbers of HCV carriers, particularly in developing countries (Hauri et al., 2004). It is most likely that HCV, which is not transmitted by arthropod vectors, evolved to ensure survival by persisting in most infected individuals prior to the recent explosion in the above iatrogenic practices.
There are at least 2.7 million individuals infected with HCV in the USA and it is thought that approximately, 8000 people die annually in that country as a result of the infection. The majority of infected persons are aged between 20 and 49 years of age. In the USA, the total costs associated with HCV infection were US$ 5.46 billion in 1997 (Leigh et al., 2001). The financial cost of HCV infection in the Australian community is correspondingly high (Shiell, 1998). There are estimated to be 16,000 new cases of HCV infection in Australia (population, 20 million per annum); most of these probably result from IDU activity (Dore et al., 2003). If these figures are extrapolated to the USA, this suggests that there are more than 200,000 new cases of HCV per annum in the US. Significantly, HCV-related mortality is predicted by The Centres for Disease Control and Prevention, Atlanta, Georgia to increase 2–3 fold within the next 10–20 years.
2. HCV molecular biology
HCV is classified in the Hepacivirus genus in the Flaviviridae; the virus genome is a single-stranded positive sense RNA molecule that contains a single open reading frame which encodes a polyprotein that is co- and post-translationally processed into the mature viral polypeptides. The open reading frame is flanked by untranslated regions at the 5′ and 3′ ends that have important cis-acting functions for replication of the virus. A diagram to show the order of the genes and the predicted sizes of the proteins is shown in Fig. 1.
Fig. 1.
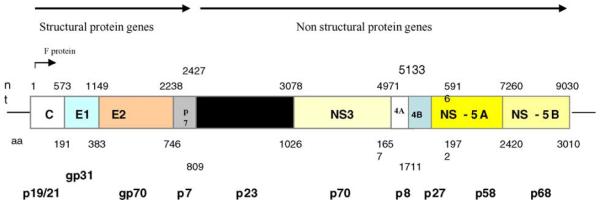
Organisation of the HCV genome. The figures immediately above the boxes depicting the HCV genes represent nucleotide positions at the 3′ end of each gene and the figures below the boxes depicts the amino acid position of a typical genotype 1 virus. The figures on the bottom line depict the electrophoretic mobility of each protein as determined by SDS-PAGE analysis. Not to scale.
There are six genotypes of hepatitis C virus (HCV), that are in turn subdivided into subtypes and isolates. Among full length sequences, the identities of the nucleotides sequences in types, subtypes and individual isolates are around 70%, 80% and >90%, respectively. The different genotypes are detected according to nucleotide differences and signature sequences in the genome specific regions of the genome. The 5′UTR, which is the most highly conserved region in the genome, is commonly used to determine the genotype but the core and NS5 regions can also be used. Because HCV cannot be cultured reproducibly in vitro, the function of some of the viral proteins is still unclear (Table 1). The proteins and peptides used to diagnose HCV infection by the detection of anti-HCV antibody are derived from genotype 1a and appear adequate to detect anti-HCV generated in response to infection with other genotypes. However, it is unclear if vaccination or immunotherapy with proteins/peptides from one genotype will generate immune responses to other genotypes. Nevertheless, recent data derived from chimpanzees suggest that immunity resulting from previous infection may show some cross protection against challenge with different genotypes (Lanford et al., 2004).
Table 1.
The functions of the HCV proteins
| Protein | Function |
|---|---|
| Core | Capsid |
| F (Frameshift) | Unknown |
| E1/E2 | Envelope glycoproteins |
| p7 | Viroporin (in vitro only) |
| NS2 | NS2/3 protease component |
| NS3 | Protease, helicase, NTPase |
| NS4A | NS3 protease co-factor |
| NS4B | Unknown-replicase component |
| NS5A | Unknown-?IFN sensitivity determinant |
| NS5B | RNA dependent RNA polymerase |
2.1. Humoral immunity in HCV infection
Although neutralizing antibodies have been described for HCV (Farci et al., 1994), the epitopes are located in hypervariable region 1 (HVR1) of the E2 protein, one of the two HCV envelope proteins. Consequently, although antibody can neutralize homotypic virus (Farci et al., 1994) and vaccination of chimpanzees with purified E1/E2 can protect against challenge with a low dose of homotypic virus, challenge with heterotypic virus resulted in infection (see Houghton, 2000 for review). Furthermore, although the rapid appearance of antibodies to the HVR1 correlated with recovery from acute hepatitis C in one study (Zibert et al., 1997), most HCV carriers have antibodies to both envelope glycoproteins (Houghton, 2000). Moreover, there is not always a direct relationship between the detection of antibodies to E1 or E2 and neutralizing antibody (Bichr et al., 2002) and there is a great need to develop a robust neutralizing antibody assay to improve our understanding of the events during infection. Recently, three publications reported such an assay, using pseudotyped retroviral particles which contain the HCV E1/E2 glycoproteins in the envelope (Bartosch et al., 2003; Drummer et al., 2003; Hsu et al., 2003). The HVR1 amino acid sequence in individual isolates of the virus is unique and mutations in this region result in the appearance and selection of antibody-resistant mutants (Kato et al., 1994). This may promote persistence, but studies in carrier chimpanzees (Bassett et al., 1999) and agammaglobulinaemic carriers, infected with HCV which had no mutations in the HVR1 (Kumar et al., 1994), clearly show that additional factors are responsible for the inability to clear the acute infection. The continued evolution of neutralizing antibody-resistant mutants indicate that resolution of persistent infection by neutralizing antibody is most unlikely.
2.2. Cell mediated immunity is associated with HCV clearance
Although neutralizing antibody may have little influence on the resolution of HCV infection, a number of other factors are associated with recovery from acute infection. Individuals with specific MHC Class II haplotypes are more likely to clear the virus (Donaldson, 1999), suggesting that antigen presentation may be an important factor. This is discussed below in more detail. In addition, a vigorous CD4+ T-cell response to epitopes contained in the core, NS3, NS4 and NS5 proteins correlate with recovery (Day et al., 2002; Diepolder et al., 1997; Lamonaca et al., 1999). In fact, loss of CD4+ T-cells was associated with recurrence of viraemia in one study (Gerlach et al., 1999). The CD4+ T-cell response is associated with a Th0/Th1 cytokine profile, suggesting that cell-mediated immunity is associated with recovery. A previous study (Tsai et al., 1997) showed that acute phase patients who cleared the virus generated a Th1 response, whereas those who developed persistent infection had a Th2 response. More recently, studies of the immune response in experimentally-infected chimpanzees or in naturally-infected patients showed that a strong CD8+ CTL response to multiple HCV proteins correlated with recovery from acute infection (Cooper et al., 1999; Day et al., 2002; Gruner et al., 2000; Lechner et al., 2000), although this finding is not universal (Thomson et al., 2003).
Thus a cell-mediated immune response is a major factor in recovery from acute HCV infection (for reviews see Houghton, 2000; Orland et al., 2001). There is a narrow window during which an effective cell-mediated immune response can achieve eventual clearance; lack of an effective response during this period results in persistence (Thimme et al., 2001). In needlestick patients who were followed prospectively (Thimme et al., 2001), individuals who mounted a sustained cell-mediated immune response were able to clear the infection whereas those who did not developed persistent infection. Most importantly, viral clearance was temporally associated with interferon-γ (IFN-γ) production that was not associated with liver disease. This led to the suggestion that the pathogenesis of liver disease might differ to the mechanism of viral clearance (Thimme et al., 2001) and a later study in chimpanzees also supported the concept of viral clearance without lysis of infected cells (Thimme et al., 2002).
Thus, although T-cell escape mutants have been described (Erickson et al., 2001; Weiner et al., 1995), the large number of T-cell epitopes which have been identified (Ward et al., 2002) leads to the conclusion that a response which targets many different epitopes is more likely to lead to virus clearance than a humoral response against a limited number of neutralizing epitopes.
2.3. Factors which promote persistence
It is likely that a defective or inappropriate immune response is responsible for the progression to persistent HCV infection and a number of factors have been suggested that may be responsible. These include:
inadequate stimulation of the immune system (Thimme et al., 2001). Individuals with clinical hepatitis in the acute phase appear to eliminate the virus with greater frequency than do asymptomatic individuals (Alter et al., 1999; Giuberti et al., 1994; Villano et al., 1999) and may be related to the vigour of the immune response. This assumes that symptomatic disease is a surrogate marker of the vigour of the immune response (Gordon, 2003);
viral escape mutations including mutations in cytotoxic T-lymphocyte (CTL) epitopes (Erickson et al., 2001; Weiner et al., 1995);
the quasispecies complexity (Farci et al., 2000);
failure to sustain the immune response (Gerlach et al., 1999);
inhibition of the immune response by one or more of the viral proteins (eg. Gale et al., 1997; Keskinen et al., 2002; Kittlesen et al., 2000);
a weak, narrow cell-mediated immune response (Cooper et al., 1999; Thimme et al., 2002);
impaired effector function of CD8+ T-cells (Wedemeyer et al., 2002);
impaired effector function of antigen presenting cells (APC) including dendritic cells (DC)-see below.
In fact it is possible that many of the above potential mechanisms (recently reviewed by Torresi et al., 2004) result from a lack of expansion of immune effector cells, although the contribution of each is unknown. Relevant to this is the finding that the frequency of HCV-specific CTL has been reported to be very low (see Bertoletti and Ferrari, 2003 for review). The cause of these weak responses may be related to impaired antigen presentation that results in a failure to induce an adequate CD4+ cell response that has recently been shown to be important for recovery (Grakoui et al., 2003). These findings highlight the importance of the following discussion on impaired DC function in HCV infection.
2.4. Impaired function of dendritic cells in HCV infection
DC are the most important of the professional APC which initiate the immunological cascade (Hart, 1997). They are speciallised to prime Th and CTL because they can internalise exogenous antigen in a manner that allows presentation through the MHC Class I and II pathways (reviewed in Cella et al., 1997). Several studies have reported a phagosome-to-cytosol pathway in APCs to permit MHC Class I presentation of exogenous, particulate proteins (reviewed in Rock, 1996). This cross presentation pathway may represent the major mechanism for priming CTL responses to antigens which are synthesised by other cell types than professional APC (Carbone et al., 1998). Other studies, designed to test the hypothesis that processing and presentation of endogenous antigens through the MHC Class I pathway in DC was more likely to lead to a Th1-type response, showed that infection of the DC with recombinant viruses (adeno-vaccinia or retro-virus) followed by adoptive cell transfer resulted in a potent CTL response that protected mice against lethal tumour challenge (reviewed in Schuler and Steinman, 1997). DC are able to activate naive CD8+ T-cells by engaging the T-cell receptor and several co-stimulatory cell surface molecules and also promote this response by secretion of cytokines, including IL-12, that are known to be vital for an effective Th1-type response (Cella et al., 1997).
DC are derived from bone marrow, peripheral blood monocytes or a lymphoid precursor and methods have been developed to culture immature and mature DC in vitro (Cella et al., 1997). Immature monocyte-derived DC (Mo-DC) have a high capacity for antigen uptake and processing but a low T-cell stimulation capacity, whereas mature DC have a low antigen processing capacity but high T-cell stimulation capacity. The availability of large numbers of in vitro-cultured DC has permitted ex vivo priming of DC with tumour antigens followed by adoptive transfer of the primed DC to mice that resulted in induction of a specific CTL response, which increased survival of the mice after tumour challenge (reviewed in Young and Inaba, 1996). Adoptive transfer of mature Mo-DC into patients with melanoma, prostate or renal cancer was reported to result in regression of metastases in various organs and proved that DC immunotherapy is safe (Nestle et al., 1998). A recent comprehensive review of DC immunotherapy trials to date suggests significant clinical efficacy in several cancer types (Nestle et al., 2001).
As one important function of DC is to stimulate Th cells, presumably through MHC Class II restricted presentation of processed antigens, the fact that a group of patients with certain MHC Class II haplotypes clear HCV infection more efficiently than the general population (see Donaldson, 1999 for review) led to the suggestion that impaired antigen presentation may be a factor which contributes to HCV persistence (Gowans, 2001). Consistent with this hypothesis, several reports have suggested that DC function may be compromised in HCV carriers. The first of these showed that Mo-DC from HCV-positive patients had reduced activity in a mixed lymphocyte reaction (MLR) compared with DC from normal human volunteers (Kanto et al., 1999), although the DC from the patients still retained potency for antigen-specific autologous T-cell stimulation. It was suggested that reduced levels of expression of IL-12 and IFN-γ were responsible for the impairment in the MLR and that this may inhibit the induction of the appropriate Th1-type of CD4+ T-cells in natural HCV infection. Similar data were reported by Bain et al. (2001) who showed that Mo-DC from HCV carriers showed normal phenotype and morphology, and a normal capacity for antigen uptake, but were also reported to have a defect in allostimulatory activity in the MLR. This defect was not detected in DC from HCV patients with a sustained response to interferon treatment, who were no longer viraemic. A more recent report (Auffermann-Gretzinger et al., 2001) also reported a defect in the MLR allostimulatory capacity of DC from HCV carriers and confirmed that this defect was repaired in patients who showed a sustained response to IFN. Additional evidence was presented to suggest that the defect was probably related to the inability of immature DC to mature in vitro in response to TNF-α. It has been suggested that HCV may infect the DC population in vivo (Bain et al., 2001), although the evidence for this is poor. However, expression of the HCV core and E1 proteins in DC derived from normal individuals, after infection of the cells with a recombinant adenovirus, resulted in a defect in the allostimulatory capacity of the DC (Sarobe et al., 2002) that mirrored the impairment in vivo. Furthermore, the DC which expressed the core/E1 proteins were unable to completely activate autologous T-cells, in contrast to DC that were infected with an adenovirus control. In addition to the reduction in the allostimulatory capacity, DC from HCV patients failed to mature in response to conventional maturation stimuli (Auffermann-Gretzinger et al., 2001; Bain et al., 2001; Sarobe et al., 2002), although maturation was achieved by the addition of LPS. Only a small proportion of DC showed impaired function and it was concluded that most DC from infected patients retain their immunostimulatory ability, particularly against non-HCV proteins (Sarobe et al., 2002). This provides an explanation for the general immune competence of HCV carriers. More recently, the same group reported that immunization of mice with immature DC which expressed the HCV core/E1 proteins showed a reduction in cell-mediated immune responses compared with mice vaccinated with control DC (Sarobe et al., 2003). Similar results were reported previously with recombinant adenovirus-infected murine DC expressing the HCV core/E1/E2 proteins (Hiasa et al., 1998). As the HCV core protein is able to bind to the cytoplasmic domain of the TNF receptor (Zhu et al., 1998), that is likely to participate in one of the DC maturation pathways, it is possible that endogenous expression of the core protein might modulate DC function as described above. In contrast, the addition of purified (exogenous) core protein to DC did not impair DC function, as DC treated in this way were able to prime CTL from naïve CD8+ cells in vitro (Jackson et al., 1999). In addition to the reports which suggest that the HCV core protein can impair DC function, it was reported that the HCV NS3 protein can also impair DC function in vitro (Dolganic et al., 2003). In this study, elevated levels of IL-10 (an immunosuppressive molecule) and decreased levels of IL-12 (a Th1 cytokine) were also noted, in addition to impaired MLR activity, reducing the likelihood of a CTL response.
These impairments in DC function were detected in HCV carriers whose infections were well established, and it is possible that the impairments represent an effect rather than a cause of persistence. In an effort to address this, DC function was examined during acute phase infection of chimpanzees (Rollier et al., 2003); the study showed that allostimulatory activity, the most common feature associated with impaired DC function in human carriers, was normal. It is unclear if this represents differences in the pathogenesis in chimpanzees and humans, or a genuine difference related to DC function in acute versus persistent HCV infection.
To further complicate matters, a recent report (Longman et al., 2003) found no evidence for a defect in Mo-DC from HCV carriers as determined by allostimulatory capacity in a MLR. These differences may be the result of differences in disease staging in the patients, differences related to genotypes or technical differences. In this respect, a comparison of the conditions used to culture the monocyte-derived DC show that cells in one study were cultured in serum-free medium (Auffermann-Gretzinger et al., 2001), while others were cultured in 10% foetal bovine serum (Bain et al., 2001; Dolganic et al., 2003) and others were cultured in 1% autologous donor plasma (Longman et al., 2003) or 10% heat inactivated human AB serum (Sarobe et al., 2002). It is well recognized that the addition of serum, with undefined concentrations of cytokines/chemokines has a great influence on the outcome of DC culture, and this range of culture conditions prevents a direct comparison of the data.
2.5. Current HCV treatment modalities
Recombinant IFN-α2b or -α2a, pegylated IFN-α2b or -α2a or a combination of recombinant IFN-α and ribavirin are the only licensed agents for the treatment of HCV infection. The optimal response to IFN-based therapy is undetectable serum HCV RNA levels 6 months following the end of therapy and is defined as a sustained virological response (SVR). A SVR is associated with clearance of hepatic HCV RNA (Marcellin et al., 1997) and improvement in histological fibrosis (Shiratori et al., 2000). Combination therapy with standard IFN plus ribavirin or pegylated IFN plus ribavirin achieves an overall SVR in 40% and 55% of chronic HCV-infected patients, respectively (Manns et al., 2001; McHutchison et al., 1998). IFN may have a direct anti-viral effect [in this regard, the HCV replicon is sensitive to IFN-α and -γ (Cheney et al., 2002)] but may also exert its effect through the immune system because it may be able to amplify an existing immune response (Thomas and Waters, 1997). It is possible that infected hepatocytes may be cured of HCV infection in a non-lytic manner (Thimme et al., 2001, 2002) as described for hepatitis B virus (HBV) (Cavanaugh et al., 1997; Guidotti and Chisari, 2001). It is possible that this or a similar mechanism accounts for the reduction/loss of viraemia without a simultaneous rise in serum ALT that has been noted in the large international trials of IFN plus ribavirin in previously untreated chronic HCV patients (Manns et al., 2001; McHutchison et al., 1998), in contrast with the flare in serum ALT levels which precedes clearance of HBV in some patients during IFN therapy.
Only a small proportion of HCV carriers have been treated with IFN-based therapy. For logistical and economic reasons, the numbers of patients who can be treated with the current schedules is unlikely to increase significantly. Furthermore, because the patients are selected carefully prior to therapy, the current cure rate is likely to fall if the selection criteria are relaxed to permit more general use. In addition, the cost of treatment is high, the side effects are numerous and a proportion of patients fail to complete the therapy. It is clear that additional, novel treatment strategies are required.
2.6. DC-based immunotherapy in persistent HCV infection
Although the evidence is sometimes inconsistent, several publications have reported that DC function and maturation are impaired in persistent HCV infection. If we assume that this results in impaired antigen presentation, which in turn fails to result in an expansion of the Th cell population, then the result will be a limited expansion of the humoral and cell-mediated immune response. Features of HCV infection include low titres of HCV-specific antibodies (Chen et al., 1999) and a low frequency of HCV-specific CTL (Rehermann and Chisari, 2000), both of which are consistent with a failure of CD4+ Th cells to induce proliferation of the B- and CTL-effector cells. IFN-based treatment of HCV carriers indicate that the frequency of HCV core-specific Th cell precursors is significantly higher in sustained responders than in transient- or non-responders (Lasarte et al., 1998) suggesting that the expansion of Th cell precursors correlates with HCV elimination.
There is therefore, a high probability that DC from HCV carriers that are loaded and matured ex vivo with HCV proteins followed by autologous transfusion will be able to prime naive T-cells and/or stimulate existing HCV-specific cellular immunity. The aim then is to change the equilibrium between the virus and the immune response in the patients that will result in viral clearance. How have we arrived at this conclusion and how might it be possible to correct the deficit in vivo?
(A) It has been suggested that the number of impaired DC is small (Sarobe et al., 2002), although the effect appears to be great. Autologous transfusion of a large number of HCV antigen-loaded, matured DC may overcome the impairment. Previous studies to treat tumours have transfused approximately 5 million DC (Heiser et al., 2002). Thus, simply increasing the number of DC, primed to activate HCV-specific T-cells, may have the desired effect.
(B) Ex vivo loading of immature DC is likely to be very much more efficient than attempting to target immature DC in vivo by vaccination. Consequently, the number of mature, HCV antigen-loaded DC will be very much greater than could be achieved otherwise.
(C) Immature DC which express or process HCV core (and perhaps NS3) may induce tolerance in common with other immature DC (Jonuleit et al., 2001; Moser, 2003). The ex vivo maturation of the immature DC, which express the HCV core or other proteins, with maturation agents that are more effective than TNF-α or CD40L may overcome the defect. Flow cytometry will be used to ensure that only mature DC are used for autotransfusion.
(D) The reports in the literature suggest that endogenous but not exogenous HCV core antigen impairs DC function. This could be overcome by the addition of purified exogenous protein or specific peptides to the DC culture ex vivo.
(E) It is theoretically possible that DC vaccination will result in liver disease through induction of a cytolytic CD8+ T-cell response. However, the fact that patients who are successfully treated with IFN-based therapy fail to show a rise in ALT, coupled with the recent findings (Thimme et al., 2001, 2002) that the HCV viral load is decreased by several orders of magnitude without a concomitant rise in ALT levels during acute, self limited infection, suggests that there is a very strong possibility of non-cytocidal elimination of virus.
(F) The objective of DC immunotherapy to treat HCV infection is to concentrate IFN-α and IFN-γ producing cells in the liver. This is expected to result in high local concentrations of IFN and provide an environment which is conducive to virus elimination without the considerable side effects associated with systemic IFN administration. Successful IFN-based therapy appears to correct the allostimulatory impairment in DC function, and although it is unclear if this is cause or effect, serious consideration should be given to the possibility that this contributes to viral clearance.
(G) Although immature DC are very much more efficient than mature DC in antigen uptake and processing, it is possible that HCV proteins could be delivered effectively ex vivo to mature DC. Mature DC infected ex vivo with a recombinant adenovirus containing an HCV gene were previously shown to induce specific CTL in mice after DC immunization (Matsui et al., 2002).
In practical terms, this may be achieved by the ex vivo culture of DC from individual HCV patients viz. by differentiation of monocytes (collected by apheresis) with appropriate cytokines (Cella et al., 1997), followed by the addition of HCV proteins to the immature DC and subsequent maturation of the cells. The mature DC are then transfused to the same patient from whom the cells were collected.
3. Previous trials of DC immunotherapy
Previous trials of DC immunotherapy have been limited to patients with tumours, including malignant melanoma, colorectal cancer, myeloma or prostate cancer. The results have been reviewed recently (Ridgway, 2003) and show that more than 1000 patients have been vaccinated. Adverse events were uncommon and generally mild. Clinical responses were reported in approximately half of the trials. The potential to treat the liver that can be readily infiltrated by activated lymphocytes is so much greater than the potential to treat solid tumours that are infiltrated very poorly.
4. Summary
Although there are a number of technical obstacles to overcome, DC immunotherapy of HCV carriers has the potential to result in an immune response with the capacity to clear the infection. Initially, the procedure is likely to be unwieldy due to the requirements of regulatory authorities that will demand good laboratory practice conditions for the preparation of the cells, and the HCV proteins or peptides which will be used in the maturation process. However, this process should be viewed as proof of principal which, if successful, could be streamlined and may eventually result in a prophylactic vaccine.
References
- Alter MJ, Kruszon-Moran D, Nainan OV, McQuillan GM, Gao F, Moyer LA, et al. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N Engl J Med. 1999;341:556–62. doi: 10.1056/NEJM199908193410802. [DOI] [PubMed] [Google Scholar]
- Auffermann-Gretzinger S, Keeffe EB, Levy S. Impaired dendritic cell function in patients with chronic, but not resolved, hepatitis C virus infection. Blood. 2001;97:3171–6. doi: 10.1182/blood.v97.10.3171. [DOI] [PubMed] [Google Scholar]
- Bain C, Fatmi A, Zoulim F, Zarski J-P, Trepo C, Inchauspe G. Impaired allostimulatory function of dendritic cells in chronic hepatitis C infection. Gastroenterology. 2001;120:512–24. doi: 10.1053/gast.2001.21212. [DOI] [PubMed] [Google Scholar]
- Bartosch B, Dubuisson J, Cosset F-L. Infectious hepatitis C virus pseudo-particles containing functional E1–E2 envelope protein complexes. J Exp Med. 2003;197:633–42. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoletti A, Ferrari C. Kinetics of the immune response during HBV and HCV infection. Hepatology. 2003;38:4–13. doi: 10.1053/jhep.2003.50310. [DOI] [PubMed] [Google Scholar]
- Bassett SE, Thomas DL, Brasky KM, Lanford RE. Viral persistence, antibody to E1 and E2, and hypervariable region 1 sequence stability in hepatitis C virus-inoculated chimpanzees. J Virol. 1999;73:1118–26. doi: 10.1128/jvi.73.2.1118-1126.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bichr S, Rende-Fournier R, Vona G, Yamamoto A-M, Depla E, Maertens G, et al. Detection of neutralizing antibodies to hepatitis C virus using a biliary cell infection model. J Gen Virol. 2002;83:1673–8. doi: 10.1099/0022-1317-83-7-1673. [DOI] [PubMed] [Google Scholar]
- Carbone FR, Kurts C, Bennett SRM, Miller JFAP, Heath WR. Cross presentation: a general mechanism for CTL immunity and tolerance. Immunol Today. 1998;19:368–73. doi: 10.1016/s0167-5699(98)01301-2. [DOI] [PubMed] [Google Scholar]
- Cavanaugh VJ, Guidotti LG, Chisari FV. Interleukin-12 inhibits hepatitis B virus replication in transgenic mice. J Virol. 1997;71:3236–43. doi: 10.1128/jvi.71.4.3236-3243.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M, Sallusto F, Lanzavecchia A. Origin, maturation and antigen presenting function of dendritic cells. Curr Top Immunol. 1997;9:10–6. doi: 10.1016/s0952-7915(97)80153-7. [DOI] [PubMed] [Google Scholar]
- Chen M, Sallberg M, Sonnerborg A, Weiland O, Mattsson L, Jin L, et al. Limited humoral immunity in hepatitis C virus infection. Gastroenterology. 1999;116:135–43. doi: 10.1016/s0016-5085(99)70237-4. [DOI] [PubMed] [Google Scholar]
- Cheney IW, Lai VCH, Zhong W, Brodhag T, Dempsey S, Lim C, et al. Comparative analysis of anti-hepatitis C virus activity and gene expression mediated by alpha, beta, and gamma interferons. J Virol. 2002;76:11148–54. doi: 10.1128/JVI.76.21.11148-11154.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper S, Erickson AL, Adams EJ, Kansopon J, Weiner A, Chien D, et al. Analysis of a successful immune response against hepatitis C virus. Immunity. 1999;10:439–49. doi: 10.1016/s1074-7613(00)80044-8. [DOI] [PubMed] [Google Scholar]
- Day CL, Lauer GM, Robbins GK, McGovern B, Wurcel AG, Gandhi RT, et al. Broad specificity of virus-specific CD4+ T-helper-cell responses in resolved hepatitis C virus infection. J Virol. 2002;76:12584–95. doi: 10.1128/JVI.76.24.12584-12595.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diepolder HM, Gerlach J-T, Zachoval R, Hoffmann RM, Jung M-C, Wierenga EA, et al. Immunodominant CD4+ T cell epitope within nonstructural protein 3 in acute hepatitis C virus infection. J Virol. 1997;71:6011–9. doi: 10.1128/jvi.71.8.6011-6019.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolganic A, Kodys K, Kopasz A, Marshall C, Do T, Romics L, et al. Hepatitis C virus core and nonstructural protein 3 proteins induce pro-and anti-inflammatory cytokines and inhibit dendritic cell differentiation. J Immunol. 2003;170:5615–24. doi: 10.4049/jimmunol.170.11.5615. [DOI] [PubMed] [Google Scholar]
- Donaldson PT. The interrelationship between hepatitis C and HLA. Eur J Clin Invest. 1999;29:280–3. doi: 10.1046/j.1365-2362.1999.00465.x. [DOI] [PubMed] [Google Scholar]
- Dore GJ, Law M, MacDonald M, Kaldor JM. Epidemiology of hepatitis C virus infection in Australia. J Clin Virol. 2003;26:171–84. doi: 10.1016/s1386-6532(02)00116-6. [DOI] [PubMed] [Google Scholar]
- Drummer HE, Maerz A, Poumbourios P. Cell surface expression of functional hepatitis C virus E1 and E2 glycoproteins. FEBS Lett. 2003;546:360–85. doi: 10.1016/s0014-5793(03)00635-5. [DOI] [PubMed] [Google Scholar]
- Erickson AL, Kimura Y, Igarashi S, Eichelberger J, Houghton M, Sidney J, et al. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity. 2001;15:883–95. doi: 10.1016/s1074-7613(01)00245-x. [DOI] [PubMed] [Google Scholar]
- Farci P, Alter HJ, Wong DC, Miller RH, Govindarajan S, Engle R, Sahpior M, Purcell RH. Prevention of hepatitis C virus infection in chimpanzees after antibody-mediated in vitro neutralisation. Proc Natl Acad Sci USA. 1994;91:7792–6. doi: 10.1073/pnas.91.16.7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farci P, Shimoda A, Coiana A, Diaz G, Peddis G, Melpolder JF, et al. The outcome of acute hepatitis C is predicted by the evolution of the viral quasispecies. Science. 2000;288:339–44. doi: 10.1126/science.288.5464.339. [DOI] [PubMed] [Google Scholar]
- Gale MJ, Jr, Korth MF, Tang NM, Tan SL, Hopkins DA, Dever TE, et al. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology. 1997;230:217–27. doi: 10.1006/viro.1997.8493. [DOI] [PubMed] [Google Scholar]
- Gerlach JT, Diepolder HM, Juing MC, Gruener NH, Schraut WW, Zachoval R, et al. Recurrence of hepatitis C virus after loss of virus-specific CD4+ T cell response in acute hepatitis C. Gastroenterology. 1999;117:933–41. doi: 10.1016/s0016-5085(99)70353-7. [DOI] [PubMed] [Google Scholar]
- Giuberti T, Marin MG, Ferrari C, Marchelli S, Schianchi C, Degli Antoni AM, et al. Hepatitis C virus viremia following clinical resolution of acute hepatitis C. J Hepatol. 1994;20:666–71. doi: 10.1016/s0168-8278(05)80358-7. [DOI] [PubMed] [Google Scholar]
- Gordon SC. New insights into acute hepatitis C. Gastroenterology. 2003;125:253–6. doi: 10.1016/s0016-5085(03)00807-2. [DOI] [PubMed] [Google Scholar]
- Gowans EJ. Crofts, Dore, Locarnini, editors. Towards a vaccine for the hepatitis C virus. Hepatitis C. IP Communications. 2001:45–55. [Google Scholar]
- Grakoui A, Shoukry NH, Woollard DJ, Han J-H, Hanson HL, Ghrayeb J, et al. HCV persistence and immune evasion in the absence of memory T cell help. Science. 2003;302:659–62. doi: 10.1126/science.1088774. [DOI] [PubMed] [Google Scholar]
- Gruner NH, Gerlach TJ, Jung M-C, Diepolder HM, Schirren CA, Schraut WW, et al. Association of hepatitis C virus-specific CD8+ T cells with viral clearance in acute hepatitis C. J Inf Dis. 2000;181:1528–36. doi: 10.1086/315450. [DOI] [PubMed] [Google Scholar]
- Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Ann Rev Immunol. 2001;19:65–91. doi: 10.1146/annurev.immunol.19.1.65. [DOI] [PubMed] [Google Scholar]
- Hart DNJ. Dendritic cells: unique leukocyte populations which control the primary immune response. Blood. 1997;90:3245–87. [PubMed] [Google Scholar]
- Hauri AM, Armstrong GL, Hutin YJ. The global burden of disease attributable to contaminated injections given in health care settings. Int J STD AIDS. 2004;15:7–16. doi: 10.1258/095646204322637182. [DOI] [PubMed] [Google Scholar]
- Heiser A, Coleman D, Dannull J, Yancey D, Maurice MA, Lallas CD, et al. Autologous dendritic cells transfected with prostate-specific antigen RNA stimulate CTL responses against metastatic prostate tumors. J Clin Invest. 2002;109:409–17. doi: 10.1172/JCI14364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiasa Y, Horiike N, Akbar SMF, Saito I, Miyamura T. Low stimulatory capacity of lymphoid dendritic cells expressing hepatitis C virus genes. Biochem Biophys Res Commun. 1998;249:90–5. doi: 10.1006/bbrc.1998.9089. [DOI] [PubMed] [Google Scholar]
- Houghton M. Strategies and prospects for vaccination against the hepatitis C viruses. Curr Top Microbiol Immunol. 2000;242:327–39. doi: 10.1007/978-3-642-59605-6_15. [DOI] [PubMed] [Google Scholar]
- Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, et al. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci USA. 2003;100:7271–6. doi: 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M, Smith B, Bevitt DJ, Steward M, Toms GL, Bassendine MF, et al. Comparison of cytotoxic T-lymphocyte responses to hepatitis C virus core protein in uninfected and infected individuals. J Med Virol. 1999;58:239–46. doi: 10.1002/(sici)1096-9071(199907)58:3<239::aid-jmv9>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Jonuleit H, Schimtt E, Steinbrink K, Enk AH. Dendritic cells as a tool to induce anergic and regulatory T-cells. Trends Immunol. 2001;22:394–400. doi: 10.1016/s1471-4906(01)01952-4. [DOI] [PubMed] [Google Scholar]
- Kanto T, Hayashi N, Takehara T, Tatsumi T, Kuzushita N, Ito A, et al. Impaired allostimulatory capacity of peripheral blood dendritic cells recovered from hepatitis C virus-infected individuals. J Immunol. 1999;162:5584–91. [PubMed] [Google Scholar]
- Kato N, Ootsuyama Y, Sekiya H, Ohkoshi O, Nakazawa T, Hijikata M, et al. Genetic drift in hypervariable region 1 of the viral genome in persistent hepatitis C virus infection. J Virol. 1994;68:1117–20. doi: 10.1128/jvi.68.8.4776-4784.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittlesen DJ, Chianese-Bullock KA, Yao ZQ, Braciale TJ, Hahn YS. Interaction between complement receptor gC1qR and hepatitis C virus core protein inhibits T-lymphocyte proliferation. J Clin Invest. 2000;106:1239–49. doi: 10.1172/JCI10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keskinen P, Melen K, Julkunen I. Expression of HCV structural proteins impairs IFN-mediated antiviral response. Virology. 2002;299:164–71. doi: 10.1006/viro.2002.1527. [DOI] [PubMed] [Google Scholar]
- Kumar U, Monjardino J, Thomas HC. Hypervariable region of hepatitis C virus envelope glycoprotein (E2/NS1) in an agammaglobulinemic patient. Gastroenterology. 1994;106:1072–5. doi: 10.1016/0016-5085(94)90770-6. [DOI] [PubMed] [Google Scholar]
- Lamonaca V, Missale G, Urbani S, Pilli M, Boni C, Mori C, et al. Conserved hepatitis C virus sequences are highly immunogenic for CD4+ T cells: implications for vaccine development. Hepatol. 1999;30:1088–98. doi: 10.1002/hep.510300435. [DOI] [PubMed] [Google Scholar]
- Lanford RE, Guerra B, Chavez D, Bigger C, Brasky KM, Wang X-H, et al. Cross-genotype immunity to hepatitis C virus. J Virol. 2004;78:1575–81. doi: 10.1128/JVI.78.3.1575-1581.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasarte JJ, Garcia-Granero M, Lopez A, Casares N, Garcia N, Civeira M-P, et al. Cellular immunity to hepatitis C virus core protein and the response to interferon in patients with chronic hepatitis C. Hepatology. 1998;28:815–22. doi: 10.1002/hep.510280332. [DOI] [PubMed] [Google Scholar]
- Lavanchy D, McMahon B. Worldwide prevalence and prevention of hepatitis C. In: Liang H, editor. Hepatitis C. Academic Press; San Diego CA: 2001. pp. 185–201. [Google Scholar]
- Lechner F, Wong DKH, Dunbar PR, Chapman R, Chung RT, Dohrenwend P, et al. Analysis of successful immune responses in persons infected with hepatitis C virus. J Exp Med. 2000;191:1499–512. doi: 10.1084/jem.191.9.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh JP, Bowlus CL, Leistikow B, Schenker M. Costs of hepatitis C. Arch Int Med. 2001;161:2231–7. doi: 10.1001/archinte.161.18.2231. [DOI] [PubMed] [Google Scholar]
- Longman RS, Talal AH, Jacobson IM, Albert ML, Rice CM. Patients chronically infected with hepatitis C virus have functional dendritic cells. Blood. 2003 doi: 10.1182/blood-2003-04-1339. First edition online October 2, 2003 DOI 10.1182/blood-2003-04-1339. [DOI] [PubMed] [Google Scholar]
- Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, et al. Peginterferon alfa-2b in combination with ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: results of a randomized trial. Lancet. 2001;358:958–65. doi: 10.1016/s0140-6736(01)06102-5. [DOI] [PubMed] [Google Scholar]
- Marcellin P, Boyer N, Gervais A, Martinot M, Pouteau M, Castelnau C, et al. Long-term histologic improvement and loss of detectable intrahepatic HCV RNA in patients with chronic hepatitis C and sustained response to interferon-alpha therapy. Ann Intern Med. 1997;127:875–81. doi: 10.7326/0003-4819-127-10-199711150-00003. [DOI] [PubMed] [Google Scholar]
- Matsui M, Moriya O, Abdel-Aziz N, Matsuura Y, Miyamura T, Akatsuka T. Induction of hepatitis C virus-specific cytotoxic T lymphocytes in mice by immunization with dendritic cells transduced with replication-defective recombinant adenovirus. Vaccine. 2002;21:211–20. doi: 10.1016/s0264-410x(02)00460-7. [DOI] [PubMed] [Google Scholar]
- McHutchison JG, Gordon SC, Schiff ER, Shiffman Ml, Lee WM, Rustgi VK, et al. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. New Engl J Med. 1998;339:1485–92. doi: 10.1056/NEJM199811193392101. [DOI] [PubMed] [Google Scholar]
- Moser M. Dendritic cells in immunity and tolerance-do they display opposite functions. Immunity. 2003;19:5–8. doi: 10.1016/s1074-7613(03)00182-1. [DOI] [PubMed] [Google Scholar]
- Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, et al. Vaccination of melanoma patients with peptide-or tumor lysate-pulsed dendritic cells. Nat Med. 1998;4:269–70. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- Nestle FO, Banchereau J, Hart D. Dendritic cells: on the move from bench to bedside. Nat Med. 2001;7:761–5. doi: 10.1038/89863. [DOI] [PubMed] [Google Scholar]
- Orland JR, Wright TL, Cooper S. Acute hepatitis C. Hepatology. 2001;33:321–7. doi: 10.1053/jhep.2001.22112. [DOI] [PubMed] [Google Scholar]
- Rehermann B, Chisari FV. Cell mediated immune response to the hepatitis C virus. Curr Top Microbiol Immunol. 2000;242:299–325. doi: 10.1007/978-3-642-59605-6_14. [DOI] [PubMed] [Google Scholar]
- Ridgway D. The first 1000 dendritic cell vaccines. Cancer Invest. 2003;21:873–86. doi: 10.1081/cnv-120025091. [DOI] [PubMed] [Google Scholar]
- Rock KL. A new foreign policy: MHC Class I molecules monitor the outside world. Immunol Today. 1996;17:131–7. doi: 10.1016/0167-5699(96)80605-0. [DOI] [PubMed] [Google Scholar]
- Rollier C, Drexhage JAR, Verstrepen BE, Verschoor EJ, Bontrop RE, Koopman G, et al. Chronic hepatitis C virus infection established and maintained in chimpanzees independent of dendritic cell impairment. Hepatology. 2003;38:851–8. doi: 10.1053/jhep.2003.50426. [DOI] [PubMed] [Google Scholar]
- Sarobe P, Lasarte JJ, Casares N, Lopez-Diaz de Cerio A, Baizeras B, Labarga P, et al. Abnormal priming of CD4+ Tcells by dendritic cells expressing hepatitis C virus core and E1 proteins. J Virol. 2002;76:5062–70. doi: 10.1128/JVI.76.10.5062-5070.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarobe P, Lasarte JJ, Zabaleta A, Arribillaga L, Arina A, Melero I, et al. Hepatitis C virus structural proteins impair dendritic cell maturation and inhibit in vivo induction of cellular immune responses. J Virol. 2003;77:10862–71. doi: 10.1128/JVI.77.20.10862-10871.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiell A. Commonwealth Dept of Health and Family Services. Canberra; 1998. Economic Analyses for the Review of the National Hepatitis C Action Plan. [Google Scholar]
- Schuler G, Steinman RM. Dendritic cells as adjuvants for immune-mediated resistance to tumors. J Exp Med. 1997;186:1183–7. doi: 10.1084/jem.186.8.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiratori Y, Imazeki F, Moriyama M, Yano M, Arakawa Y, Yokosuka O, et al. Histologic improvement of fibrosis in pateints with hepatitis C who have sustained response to interferon therapy. Ann Intern Med. 2000;132:517–24. doi: 10.7326/0003-4819-132-7-200004040-00002. [DOI] [PubMed] [Google Scholar]
- Thimme R, Oldach D, Chang K-M, Steiger C, Ray SC, Chisari FV. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J Exp Med. 2001;194:1395–406. doi: 10.1084/jem.194.10.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimme R, Jubkh J, Spangenberg HC, Weiland S, Pemberton J, Steiger C, et al. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc Natl Acad Sci USA. 2002;99:15661–8. doi: 10.1073/pnas.202608299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas HC, Waters JA. Future approaches to treatment of chronic hepatitis B and hepatitis C infection. J Viral Hep. 1997;4(Suppl 2):92–7. doi: 10.1111/j.1365-2893.1997.tb00186.x. [DOI] [PubMed] [Google Scholar]
- Thomson M, Nascimbeni M, Havert MB, Major M, Gonzales S, Alter H, et al. The clearance of hepatitis C virus infection in chimpanzees may not necessarily correlate with the appearance of acquired immunity. J Virol. 2003;77:862–70. doi: 10.1128/JVI.77.2.862-870.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torresi J, Bharadwaj M, Jackson DC, Gowans EJ. Neutralising antibody, CTL and dendritic cell responses to hepatitis C virus: a preventative vaccine strategy. Curr Drug Targets. 2004;5:41–56. doi: 10.2174/1389450043490677. [DOI] [PubMed] [Google Scholar]
- Tsai SL, Liaw YF, Chen MH, Huang Cy, Kuo GC. Detection of type 2-like T-helper cells in hepatitis C virus infection: implications for hepatitis C virus chronicity. Hepatol. 1997;25:449–58. doi: 10.1002/hep.510250233. [DOI] [PubMed] [Google Scholar]
- Villano SA, Vlahov D, Nelson KE, Cohn S, Thomas DL. Persistence of viremia and the importance of long-term follow up after acute hepatitis C infection. Hepatology. 1999;29:908–14. doi: 10.1002/hep.510290311. [DOI] [PubMed] [Google Scholar]
- Ward SM, Lauer G, Isba R, Walker B, Klenerman P. Cellular immune responses against hepatitis C virus the evidence base, 2002. Clin Exp Immunol. 2002;128:195–203. doi: 10.1046/j.1365-2249.2002.01840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedemeyer H, He X-S, Nascimbeni M, Davis AR, Greenberg HB, Hoofnagle JH, et al. Impaired effector function of hepatitis C virus-specific CD8+ T cells in chronic hepatitis C virus infection. J Immunol. 2002;169:3447–58. doi: 10.4049/jimmunol.169.6.3447. [DOI] [PubMed] [Google Scholar]
- Weiner A, Erickson Al, Kansopon J, Crawford K, Muchmore E, Hughes AL, et al. Persistent hepatitis C virus infection in a chimpanzee is associated with emergence of a cytotoxic T lymphocyte escape variant. Proc Natl Acad Sci USA. 1995;92:2755–9. doi: 10.1073/pnas.92.7.2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JW, Inaba K. Dendritic cells as adjuvants for Class I major histocompatibility complex-restricted antitumor activity. J Exp Med. 1996;183:7–11. doi: 10.1084/jem.183.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N, Khoshnan A, Schneider R, Matsumoto M, Dennert G, Ware C, et al. Hepatitis C virus core protein binds to the cytoplasmic domain of tumor necrosis factor (TNF) receptor 1 and enhances TNF-induced apoptosis. J Virol. 1998;72:3691–7. doi: 10.1128/jvi.72.5.3691-3697.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zibert A, Meisel H, Kraas W, Schulz A, Jung G, Roggendorf M. Early antibody against hypervariable region 1 is associated with acute self limited infections of hepatitis C virus. Hepatology. 1997;25:1245–9. doi: 10.1002/hep.510250530. [DOI] [PubMed] [Google Scholar]