

Abstract
Suppressive myeloid cells represent a significant barrier to the generation of productive antitumor immune responses to many solid tumors. Eliminating or reprogramming suppressive myeloid cells to abrogate tumor-associated immune suppression is a promising therapeutic approach. We asked whether treatment of established aggressive disseminated pancreatic cancer with the immunotherapeutic attenuated Toxoplasma gondii vaccine strain CPS would trigger tumor-associated myeloid cells to generate therapeutic antitumor immune responses. CPS treatment significantly decreased tumor-associated macrophages and markedly increased dendritic cell infiltration of the pancreatic tumor microenvironment. Tumor-resident macrophages and dendritic cells, particularly cells actively invaded by CPS, increased expression of co-stimulatory molecules CD80 and CD86 and concomitantly boosted their production of IL12. CPS treatment increased CD4+ and CD8+ T-cell infiltration into the tumor microenvironment, activated tumor-resident T cells, and increased IFNγ production by T-cell populations. CPS treatment provided a significant therapeutic benefit in pancreatic tumor-bearing mice. This therapeutic benefit depended on IL12 and IFNγ production, MyD88 signaling, and CD8+ T-cell populations. Although CD4+ T cells exhibited activated effector phenotypes and produced IFNγ, CD4+ T cells as well as NK cells were not required for the therapeutic benefit. In addition, CD8+ T cells isolated from CPS-treated tumor-bearing mice produced IFNγ after re-exposure to pancreatic tumor antigen, suggesting this immunotherapeutic treatment stimulated tumor cell antigen-specific CD8+ T-cell responses. This work highlights the potency and immunotherapeutic efficacy of CPS treatment and demonstrates the significance of targeting tumor-associated myeloid cells as a mechanism to stimulate more effective immunity to pancreatic cancer.
Keywords: Pancreatic cancer, immunotherapy, CD8 T cells, interleukin12, interferonγ, attenuated Toxoplasma gondii
Introduction
Pancreatic ductal adenocarcinoma (PDA) is an aggressive, lethal disease and is the fourth deadliest cancer for both men and women in the United States (1). Currently, early tumor resection is the optimal treatment. Unfortunately, few patients are eligible and there is a high incidence (~70%) of tumor recurrence (2–4). Classical cancer therapeutic approaches are relatively ineffective in eliminating PDA. Consequently, immunotherapeutic approaches that boost the ability of the immune system to recognize and kill tumor cells may be necessary to combat pancreatic cancer.
Although antitumor CD8+ T-cell populations develop spontaneously to PDA, active suppression mediated by tumor-associated myeloid cells negate the ability of T cells to eradicate tumor cells (5–7). Suppressive myeloid cells dampen the effectiveness of the antitumor immune response in multiple murine models of PDA (8, 9), and higher levels of suppressive macrophages correlate to shorter overall survival of pancreatic cancer patients (10). Targeting suppressive myeloid cells in PDA has emerged as a promising immunotherapeutic strategy to stimulate CD8+ T-cell populations with improved potential to kill pancreatic tumor cells (11, 12).
One remarkably potent microbial stimulator of the immune system is an attenuated strain of Toxoplasma gondii, an obligate intracellular parasite that uses secretory mechanisms and a specialized form of gliding motility to actively penetrate host cells (13). This avirulent non-replicating vaccine strain (CPS) was developed by genetic disruption of the de novo pyrimidine synthesis pathway to establish a uracil auxotroph (14, 15). CPS invades and replicates in cells exogenously supplemented with uracil in vitro. In the absence of uracil in vitro and during in vivo infection in normal or immune deficient mice (14–19), CPS actively invades cells but fails to replicate making this vaccine strain avirulent and safe. CPS vaccination elicits strong induction of interleukin-12 (IL12) and local interferon-gamma (IFNγ) that drives development of a potent CD8+ T-cell immunity and memory against T. gondii infection (14, 15, 17, 19–22). Immunotherapeutic CPS treatment of mice bearing established aggressive ovarian cancer or B16 melanoma recently was shown to stimulate potent antitumor responses and tumor-free survival (16, 23–25).
In this study, we investigated CPS immunotherapy using a highly aggressive, non-immunogenic disseminated peritoneal PDA model. We demonstrated CPS treatment prolonged survival of mice bearing disseminated pancreatic tumors and examined the mechanisms underlying this effective immunotherapeutic treatment. CPS treatment rapidly increased expression of co-stimulatory molecules and IL12 production by tumor-associated macrophages and dendritic cells (DC), particularly in myeloid cells actively invaded by CPS. Subsequently, T-cell populations exhibited activated phenotypes and CD8+ T cells produced IFNγ in response to pancreatic tumor antigens. The therapeutic benefit of CPS treatment relied on invasive parasites, IL12 and IFNγ production, MyD88 signaling, and CD8+ T cells. Our findings demonstrate immunotherapy with the attenuated CPS vaccine strain neutralized suppressive myeloid-cell mechanisms in PDA and stimulated effective antitumor T-cell responses. These results highlight the significance of targeting suppressive myeloid-cell populations as an effective immunotherapeutic mechanism to combat pancreatic cancer.
Materials and Methods
Mice and cell lines
6–8 week old female C57BL/6 (000664), IL12p35−/− (002692), IFNγ−/− (002287), MyD88−/− (009088) and CD8a−/− (002665) were purchased from Jackson Laboratory. All animal work was performed at the Dartmouth Hitchcock Medical Center animal facility with Dartmouth IACUC approval. The murine pancreatic adenocarcinoma Pan02 cell line, also known as Panc02 (26), was acquired from the Division of Cancer Treatment Tumor Repository (NCI). Pan02 cells were maintained in high glucose Roswell Park Memorial Institute (RPMI) 1640 media. ID8-GFP cells (27) were maintained in high glucose Dulbecco's Modified Eagle Medium (DMEM). Human foreskin fibroblasts (HFF) (28) cultures were maintained in Eagle's Minimum Essential Medium (EMEM). All cell culture media was supplemented with 10% FBS, L-glutamine, and penicillin/streptomycin.
Parasites
Tachyzoites of the CPS vaccine strain were grown in HFF cells supplemented with 300 μM of uracil (14, 15). Tachyzoites were purified through a 3.0 μm nuclepore membrane and washed twice with phosphate buffer saline (PBS) prior to treatment of tumor-bearing mice. For experiments tracking cell types invaded by CPS, tachyzoites were labeled with carboxyfluorescein succinimidyl ester (CFSE) (16).
Tumor inoculation and treatment of pancreatic tumors
All studies used 106 Pan02 cells injected intraperitoneally (i.p.) in 200 μL of PBS. All CPS treatments used 2.0 × 106 tachyzoites injected i.p. For survival studies, mice were treated with CPS using a 2-dose (7 and 19 d), 3-dose (7, 19, 31 d), or 6-dose (7, 8, 11, 12, 24, and 36 d) schedule. For cytokine analysis, mice were treated once at 7 d. For all cellular analysis studies, mice were treated with CPS at 14 d.
Tissue and cell isolation
For spleen and mesenteric lymph node isolations, tissues were homogenized with DMEM in 10% FBS and single-cell suspensions were obtained by disrupting the organs using a cell strainer (40 μm). Peritoneal cells were harvested by lavage at the time of sacrifice. Red blood cells were lysed in cell suspensions using red blood cell lysis buffer (eBioscience). Serum and peritoneal fluid was stored at −80°C.
Cellular analysis and flow cytometry
For intracellular staining studies, cells were incubated with Brefeldin A for 5 h at 37°C. Antibody reagents were obtained from Biolegend: AF647-conjugated anti-mouse CD45 (30-F11), PE-conjugated and AF647-conjugated anti-mouse CD11b (M1/70), Brilliant Violet 421-conjugated anti-mouse CD11c (N418), PE-Cy7-conjugated anti-mouse CD4 and PE-conjugated anti-mouse CD4 (GK1.5), AF647-conjugated anti-mouse CD19 (6D5), PE-Cy7-conjugated anti-mouse F4/80 (BM8), Brilliant Violet 421-conjugated anti-mouse CD3 (17A2), PE-Cy7-conjugated anti-mouse Gr-1 (RB6-8C5), PE-conjugated and AF647-conjugated anti-mouse CD8b (YTS156.7.7), AF488-conjugated anti-mouse/human CD44 (IM7), PE-Cy7-conjugated anti-mouse CD62L (MEL-14), AF647-conjugated anti-mouse CD69 (H1.2F3), PE-conjugated anti-mouse IFNγ (XMG1.2), PE-conjugated anti-mouse IL12/IL23p40 (C15.6), PE-conjugated anti-mouse CD86 (GL-1), PE-conjugated anti-mouse CD80 (16-10A1), AF647-conjugated anti-mouse FoxP3 (MF-14), PE-conjugated anti-mouse CD25 (PC61) and CD16/32 blocking antibody (93). IL12p35 (4D10p35) eflour660 was obtained from eBioscience. FACS analysis was performed using a Miltenyi 8-color MACSQuant and data were analyzed using FlowJo (TreeStar).
Depleting Antibodies
Purified anti-CD4 (GK1.5), anti-CD8 (2.43), and isotype control (rat IgG2a) antibodies were purchased from BioXCell. 500 μg of antibody was administered i.p.1 d prior to and 250 μg of antibody was administered i.p. 0 and 3 d after each CPS treatment. 50 μg of anti-NK1.1 (PK136) was administered i.p. 2 d prior to and 0 and 3 d after CPS treatment. In all experiments target-cell populations were depleted by greater than 99%.
Cytokine measurements
IFNγ, IL12p40, and IL12p70 levels in serum and peritoneal fluid were determined using OptEIA ELISA kits and reagent sets (BD Biosciences).
IFNγ ELISPOT
CD8+ T cells were isolated from splenic tissue 10 d after CPS treatment. CD8+ T cells were purified using EasySep Mouse CD8+ T cell Enrichment Kit (Stem Cell Technologies). Target cells (Pan02, HFF, or ID8-GFP) and splenocytes (5.0 × 104) were irradiated (300 Rads). CD8+ T cells were plated at a 10:1 ratio of T cells to irradiated target cells in the presence of 5.0 × 104 irradiated splenocytes.
Statistics
Statistical analysis was performed using Graphpad Prism 5 software. The log-rank Mantel-Cox test was used for survival analysis. Bar graph samples were compared using the student t-test. Error bars show the SEM. P values of less than 0.05, 0.01, 0.001, or 0.0001 are indicated by *, **, ***, ****, respectively, and statistically non-significant differences are indicated by “n.s.”.
Results
CPS treatment alters myeloid cell populations in the tumor microenvironment
Myeloid cell suppression of immune responses systemically and within the pancreatic tumor neutralizes the effectiveness of antitumor CD8+ T cells (9). The non-replicating Toxoplasma CPS vaccine strain is a potent stimulator of DCs and macrophages (20, 23–25, 29). We established disseminated, peritoneal PDA tumors by injection of Pan02 cells, a model that establishes pancreatic tumor on the pancreas and in distal locations throughout the peritoneum (30), and asked whether CPS treatment of disseminated pancreatic tumors would stimulate tumor-associated myeloid cells. The cellular compositions of the local tumor microenvironment (TME), the spleen, and the mesenteric lymph node were analyzed at 1, 2, 4, 7, and 10 d after CPS treatment. Treatment significantly increased the population of DCs (CD45+CD11b+CD11c+F4/80−) in the TME for at least 4 days (Figure 1A). Additionally, the number of DCs at systemic locations in the mesenteric lymph node and spleen were increased significantly in the first few days after CPS treatment (Supplemental Figure 1A and C). In contrast, macrophage populations (CD45+CD11b+CD11c−F4/80+) in the TME sharply decreased immediately following CPS treatment (Figure 1B). This reduction in macrophages was not observed systemically (Supplemental Figure 1B and D).
Figure 1. CPS treatment alters numbers and activation of dendritic cells and macrophages in the pancreatic tumor microenvironment.

1.0 × 106 Pan02 cells were injected i.p. and mice were CPS treated (n=4) or PBS was administered (n=4) 14 d post-tumor inoculation. A, At indicated time-points peritoneal cells were analyzed by flow cytometry staining for CD45+CD11b+CD11c+F4/80− dendritic cells. B, Peritoneal cells isolated from the same mice as in A were analyzed at the same time for CD45+CD11b+CD11c−F4/80+Gr1− macrophage populations. C and D, Dendritic cells and macrophages from CPS-treated and untreated Pan02 tumor-bearing mice were analyzed 1 d post-CPS treatment CD80 and CD86 expression (mean fluorescence index). CPS-invaded and CPS treated non-invaded cells were tracked using CFSE labeled CPS parasites. *=P<0.05, **=P<0.01, ***=P<0.001.
CPS activates invaded myeloid cells in the tumor microenvironment
CPS was shown recently to stimulate strong Th1 and CD8+ T-cell responses by preferential invasion and activation of myeloid cells (20). To track CPS-invaded cells in the pancreatic TME, CPS was CFSE-labeled to identify actively invaded myeloid cells (CFSE+), in contrast to exposed but not actively invaded myeloid cells (CFSE−). Remarkably, ~25% of DCs and ~45% of macrophages present in the TME were actively invaded by CPS (Supplemental Figure 2A–B). CPS-invaded cells were rarely observed at systemic locations in the mesenteric lymph node or in the spleen (Supplemental Figure 2A–B).
Treatment of the Pan02 tumor using adenovirus expressing IL12 and CD80 (B7.1) provided significant antitumor efficacy suggesting that antigen-presenting cells in the Pan02 TME may lack co-stimulatory CD80/86 expression that contributes to the poor immunogenicity of this tumor (31). CPS-invaded DCs and macrophages significantly upregulated their expression of CD80/86 within 18 h, while a slight increase in expression of CD80/86 was also observed in CPS-exposed but non-invaded myeloid cells (Figure 1C–D; Supplemental Figure 2C).
IL12 production is stimulated in CPS-invaded myeloid cells in the tumor microenvironment
Production of IL12 is required for CD8+ T cell-dependent immunity to T. gondii (17, 22). In cancer patients, systemic administration of IL12 slows tumor growth but is associated with severe systemic toxicity (32, 33). Recruitment and activation of myeloid cells in the TME suggested CPS treatment could activate production of IL12 by DCs and macrophages. We examined IL12 production locally, systemically, and at the cellular level in Pan02 tumor-bearing mice following CPS treatment. IL12p40 and IL12p70 levels were significantly increased in the TME after CPS treatment (Figure 2A–B), and IL12p70 was induced at a higher level than IL12p40. Additionally systemic IL12p70 levels increased following CPS treatment (Supplemental Figure 3B), whereas systemic IL12p40 levels were unchanged (Supplemental Figure 3A).
Figure 2. Treatment with CPS increases IL12 production by myeloid cells.

1.0 × 106 Pan02 cells were injected i.p. and tumors were established for 7 d in mice. A and B, Mice were CPS treated (n=4) at 7 d (untreated mice [n=4] received PBS control). Peritoneal supernatant was collected at the indicated days after CPS treatment. IL12p40 and IL12p70 production was measured by ELISA. C and D, 18 and 48 h post-CPS treatment, peritoneal cells were harvested from CPS-treated (n=4) and untreated (PBS) mice (n=4). Cells were analyzed via flow cytometry with CD45, CD11b, CD11c, F4/80 and IL12p40 to identify IL12-producing dendritic cell and macrophage populations (representative panels are shown). Quantification for each experiment is shown below. E. CD45+CD11b+CD11c+F4/80− dendritic cells were analyzed via flow cytometry for expression of IL12p40+ following treatment with CFSE-labeled CPS parasites to track invaded and non-invaded subpopulations. F. CD45+CD11b+CD11c−F4/80+Gr-1− macrophages were analyzed via flow cytometry for expression of IL12p40+ following treatment with CFSE-labeled CPS parasites to track invaded and non-invaded subpopulations. *=P<0.05, **=P<0.01, ***=P<0.001.
CPS was labeled with CFSE to track the production of IL12p40 by invaded DC and macrophage populations. Percentages of IL12-producing DCs were unchanged in the total cell population 18 and 48 h after CPS treatment (Figure 2C). In contrast, the total peritoneal macrophage population rapidly increased IL12p40 production within 18 h after CPS treatment, and this marked increase in IL12 in the TME was also present at 48 h after treatment (Figure 2D). The percentage of IL12-producing DCs significantly increased within 48 h after treatment specifically in the population of CPS-invaded DCs (Figure 2E). Similarly, CPS-invaded macrophages exhibited dramatic increases in the percentage of IL12p40-expressing macrophages at both 18 and 48 h after treatment (Figure 2F). In addition, CPS-invaded myeloid cells highly upregulated expression of IL12p35 (Supplemental Figure 3C–D). These results highlight the potent ability of CPS to invade myeloid cell populations, stimulating biologically active IL12 production in the pancreatic TME.
Active invasion and IL12 production are required for the therapeutic benefit
To investigate whether myeloid cell responses triggered by CPS provided a therapeutic benefit, we established Pan02 tumors for 7 d and tumor-bearing mice were treated using a 3-dose or a 6-dose strategy according to the schedule shown in Figure 3A. CPS treatment resulted in significantly increased survival time compared to that of untreated tumor-bearing mice (Figure 3B). Remarkably, 15% of CPS-treated tumor-bearing mice survived long-term.
Figure 3. The therapeutic benefit of CPS treatment depends on IL12 production and active invasion.
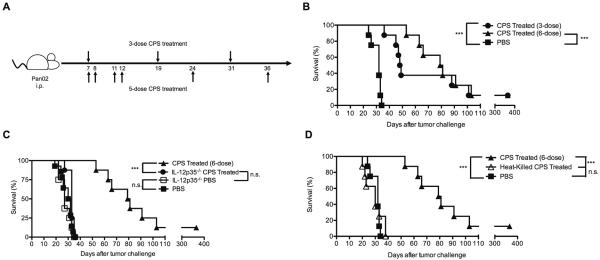
A, One week after injection of 1.0 × 106 Pan02 cells i.p., mice were treated with CPS using a 3-dose (top) or 6-dose (bottom) treatment schedule. B, Mice (n=8 per group) bearing Pan02 tumors for 7 d were CPS treated using a 3-dose or 6-dose treatment schedule. C, IL12p35−/− mice (n=8 per group) were CPS treated using the 3-dose treatment schedule or were administered PBS. D, Mice (n=8 per group) were treated with non-invasive heat-killed CPS using the 3-dose treatment schedule. Survival experiments represent cumulative percentages of two independent experiments. *** = P<0.001
In view of increased IL12 production by CPS-activated DCs and macrophages, we examined whether IL12 production was essential for the efficacy of this treatment. Survival of untreated IL12p35−/− mice and wild-type untreated mice was indistinguishable (Figure 3C). As well, CPS-treated IL12p35−/− mice failed to gain any detectable therapeutic benefit (Figure 3C). Since MyD88 signaling plays a crucial role in protection against T. gondii infection through IL12 production (34), we examined whether MyD88 signaling was required for the therapeutic benefit. MyD88−/− mice exhibited no detectable increase in survival following CPS treatment (Supplemental Figure 4).
We also examined whether active parasite invasion of host cells, as opposed to phagocytosis of CPS by phagocytic cell types, was required for the therapeutic benefit. Heat-inactivated CPS tachyzoites incapable of cellular invasion (data not shown) failed to elicit any detectable therapeutic benefit (Figure 3D). Collectively, these results revealed mechanistic requirements for active invasion of cells by CPS, IL12 production by CPS-invaded myeloid cells, and MyD88 signaling for stimulation of the therapeutic benefit provided by CPS treatment.
Activated T cells accumulate in the tumor microenvironment following CPS treatment
Activation of myeloid cells and increased IL12 production mediated by CPS treatment suggested increased infiltration of the TME by T cells, which is a positive prognostic marker for antitumor responses in the premalignant phase of cancer (35, 36). Rapid recruitment of CD4+ and CD8+ T cells is also a key feature of the host response to CPS vaccination (17). We examined the recruitment of T cells to the spleen, mesenteric lymph node, and pancreatic TME at 1, 2, 4, 7, and 10 d after CPS treatment of tumor-bearing mice. The numbers of CD4+ and CD8+ T cells in the TME were significantly increased by CPS treatment (Figure 4A–B). CD8+ T-cell populations were also increased in the mesenteric lymph node (Supplemental Figure 5B) but were relatively unchanged in the spleen (Supplemental Figure 5D). In contrast, CD4+ T-cell numbers fluctuated only modestly in the mesenteric lymph node and in the spleen (Supplemental Figure 5A and C).
Figure 4. T cells accumulate and are activated within the tumor microenvironment.

1.0 × 106 Pan02 cells were injected i.p. and mice were treated 14 d post-tumor inoculation with CPS or PBS (n=4 for each group). A, At indicated times post-CPS treatment peritoneal cells were analyzed by flow cytometry staining for CD4+ T cells (CD3+CD4+CD8b−). B, At indicated times post-CPS treatment peritoneal cells were analyzed by flow cytometry staining for CD8+ T cells (CD3+CD4−CD8b+). C, Seven days post-CPS treatment, peritoneal cells from CPS-treated and PBS mice were stained for CD3, CD4, CD62L, CD44, and CD69. Totals gated on CD3+CD4+ T cells. Shaded histogram = CPS-treated mice; white histogram = PBS untreated mice. Quantification of activated cells is shown below. D, Seven days post-CPS treatment, peritoneal cells from CPS-treated and PBS mice were stained for CD3, CD8, CD62L, CD44, and CD69. Totals gated on CD3+CD8+ T cells. Shaded histogram = CPS-treated mice; white histogram = PBS untreated mice. Quantification of activated cells is shown below. *=P<0.05, **=P<0.01, ***=P<0.001.
To determine whether recruited CD4+ and CD8+ T cells were activated to effector phenotypes, we examined tumor-resident CD4+ and CD8+ T cells 7 d after CPS treatment for expression levels of CD62L, CD44, and CD69. Resident CD4+ and CD8+ T cells in CPS-treated tumor-bearing mice displayed a CD62Llo phenotype, exhibiting a down-regulation of CD62L (Figure 4C–D). Further evaluation of the CD62Llo CD4+ and CD62Llo CD8+ T-cell populations revealed an increase in T-cell populations co-expressing CD44 and CD69, markers of phenotypic antigen-experience and T-cell activation (Figure 4C–D). In addition, CPS treatment suppressed regulatory T cells (CD4+CD25+FoxP3+) and markedly increased the CD8+ T cell to regulatory T cell ratio in the TME (Supplementary Figure 5E–F). These results indicate that CPS treatment activated CD4+ and CD8+ T-cell populations in the pancreatic tumor microenvironment.
IFNγ production is required for the therapeutic benefit
IFNγ plays a significant role in the generation of antitumor responses (37, 38). Therefore, we investigated IFNγ levels within the TME and systemically. Interestingly, systemic IFNγ levels were not significantly altered following CPS treatment (Supplemental Figure 6). In contrast, IFNγ levels were elevated significantly in the TME within 7 d after CPS treatment (Figure 5A). We hypothesized that the increased IFNγ arose from activated T cells present in the TME, although IFNγ may also arise from activated NK cells in response to T. gondii (39). NK cell populations did not significantly change following CPS treatment and depletion of NK cells did not diminish the therapeutic benefit of CPS treatment (Supplemental Figure 7).
Figure 5. IFNγ production by tumor-resident T cells is required for the therapeutic benefit.

Pan02 tumors were established in mice for 7 d and were treated (n=4) with CPS (untreated mice [n=4] received PBS control). A, Peritoneal supernatant was collected after CPS treatment and IFNγ levels were measured using ELISA. B–E, Seven days post-CPS treatment, peritoneal cells were harvested from treated (black bars; n=4) and untreated mice (white bars; n=4) and analyzed via flow cytometry. B and C, gated on CD3+CD4+ T cells. D and E, gated on CD3+CD8+ T cells. F, IFNγ−/− mice (n=8 for each group) were treated using the 3-dose treatment schedule. Survival experiments represent cumulative percentages of two independent experiments. *=P<0.05, **=P<0.01, ***=P<0.001.
To identify the potential T-cell source of IFNγ production, we tracked cellular production of IFNγ by CD4+ and CD8+ T cells. The number and frequency of IFNγ-producing CD4+ T cells significantly increased following CPS treatment (Figure 5B and 5C; Supplementary Figure 6B). While the number of IFNγ+ CD8+ T cells significantly increased following CPS treatment, the frequency of IFNγ+ CD8+ T cells was unchanged (Figure 5D and 5E; Supplementary Figure 6C). To address the functional importance of IFNγ in the TME, we examined CPS treatment of tumor-bearing IFNγ−/− mice. The therapeutic benefit of CPS treatment was fully dependent on IFNγ production (Figure 5F). These results suggested that active production of IFNγ by infiltrating T cells in CPS-treated tumor-bearing mice played a critical role in stimulating potent antitumor responses.
CD8+ T cells are specifically required for the therapeutic benefit
To investigate the functional importance of recruited and activated T-cell populations, tumor-bearing mice were treated by depleting with αCD4 or αCD8 antibody prior to and following each CPS treatment (Figure 6A). The absence of CD4+ T cells did not significantly affect the therapeutic benefit (Figure 6B). In contrast, the absence of CD8+ T cells via antibody depletion (Figure 6C) or genetic knockout (Figure 6D) led to a complete loss of the therapeutic benefit. Additionally, tumor-bearing CD8α−/− mice succumbed faster to pancreatic cancer than wild-type mice, suggesting that CD8+ T cells naturally respond to this tumor (Figure 6D). Collectively, these findings point to the key functional significance of CD8+ T cells in controlling pancreatic cancer and expose a key role of the CD8+ T-cell population in mediating the therapeutic benefit provided by CPS treatment.
Figure 6. CD8+ T cells are required for the therapeutic benefit of CPS treatment.

A, Mice were injected i.p. with 1.0 × 106 Pan02 cells and tumor-bearing mice were treated with CPS 7 d later as indicated. B, CD4 or isotype control antibody was injected on days indicated in the schedule shown in panel A. Depletion of CD4+ T cells was verified >99.9%. C, CD8b or isotype control antibody was injected on days indicated in the schedule shown in panel A. Depletion of CD8+ T cells was verified >99.9%. D, Pan02-tumor-bearing CD8α−/− mice were treated with CPS (n=4) or were treated with PBS (n=4) using the 3-dose treatment schedule. Survival experiments represent cumulative percentages of two independent experiments. ***=P<0.001
CD8+ T cells produce IFNγ in response to pancreatic tumor antigen
We next analyzed the capacity of CD8+ T-cell populations in CPS treated tumor-bearing mice to produce IFNγ in response to re-stimulation by Pan02 tumor cells. CD8+ T cells harvested from the spleens of mice were re-exposed to irradiated Pan02 cells for 48 h and cellular production of IFNγ was measured by ELISPOT analysis (Figure 7). CD8+ T cells from CPS-treated mice produced significantly higher levels of IFNγ in response to Pan02 antigens. To determine if this response was driven by Pan02 tumor-antigen specificity, an ovarian cancer cell line (ID8-GFP) or an unrelated human fibroblast (HFF) cell line was used as the target cell. The IFNγ response by CD8+ T cells to Pan02 cells was elevated in comparison to the response to ID8-GFP cells or to HFF cells (Figure 7), suggesting CPS treatment initiated an antigen-specific antitumor CD8+ T-cell response.
Figure 7. CD8+ T cells produce IFNγ in response to Pan02 tumor cells.

Mice were injected i.p. with 1.0 × 106 Pan02 cells and 14 d later tumor-bearing mice were treated with CPS. 10 days post-CPS treatment, CD8+ T cells were isolated from the spleen and analyzed via ELISPOT for IFNγ production after exposure to irradiated Pan02 cells, ID8-GFP cells, or HFF cells. *=P<0.05, **=P<0.01.
Discussion
In this study, we explored therapeutic vaccination with attenuated Toxoplasma gondii to disrupt pancreatic tumor-associated immune suppression. The complex network of immunosuppression established by pancreatic cancer abrogates natural immunity and hinders the development of successful immunotherapeutic strategies (9). In murine genetic models of pancreatic cancer as well as in the Pan02 model of PDA explored here, suppressive populations of myeloid cells infiltrate the tumor microenvironment and promote tumor growth while inhibiting immune responses that could halt tumor growth (5, 40–43). Previous therapeutics based on enzyme activation (44), magnetic nanoparticles (45), drug-eluting beads (46), and stem cells (47) in the disseminated intraperitoneal Pan02 PDA model failed to provide significant long-term survival benefits. CPS immunotherapy is the first therapeutic treatment that has provided a long-term survival benefit.
Monocytes and myeloid cells are recruited quickly in response to malignancy and these cell types are rapidly reprogrammed to suppress or be suppressed within the pancreatic TME (9). Treatment of Pan02 tumor-bearing mice with CPS altered the dynamics and functions of myeloid cells within the TME. CPS therapy did not simply alter the numbers of myeloid cells types. CPS selectively accessed myeloid cells in the TME and reprogrammed these cells to promote cellular activation and dynamic changes in tumor-associated cell populations. Compared to the dormant/pro-tumor myeloid cells in untreated tumor-bearing mice, DCs and macrophages in CPS-treated mice up-regulated expression of co-stimulatory molecules CD80 and CD86, particularly in CPS-invaded cells.
Although the highest levels of co-stimulatory molecule expression were found on CPS-invaded myeloid cells, up-regulation of CD80 and CD86 expression also detectably occurred on DCs and macrophages not invaded by CPS. While toll-like receptors (TLR) may trigger up-regulation of CD80 and CD86, recent work has revealed an alternative mechanism for cellular activation based on the injection of specialized parasite-secreted molecules directly into host cells contacted by T. gondii (48). More significantly, our results demonstrate active invasion by CPS is essential for the therapeutic benefit in view that non-invasive heat-killed parasites failed to stimulate any therapeutic benefit, even though parasite molecules recognized through TLRs are present. Thus, our results reinforce the emerging view that Toxoplasma gains preferential access to myeloid cell populations and actively manipulates myeloid cell types from “within” via specialized parasite-secreted molecules (49).
Concurrent with high-level induction of CD80 and CD86, high-level expression of IL12 was observed in CPS-invaded DCs and macrophages. CPS-invasion of tumor-associated myeloid cells activated these cell types in the TME, and could represent the predominant source of co-stimulation and IL12 production within the pancreatic tumor microenvironment. Our results in pancreatic tumor-bearing mice parallels recent results observed in naïve mice and ovarian cancer-bearing mice highlighting the critical importance of myeloid cell invasion by Toxoplasma in stimulating up-regulation of co-stimulatory molecules (20, 23, 50), and activation of T-cell immunity (20).
While systemic administration of IL12 is beneficial to cancer patients, the treatment is highly toxic (33). CPS treatment selectively increased production of IL12 by macrophages and DCs present in the tumor microenvironment. In addition, while CPS induced IL12p70 production systemically no systemic increase in pro-inflammatory IL12p40 was observed and no adverse effects were observed within treated mice.
The alterations in tumor-associated myeloid cells and IL12 production triggered by CPS treatment suggested the renewed potential for CD4+ and CD8+ T cells to gain access to a less suppressive and immune activated pancreatic tumor microenvironment. While both CD4+ and CD8+ T cells infiltrate the Pan02 TME and display activation, CD4+ T cells did not play a crucial role in protection against pancreatic tumor. In contrast, CD8+ T cells were critical in protection against progressive disease. Depletion or genetic loss of the CD8+ T-cell subset resulted in loss of therapeutic benefit in CPS-treated mice. The potency of CD8+ T cells as a protective subset in disease is also highlighted in untreated Pan02 tumor-bearing animals. In the absence of CD8+ T cells, such as in antibody-depleted or in genetically knocked out CD8−/− mice, the Pan02 tumor progressed at a faster rate.
Induction of IFNγ results in potent antitumor effects and increased production of this cytokine correlates with improved antitumor therapeutic benefits (38). Treatment of the Pan02 disseminated tumor with CPS resulted in strong local induction of IFNγ, but only modest changes were observed in systemic IFNγ levels. IFNγ production was required for the therapeutic efficacy of CPS treatment. Additionally, CD8+ T cells were significant producers of IFNγ within the pancreatic TME, and were functionally required to mediate the therapeutic benefit provided by CPS treatment. Furthermore, CPS treatment elicited a population of CD8+ T cells that recognized pancreatic tumor-cell antigen, suggesting this treatment activated T-cell immunity to pancreatic cancer.
In models of melanoma and ovarian cancer, CPS treatment has provided a significant survival benefit to tumor-bearing mice (16, 23–25). Interestingly, there are variations in the immune cells and mechanisms required for CPS-induced therapeutic benefit in different tumor models. In the B16 melanoma tumor model, NK cell production of IFNγ was essential for the therapeutic benefit provided by CPS treatment (16), whereas NK cells were not required for the therapeutic benefit provided by CPS treatment of the disseminated pancreatic tumor model. In the aggressive ovarian cancer ID8 cell tumor model as well as the B16 melanoma model, MyD88 expression was not required for the therapeutic benefit provided by CPS treatment (16, 23–25). Here, we identified a key role for MyD88 signaling in the antitumor response to pancreatic cancer. In all solid tumor models examined to date, effective CPS therapy depends on activation of myeloid cell populations that stimulate tumor antigen-specific CD8+ T-cell populations (16, 23–25). In the absence of engineered exogenous antigen expression, CPS treatment induced host recognition of aberrant tissue that leads to the amplification of antigen-specific antitumor CD8+ T-cell responses.
Generation of tumor-specific responses that extend survival is a prime goal of immunotherapeutic treatment. CPS treatment stimulates the removal of immunosuppressive barriers and generates tumor-specific CD8+ T cells. CPS activated pro-inflammatory myeloid cell populations, reduced regulatory T-cell populations, and promoted the infiltration of T cells into the tumor microenvironment. The increase in infiltrating T cells and the generation of antitumor specific responses are all key elements known to be important in tumor immunotherapy. CPS accomplishes all of these goals without the need for secondary chemotherapeutic or small molecule treatments, although co-administration of these or other treatments may synergize with CPS treatment to provide even more effective antitumor therapy or increased immunity to tumor recurrences. The effect of CPS treatment on disseminated pancreatic tumors reveals the high potency and potential of CPS as an immunotherapeutic treatment for metastatic/advanced pancreatic cancer. In conclusion, this study shows selective targeting and immune activation of tumor-associated myeloid cells by CPS provides an effective immunotherapeutic mechanism to trigger immunity to pancreatic cancer.
Supplementary Material
Acknowledgements
We would like to thank DartLab for flow cytometry instrument resources. The authors would also like to thank Charles Sentman and Edward Usherwood for advice throughout the project.
Support: This work was supported by grants from the National Institute of Health: NIH AI041930 (D.J.B.) and K.L.S. was a trainee on NIH training grants 5T32AI007363-23 and 2T32A1007519
Footnotes
Conflict of interest: No potential conflicts of interest were disclosed by the authors.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer Statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Mayo SC, Nathan H, Cameron JL, Olino K, Edil BH, Herman JM, et al. Conditional survival in patients with pancreatic ductal adenocarcinoma resected with curative intent. Cancer. 2012;118:2674–81. doi: 10.1002/cncr.26553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oettle H, Post S, Neuhaus P, Gellert K, Langrehr J, Ridwelski K, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA. 2007;297:267–77. doi: 10.1001/jama.297.3.267. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki N, Hazama S, Ueno T, Matsui H, Shindo Y, Iida M, et al. A Phase I Clinical Trial of Vaccination With KIF20A-derived Peptide in Combination With Gemcitabine For Patients With Advanced Pancreatic Cancer. J Immunother. 2014;37:36–42. doi: 10.1097/CJI.0000000000000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–35. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmitz-Winnenthal FH, Escobedo LV, Beckhove P, Schirrmacher V, Bucur M, Ziouta Y, et al. Specific immune recognition of pancreatic carcinoma by patient-derived CD4 and CD8 T cells and its improvement by interferon-gamma. Int J Oncol. 2006;28:1419–28. doi: 10.3892/ijo.28.6.1419. [DOI] [PubMed] [Google Scholar]
- 7.Schmitz-Winnenthal FH, Volk C, Z'Graggen K, Galindo L, Nummer D, Ziouta Y, et al. High frequencies of functional tumor-reactive T cells in bone marrow and blood of pancreatic cancer patients. Cancer Res. 2005;65:10079–87. doi: 10.1158/0008-5472.CAN-05-1098. [DOI] [PubMed] [Google Scholar]
- 8.Sideras K, Braat H, Kwekkeboom J, van Eijck CH, Peppelenbosch MP, Sleijfer S, et al. Role of the immune system in pancreatic cancer progression and immune modulating treatment strategies. Cancer Treat Rev. 2014;40:513–22. doi: 10.1016/j.ctrv.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 9.Wormann SM, Diakopoulos KN, Lesina M, Algul H. The immune network in pancreatic cancer development and progression. Oncogene. 2014;33:2956–67. doi: 10.1038/onc.2013.257. [DOI] [PubMed] [Google Scholar]
- 10.Kurahara H, Shinchi H, Mataki Y, Maemura K, Noma H, Kubo F, et al. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J Surg Res. 2011;167:e211–9. doi: 10.1016/j.jss.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 11.Panni RZ, Linehan DC, DeNardo DG. Targeting tumor-infiltrating macrophages to combat cancer. Immunotherapy. 2013;5:1075–87. doi: 10.2217/imt.13.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wesolowski R, Markowitz J, Carson WE., 3rd Myeloid derived suppressor cells - a new therapeutic target in the treatment of cancer. J Immunother Cancer. 2013;1:10. doi: 10.1186/2051-1426-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sibley LD. Intracellular parasite invasion strategies. Science. 2004;304:248–53. doi: 10.1126/science.1094717. [DOI] [PubMed] [Google Scholar]
- 14.Fox BA, Bzik DJ. De novo pyrimidine biosynthesis is required for virulence of Toxoplasma gondii. Nature. 2002;415:926–9. doi: 10.1038/415926a. [DOI] [PubMed] [Google Scholar]
- 15.Fox BA, Bzik DJ. Avirulent uracil auxotrophs based on disruption of orotidine-5'-monophosphate decarboxylase elicit protective immunity to Toxoplasma gondii. Infect Immun. 2010;78:3744–52. doi: 10.1128/IAI.00287-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baird JR, Byrne KT, Lizotte PH, Toraya-Brown S, Scarlett UK, Alexander MP, et al. Immune-mediated regression of established B16F10 melanoma by intratumoral injection of attenuated Toxoplasma gondii protects against rechallenge. J Immunol. 2013;190:469–78. doi: 10.4049/jimmunol.1201209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gigley JP, Fox BA, Bzik DJ. Cell-mediated immunity to Toxoplasma gondii develops primarily by local Th1 host immune responses in the absence of parasite replication. J Immunol. 2009;182:1069–78. doi: 10.4049/jimmunol.182.2.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaw MH, Freeman GJ, Scott MF, Fox BA, Bzik DJ, Belkaid Y, et al. Tyk2 negatively regulates adaptive Th1 immunity by mediating IL-10 signaling and promoting IFN-gamma-dependent IL-10 reactivation. J Immunol. 2006;176:7263–71. doi: 10.4049/jimmunol.176.12.7263. [DOI] [PubMed] [Google Scholar]
- 19.Sukhumavasi W, Egan CE, Warren AL, Taylor GA, Fox BA, Bzik DJ, et al. TLR adaptor MyD88 is essential for pathogen control during oral toxoplasma gondii infection but not adaptive immunity induced by a vaccine strain of the parasite. J Immunol. 2008;181:3464–73. doi: 10.4049/jimmunol.181.5.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dupont CD, Christian DA, Selleck EM, Pepper M, Leney-Greene M, Harms Pritchard G, et al. Parasite fate and involvement of infected cells in the induction of CD4+ and CD8+ T cell responses to Toxoplasma gondii. PLoS Pathog. 2014;10:e1004047. doi: 10.1371/journal.ppat.1004047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jordan KA, Wilson EH, Tait ED, Fox BA, Roos DS, Bzik DJ, et al. Kinetics and phenotype of vaccine-induced CD8+ T-cell responses to Toxoplasma gondii. Infect Immun. 2009;77:3894–901. doi: 10.1128/IAI.00024-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson DC, Grotenbreg GM, Liu K, Zhao Y, Frickel EM, Gubbels MJ, et al. Differential regulation of effector- and central-memory responses to Toxoplasma gondii Infection by IL-12 revealed by tracking of Tgd057-specific CD8+ T cells. PLoS Pathog. 2010;6:e1000815. doi: 10.1371/journal.ppat.1000815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baird JR, Fox BA, Sanders KL, Lizotte PH, Cubillos-Ruiz JR, Scarlett UK, et al. Avirulent Toxoplasma gondii generates therapeutic antitumor immunity by reversing immunosuppression in the ovarian cancer microenvironment. Cancer Res. 2013;73:3842–51. doi: 10.1158/0008-5472.CAN-12-1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fox BA, Sanders KL, Bzik DJ. Non-replicating reverses tumor-associated immunosuppression. Oncoimmunology. 2013;2:e26296. doi: 10.4161/onci.26296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fox BA, Sanders KL, Chen S, Bzik DJ. Targeting tumors with nonreplicating Toxoplasma gondii uracil auxotroph vaccines. Trends Parasitol. 2013;29:431–7. doi: 10.1016/j.pt.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corbett TH, Roberts BJ, Leopold WR, Peckham JC, Wilkoff LJ, Griswold DP, Jr, et al. Induction and chemotherapeutic response of two transplantable ductal adenocarcinomas of the pancreas in C57BL/6 mice. Cancer Res. 1984;44:717–26. [PubMed] [Google Scholar]
- 27.Zhang L, Yang N, Garcia JR, Mohamed A, Benencia F, Rubin SC, et al. Generation of a syngeneic mouse model to study the effects of vascular endothelial growth factor in ovarian carcinoma. Am J Pathol. 2002;161:2295–309. doi: 10.1016/s0002-9440(10)64505-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Combe CL, Moretto MM, Schwartzman JD, Gigley JP, Bzik DJ, Khan IA. Lack of IL-15 results in the suboptimal priming of CD4+ T cell response against an intracellular parasite. Proc Natl Acad Sci U S A. 2006;103:6635–40. doi: 10.1073/pnas.0506180103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patil V, Zhao Y, Shah S, Fox BA, Rommereim LM, Bzik DJ, et al. Co-existence of classical and alternative activation programs in macrophages responding to Toxoplasma gondii. Int J Parasitol. 2014;44:161–4. doi: 10.1016/j.ijpara.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wennier ST, Liu J, Li S, Rahman MM, Mona M, McFadden G. Myxoma virus sensitizes cancer cells to gemcitabine and is an effective oncolytic virotherapeutic in models of disseminated pancreatic cancer. Mol Ther. 2012;20:759–68. doi: 10.1038/mt.2011.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Putzer BM, Rodicker F, Hitt MM, Stiewe T, Esche H. Improved treatment of pancreatic cancer by IL-12 and B7.1 costimulation: antitumor efficacy and immunoregulation in a nonimmunogenic tumor model. Mol Ther. 2002;5:405–12. doi: 10.1006/mthe.2002.0570. [DOI] [PubMed] [Google Scholar]
- 32.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–46. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 33.Atkins MB, Robertson MJ, Gordon M, Lotze MT, DeCoste M, DuBois JS, et al. Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin Cancer Res. 1997;3:409–17. [PubMed] [Google Scholar]
- 34.Scanga CA, Aliberti J, Jankovic D, Tilloy F, Bennouna S, Denkers EY, et al. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J Immunol. 2002;168:5997–6001. doi: 10.4049/jimmunol.168.12.5997. [DOI] [PubMed] [Google Scholar]
- 35.Boon T, Coulie PG, Van den Eynde BJ, van der Bruggen P. Human T cell responses against melanoma. Annu Rev Immunol. 2006;24:175–208. doi: 10.1146/annurev.immunol.24.021605.090733. [DOI] [PubMed] [Google Scholar]
- 36.Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001;411:380–4. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 37.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6:836–48. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- 38.Zaidi MR, Merlino G. The two faces of interferon-gamma in cancer. Clin Cancer Res. 2011;17:6118–24. doi: 10.1158/1078-0432.CCR-11-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sher A, Oswald IP, Hieny S, Gazzinelli RT. Toxoplasma gondii induces a T-independent IFN-gamma response in natural killer cells that requires both adherent accessory cells and tumor necrosis factor-alpha. J Immunol. 1993;150:3982–9. [PubMed] [Google Scholar]
- 40.Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67:9518–27. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- 41.Porembka MR, Mitchem JB, Belt BA, Hsieh CS, Lee HM, Herndon J, et al. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol Immunother. 2012;61:1373–85. doi: 10.1007/s00262-011-1178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21:836–47. doi: 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. 2014;63:1769–81. doi: 10.1136/gutjnl-2013-306271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Basel MT, Balivada S, Shrestha TB, Seo GM, Pyle MM, Tamura M, et al. A cell-delivered and cell-activated SN38-dextran prodrug increases survival in a murine disseminated pancreatic cancer model. Small. 2012;8:913–20. doi: 10.1002/smll.201101879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Basel MT, Balivada S, Wang H, Shrestha TB, Seo GM, Pyle M, et al. Cell-delivered magnetic nanoparticles caused hyperthermia-mediated increased survival in a murine pancreatic cancer model. Int J Nanomedicine. 2012;7:297–306. doi: 10.2147/IJN.S28344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yagublu V, Caliskan N, Lewis AL, Jesenofsky R, Gasimova L, Lohr JM, et al. Treatment of experimental pancreatic cancer by doxorubicin-, mitoxantrone-, and irinotecan-drug eluting beads. Pancreatology. 2013;13:79–87. doi: 10.1016/j.pan.2012.11.305. [DOI] [PubMed] [Google Scholar]
- 47.Doi C, Maurya DK, Pyle MM, Troyer D, Tamura M. Cytotherapy with naive rat umbilical cord matrix stem cells significantly attenuates growth of murine pancreatic cancer cells and increases survival in syngeneic mice. Cytotherapy. 2010;12:408–17. doi: 10.3109/14653240903548194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koshy AA, Dietrich HK, Christian DA, Melehani JH, Shastri AJ, Hunter CA, et al. Toxoplasma co-opts host cells it does not invade. PLoS Pathog. 2012;8:e1002825. doi: 10.1371/journal.ppat.1002825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Denkers EY, Bzik DJ, Fox BA, Butcher BA. An inside job: hacking into Janus kinase/signal transducer and activator of transcription signaling cascades by the intracellular protozoan Toxo plasma gondii. Infect Immun. 2012;80:476–82. doi: 10.1128/IAI.05974-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morgado P, Ong YC, Boothroyd JC, Lodoen MB. Toxoplasma gondii induces B7-2 expression through activation of JNK signal transduction. Infect Immun. 2011;79:4401–12. doi: 10.1128/IAI.05562-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.