

Abstract
Mutant isocitrate dehydrogenase 1 (IDH1) catalyzes the production of 2-hydroxyglutarate but also elicits additional metabolic changes. Levels of both glutamate and pyruvate dehydrogenase (PDH) activity have been shown to be affected in U87 glioblastoma cells or normal human astrocyte (NHA) cells expressing mutant IDH1, as compared to cells expressing wild-type IDH1. In this study, we show how these phenomena are linked through the effects of IDH1 mutation, which also reprograms pyruvate metabolism. Reduced PDH activity in U87 glioblastoma and NHA IDH1 mutant cells was associated with relative increases in PDH inhibitory phosphorylation, expression of pyruvate dehydrogenase kinase-3 and levels of hypoxia inducible factor-1α. PDH activity was monitored in these cells by hyperpolarized 13C-magnetic resonance spectroscopy (13C-MRS), which revealed a reduction in metabolism of hyperpolarized 2-13C-pyruvate to 5-13C-glutamate, relative to cells expressing wild-type IDH1. 13C-MRS also revealed a reduction in glucose flux to glutamate in IDH1 mutant cells. Notably, pharmacological activation of PDH by cell exposure to dichloroacetate (DCA) increased production of hyperpolarized 5-13C-glutamate in IDH1 mutant cells. Further, DCA treatment also abrogated the clonogenic advantage conferred by IDH1 mutation. Using patient-derived mutant IDH1 neurosphere models, we showed that PDH activity was essential for cell proliferation. Taken together, our results established that the IDH1 mutation induces an MRS-detectable reprogramming of pyruvate metabolism which is essential for cell proliferation and clonogenicity, with immediate therapeutic implications.
Keywords: mutant IDH1, glioma, pyruvate dehydrogenase, metabolic reprogramming, magnetic resonance spectroscopy
INTRODUCTION
Metabolic reprogramming is increasingly emerging as a new hallmark of cancer (1). This reprogramming is frequently controlled by oncogenic signaling pathways in ways that support cell growth and proliferation, and that are likely essential for malignant transformation (2,3). The most extensively studied metabolic alteration is the “Warburg effect”(4) characterized by an increase in glucose uptake and glucose metabolism to lactate rather than full oxidation via the tricarboxylic acid (TCA) cycle, even under conditions of high oxygen tension. The elevated levels of lactate produced by cancer cells play an essential role in tumor progression (5). Furthermore, recent studies in tumor cells have shown that intermediates of the glycolytic pathway are channeled toward the synthesis of macromolecules that are essential for cell growth and proliferation (6).
Beyond the metabolic reprogramming that occurs downstream of oncogenic signaling, a powerful illustration of the role of altered metabolism as a potential driver of tumorigenesis is the discovery of tumors with somatic mutations in the metabolic enzyme isocitrate dehydrogenase (IDH) (7). Specifically, the cytosolic isoform of IDH, IDH1, is mutated in 70-90% of low-grade gliomas and secondary glioblastoma (GBM) and mitochondrial IDH2 is mutated in ~20% of acute myeloid leukemia cases (8). Wild-type IDH1 catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG). The IDH1 mutation, which commonly occurs at the R132 residue in the active site, leads to a loss of wild-type enzyme activity and to a neomorphic activity that catalyzes the reduction of α-KG to 2-hydroxyglutarate (2-HG) (9). As a result, IDH1 mutant gliomas show significantly elevated levels of 2-HG. In turn, 2-HG inhibits the activity of several α-KG-dependent dioxygenases, including the Jmjc domain histone demethylases and the TET family of 5-methylcytosine hydroxylases, which play an essential role in epigenetic modulation of gene expression. The epigenetic perturbations induced by 2-HG interfere with cellular differentiation and proliferation, ultimately leading to tumorigenesis (8).
Interestingly, several studies have described additional changes in cellular metabolism, beyond production of 2-HG (10-17). Reitman et al. were the first to perform a metabolomic analysis of genetically engineered IDH1 mutant cells and associated wild-type controls. This revealed increased levels of several amino acids and reduced levels of TCA cycle intermediates including glutamate (10). Similar findings were made in a study of patient samples (11). Furthermore, a noninvasive 1H-MRS investigation of mutant IDH1-glioma patients reported that glutamate levels were lower compared to normal brain (12). In line with these findings, the activity of three enzymes – branched chain amino acid transferase 1 (BCAT-1), aspartate transaminase (AST), and glutamate dehydrogenase (GDH) – that catalyze the conversion of α-KG to glutamate, was shown to be lower in IDH1 mutant cells (13). Reitman et al. also noted a drop in phosphocholine (PC) and an increase in glycerophosphocholine (GPC) in IDH1 mutant cells (10) and analysis of ex vivo tumor samples and animal models by MRS confirmed the increase of GPC (14). This observation is counter to the increase in PC and drop in GPC typically observed in cancer (18) and possibly points to metabolic alterations unique to mutant IDH1 tumors. Consistent with this idea, the lactate dehydrogenase (LDHA) gene, responsible for lactate production and typically overexpressed in cancer, is silenced in IDH1 mutant gliomas (15) and IDH1 mutant cells appear to have a greater dependence on the TCA cycle compared to wild-type cells (16,17).
In our laboratory, we have studied two genetically engineered cell models that overexpress either wild-type IDH1 or mutant IDH1: a U87 GBM-derived model and an immortalized normal human astrocyte (NHA)-derived model. We used 1H-MRS to analyze the metabolomic signature associated with the IDH1 mutation and, consistent with previous work, found that it was associated with an MRS-detectable increase in GPC and drop in PC, lactate and glutamate (19). We also used hyperpolarized 13C-MRS, a novel metabolic imaging approach that can rapidly monitor metabolic fluxes (20-23) and showed that we could detect elevated flux from α-KG to 2-HG (24) and reduced flux from α-KG to glutamate (13) in mutant IDH1 tumors compared to wild-type. In a separate study, we observed that the activity of PDH, the enzyme that catalyzes the decarboxylation of pyruvate to acetyl CoA prior to entry into the TCA cycle, was also reduced in IDH1 mutant cells (25). This led us to question the role of PDH in IDH1 mutant cells.
Here, we investigated our two genetically engineered cell models and first confirmed that down-regulation of PDH activity is mediated in both our models by a 2-HG-dependent increase in hypoxia inducible factor-1α (HIF-1α) levels. Using 13C-MRS and hyperpolarized 13C-MRS we then confirmed that glucose flux via PDH was reduced in IDH1 mutant cells compared to wild-type. Importantly, we found that pharmacological activation of PDH not only altered metabolism but also abrogated the mutant IDH1-mediated clonogenicity of our cells and inhibited proliferation of patient-derived mutant IDH1 neurospheres. Our results thus suggest that the IDH1 mutation induces an MRS-detectable reprogramming of pyruvate metabolism via PDH that is essential for tumorigenesis and that could serve as a possible target for treatment of mutant IDH1 tumors.
MATERIALS AND METHODS
Cell culture
U87 and NHA cell lines expressing wild-type IDH1 (U87IDHwt and NHAIDHwt) or IDH1 R132H mutant gene (U87IDHmut and NHAIDHmut) were generated and maintained as described previously (19,24). All cell lines were authenticated by single nucleotide polymorphism fingerprinting and IDH1 mutational status was verified by western blotting as described earlier (19). BT54 and BT142 cells were grown as neurospheres in serum-free medium (NeuroCult, Stemcell technologies) as described previously (26,27). To probe the effect of DCA, cells were treated with 10mM DCA (Sigma-Aldrich) for 24h. To probe the role of 2-HG, NHAIDHwt cells were treated with 10mM 2-HG (Sigma-Aldrich) for 5 days and U87IDHwt cells were permeabilized with 0.01% digitonin (Sigma-Aldrich) in phosphate buffered saline (PBS) for 10min prior to treatment with 30mM 2HG for 24h.
PDH activity and phosphorylation assays
PDH activity was measured using a spectrophotometric assay (Abcam) and normalized to cell number. PDH phosphorylation was measured using an ELISA kit (Abcam) (25) and normalized to cell number.
Expression analysis
mRNA levels were measured by quantitative RT-PCR analysis and normalized to the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transcript (25). For western blotting, cellular protein (~20 µg) was probed for PDK1 (Abcam), PDK3 (Proteintech) and HIF-1α (Cell Signaling) and visualized using an enhanced chemiluminescence substrate kit (Thermo Scientific). Actin (Cell Signaling) and β-tubulin (Cell Signaling) were used as loading controls. Nuclear extracts were prepared using a kit (Thermo Scientific) for HIF-1α detection.
Clonogenicity
Clonogenicity of U87 and NHA cells was measured using a soft agar assay (28) in the presence of 10mM DCA or water (vehicle control).
Proliferation of mutant IDH1 glioma lines
BT54 and BT142 neurospheres were dissociated and 2.5×105 cells/well placed in 6-well plates with medium containing 10mM DCA or water for 12 and 7 days respectively. Media and drug were replenished every 72h. At the end of the study neurospheres were dissociated and number of cells determined.
MRS of live cells
U87 and NHA cells were grown on beads, BT142 neurospheres were encapsulated in agarose, and all MRS studies were performed using an MR-compatible cell perfusion system as described (29-31). All studies were performed on a 500MHz INOVA spectrometer (Agilent Technologies). For 13C-MRS studies glucose or glutamine in the medium were replaced with 5mM 1-13C-glucose or 2mM 3-13C-glutamine (Sigma-Aldrich) and spectra acquired using 60° flip angle (FA), 6s repetition time (TR) and 15min acquisition time. For hyperpolarized 13C-MRS, 2-13C-pyruvic acid (Sigma-Aldrich) was hyperpolarized using the Hypersense DNP polarizer (Oxford Instruments), injected into the perfusion system to a final concentration of 5mM pyruvate, and dynamic sets of 13C-MRS spectra acquired using 5° FA and 3s TR over 300s. All peak integrals were quantified using Mnova (Mestrelab Research). For thermally polarized 13C-MRS peaks, were normalized to cell number and initial 13C-substrate concentrations. For hyperpolarized 13C-MRS studies, total glutamate signal was normalized to total pyruvate signal and cell number.
MRS of cell extracts
U87 and NHA cells were grown in media containing 1-13C-glucose (5mM) or 3-13C-glutamine (2mM) for 24h. BT54 and BT142 neurospheres were grown in Neurocult media with 35mM 1-13C-glucose for 15 days. Metabolites were extracted by dual phase extraction (19). 1H-MRS spectra (1D water presaturation ZGPR sequence, 90° FA, 3s TR, 256 acquisitions) and proton-decoupled 13C-MRS spectra (30° FA, 3s TR, 2048 acquisitions) were acquired using a 500MHz Avance spectrometer (Bruker BioSpin), equipped with a Triple Resonance CryoProbe. Metabolites were quantified by normalizing to a trimethylsilyl propanoic acid reference of known concentration and correcting for saturation and Nuclear Overhauser effect.
Statistical analysis
All studies were performed at least 3 times and results are expressed as mean±standard deviation. Statistical significance was evaluated using a two-tailed Student’s t-test assuming unequal variance with p<0.05 considered significant.
RESULTS
PDH activity is down-regulated in IDH1 mutant cells in a 2-HG-dependent manner
PDH is a rate limiting metabolic checkpoint for oxidation of glucose via the TCA cycle (32). We previously found a significant reduction in PDH activity in NHAIDHmut cells compared to NHAIDHwt (130±16×10−10 OD/hr/cell in NHAIDHwt to 28±5×10−10 OD/hr/cell in NHAIDHmut, p<0.005, Table S1), associated with inhibitory phosphorylation of PDH (25). To assess the importance of PDH in IDH1 mutant cells, we first sought to confirm our findings in a second genetically engineered model.
We found that PDH activity was significantly reduced in U87IDHmut cells compared to U87IDHwt (75±6%, p<0.005; Fig. 1A and Table S1). Furthermore, and similar to the NHA model (25), this was associated with increased phosphorylation of Ser293 (93±26%, p<0.05) and Ser300 (71±19%, p<0.005) (Fig. 1B). Phosphorylation of Ser232 was comparable. Given that PDH activity is regulated by inhibitory phosphorylation via pyruvate dehydrogenase kinases (PDKs) (32), we examined mRNA and protein levels of all four PDKs (PDK1-4). Again, similar to the NHA model (25), we found that PDK1 and PDK3 mRNA levels were significantly higher in U87IDHmut cells relative to U87IDHwt by 183± 82% (p<0.05) for PDK1 and 100±26% (p<0.05) for PDK3 (Fig. 1C). A small 18±2% (p<0.05) drop was observed in PDK2 mRNA, but no change was detected in the NHA model and hence was not investigated further. PDK4 mRNA was below detection in both models.
Figure 1. PDH activity is down-regulated in IDH1 mutant cells as a result of PDK-mediated phosphorylation.
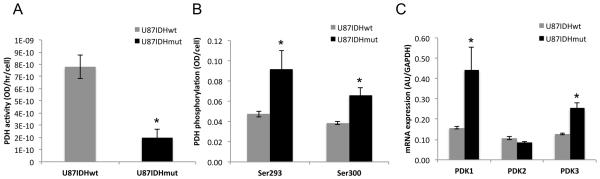
PDH activity (A), PDH phosphorylation at Ser293 and Ser300 (B) and PDK1-3 mRNA levels in U87IDHwt and U87IDHmut cells (C).
Next, we examined protein expression for PDK1 and PDK3 by western blotting. PDK3 expression was higher in IDH1 mutant cells compared to IDH1 wild-type for both U87 and NHA models (Fig. 2A and 2B; 56±16%, p<0.01 for U87 and 71±3%, p<0.001 for NHA), consistent with increased phosphorylation of Ser293 and Ser300, the targets of PDK3. Expression of PDK1, known to phosphorylate Ser232, was unchanged in both models, consistent with the unchanged Ser232 phosphorylation.
Figure 2. 2-HG-mediated increase in HIF-1α and PDK3 levels result in reduced PDH activity.

Western blots and quantification for PDK3 (A,B) and HIF-1α (C,D) in U87 and NHA models. PDH activity in IDH1 wild-type cells treated with 2-HG in U87 (E) and NHA (F) models. Western blots and quantification for PDK3 (G,H) and HIF-1α (I,J) in U87 and NHA IDH1 wild-type cells treated with 2-HG.
In cancer, PDK3 expression is typically up-regulated by HIF-1α leading to a drop in PDH activity and contributing to the switch from oxidative phosphorylation to glycolysis (33). We therefore determined HIF-1α expression in our cells. Our results (Fig. 2C and 2D) indicated that HIF-1α levels were higher in IDH1 mutant cells compared to wild-type for both U87 and NHA models (32±4%, p<0.005 for U87 and 42±21%, p<0.05 for NHA).
To confirm that increased HIF-1α and PDK3 levels and reduced PDH activity were associated with mutant IDH1-induced 2-HG production, we measured PDH activity in NHAIDHwt and U87IDHwt cells treated with exogenous 2-HG, previously shown to model the effect of the IDH1 mutation (10,34,35). The presence of 2-HG did not alter cell proliferation in either the U87 or NHA models (Fig. S1). Importantly, 2-HG treatment did induce a significant reduction in PDH activity by 48±18% (p<0.05) in the U87 model (Fig. 2E) and by 45±11% (p<0.05) in the NHA model (Fig. 2F). Furthermore, western blotting revealed increased levels of PDK3 (Fig. 2G and 2H, 83±36, p<0.05 for U87 and 75±9%, p<0.0005 for NHA) and HIF-1α (Fig. 2I and 2J, 41±8%, p<0.005 for U87 and 82±35%, p<0.05 for NHA) in 2-HG-treated cells. Taken together, our findings suggested that higher HIF-1α levels in IDH1 mutant cells lead to elevated expression of PDK3, likely resulting in reduced PDH activity.
Reduced PDH activity results in a 13C-MRS detectable change in hyperpolarized pyruvate metabolism
To confirm that the drop in PDH activity also results in a drop in pyruvate flux, we used 13C-MRS to investigate live cells and dynamically probe the fate of hyperpolarized 2-13C-pyruvate, which is metabolized via PDH, enters the TCA cycle and is converted into 5-13C-glutamate (36). Build-up of hyperpolarized 5-13C-glutamate was readily detectable in our cells (Fig. 3A and 3B) and its quantification indicated that it was significantly reduced in IDH1 mutant cells compared to wild-type. 5-13C-glutamate dropped by 24±16% (p<0.001) in U87IDHmut cells (Fig. 3C) and by 76±22% (p<0.0001) in NHAIDHmut cells (Fig. 3D). This confirmed that the drop in PDH activity reduced pyruvate flux into the TCA cycle and pointed to a possible role for PDH in modulating cellular glutamate levels.
Figure 3. Hyperpolarized 13C-MRS can detect reduced PDH activity in IDH1 mutant cells.

Dynamic 13C-MRS array showing metabolism of hyperpolarized 2-13C-pyruvate in U87 (A) and NHA (B) models; insets illustrate the sum of all spectra within dotted region of the array. Hyperpolarized 5-13C-glutamate production in mutant and wild-type U87 (C) and NHA (D) models. 2-13C-PYR: 2-13C-pyruvate, 2-13C-PYR Hydrate: 2-13C-pyruvate hydrate, 1-13C-PYR: 1-13C-pyruvate, 1-13C-PYR Hydrate: 1-13C-pyruvate hydrate, 2-13C-LAC: 2-13C-lactate, 5-13C-GLU: 5-13C-glutamate.
Glutamate production from glucose is reduced in IDH1 mutant cells
To assess the effect of reduced PDH activity on glutamate levels, we probed the synthesis of glutamate from 1-13C-glucose and 3-13C-glutamine – its two main metabolic precursors (Fig. 4A). Following 3-13C-glutamine perfusion, we detected build-up of 3-13C-glutamate (27.8 ppm) and 3-13C-2-HG (31.6 ppm) in U87IDHmut cells, whereas, as expected, only glutamate labeling was detected in U87IDHwt cells (Fig. 4B). Following 1-13C-glucose perfusion, we detected production of 3-13C-lactate (21.0 ppm) in both IDH1 wild-type and mutant cells, but no difference in 3-13C-lactate production was observed between U87IDHwt and U87IDHmut cells. We also detected a signal at 34.2 ppm, which based on chemical shift, could represent either 4-13C-glutamate, 4-13C-2-HG, or both (Fig. 4C). Because the resolution of the 13C-MRS data from cells was not sufficient to interpret the peak at 34.2 ppm, we turned to extracting our cells following incubation with 1-13C-glucose and used this data to determine the buildup of glutamate (Fig. 4C right inset). The extracts demonstrated that 1-13C-glucose was incorporated into both 4-13C-glutamate and 4-13C-2-HG (at a ratio of 69±13%) in the U87IDHmut cells (Fig. 4C left inset) but, as expected, only 4-13C-glutamate was labeled in U87IDHwt cells.
Figure 4. Glucose and glutamine flux to glutamate is reduced in U87 IDH1 mutant cells.

Schematic representation of 13C-labeling of glutamate and 2-HG derived from 1-13C-glucose and 3-13C-glutamine (A). Build-up of 3-13C-glutamate (3-13C-GLU) and 3-13C-2-HG following perfusion of U87IDHwt and U87IDHmut cells with 3-13C-glutamine (3-13C-GLN) with quantification of 3-13C-glutamate production shown in the inset (B). Build-up of 3-13C-lactate (3-13C-LAC), 4-13C-glutamate (4-13C-GLU) and a combined signal of 4-13C-glutamate and 4-13C-2-HG in U87IDHwt and U87IDHmut cells with quantification of the build-up of glutamate in right inset. Left inset illustrates data from cell extracts resolving 2-HG and glutamate (C).
We also quantified the total cellular concentration of glutamate using 1H-MRS and compared to the 13C-MRS data at the end of our study. As illustrated in Fig. 5A, the sum total of glutamate produced from glucose and glutamine, as determined from 13C-MRS data, was within experimental error of the total pool of glutamate determined by 1H-MRS in both U87IDHwt (5.9±0.8 fmol/cell) and U87IDHmut (2.5±0.7 fmol/cell) cells. In U87IDHwt cells, 1.0±0.2 fmol/cell of glutamate was derived from 1-13C-glucose and 4.9±0.8 fmol/cell was derived from 3-13C-glutamine. In U87IDHmut cells 0.20±0.01 fmol/cell was derived from 1-13C-glucose, and 2.2±0.4 fmol/cell from 3-13C-glutamine.
Figure 5. Glutamate production from glucose is reduced in both U87 and NHA IDH1 mutant cells.
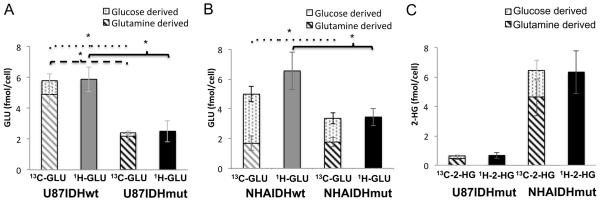
Quantification of glutamate produced from 1-13C-glucose and 3-13C-glutamine (13C-GLU) and total glutamate levels (1H-GLU) in U87 (A) and NHA (B) models. Quantification of 2-HG produced from 1-13C-glucose and 3-13C-glutamine (13C-2-HG) and total 2-HG levels (1H-2-HG) in both models (C).
To analyze the precursors of glutamate in the NHA model, we directly resorted to investigating extracts of cells incubated with 1-13C-glucose and 3-13C-glutamine and compared our findings to the total glutamate pool determined by 1H-MRS. In NHAIDHwt cells, of 6.6±1.2 fmol/cell glutamate, 3.3±0.5 fmol/cell was produced from 1-13C-glucose and 1.7±0.5 fmol/cell from 3-13C-glutamine. In NHAIDHmut cells, of 3.5±0.6 fmol/cell, 1.6±0.4 fmol/cell was produced from 1-13C-glucose while 1.8±0.3 fmol/cell was produced from 3-13C-glutamine (Fig. 5B). Similar to the U87 model, the sum of the 13C-labeled pools accounted for the total pool of glutamate in both NHAIDHwt and NHAIDHmut cells.
When considering the contribution of labeled glutamine and glucose to the total pool of glutamate, we found that U87IDHmut cells displayed a significant drop in glutamine-derived glutamate compared to U87IDHwt cells (4.9±0.8 versus 2.2±0.4 fmol/cell, p<0.05, Fig. 5A) but no such reduction was observed in the NHA model (1.7±0.5 versus 1.8±0.3 fmol/cell, p=0.4, Fig. 5B). In contrast, a significant reduction in glucose-derived glutamate occurred in both U87 (1.0±0.2 versus 0.20±0.01 fmol/cell, p<0.01, Fig. 5A) and NHA (3.3±0.5 versus 1.6±0.4 fmol/cell, p<0.05, Fig. 5B) models. We cannot rule out small contributions to the total glutamate pool from another source. For example, glutamate, which has been shown to enhance the growth of IDH1 mutant gliomas (37), could be imported into the cell via the excitatory amino acid transporter-2. However, the drop in glucose-derived glutamate was sufficient, within experimental error, to explain the full drop in cellular glutamate levels in the NHA model. This points to a role for glucose-derived glutamate, and thus PDH, in explaining the drop in glutamate levels in IDH1 mutant cells.
Glucose contributes to 2-HG synthesis
Previous work indicated glutamine as the major source of 2-HG (38). Interestingly, our data indicated that glucose also contributes significantly to 2-HG production (Fig. 5C). Specifically, in U87IDHmut, of 0.67±0.19 fmol/cell total 2-HG, 0.11±0.01 fmol/cell was derived from 1-13C-glucose while 0.49±0.11 fmol/cell was derived from 3-13C-glutamine. In NHAIDHmut cells, of 6.3±1.5 fmol/cell 2-HG, 1.3±0.7 fmol/cell was derived from 1-13C-glucose and 4.7±1.3 fmol/cell from 3-13C-glutamine. In both U87IDHmut and NHAIDHmut cells the sum of glucose- and glutamine-derived fractions accounted for the total 2-HG pool.
DCA inhibits clonogenicity of IDH1 mutant cells
Next we wanted to assess whether the reduction in PDH activity in IDH1 mutant cells has any biological significance. We used DCA – a PDK inhibitor that activates PDH activity – to assess the effect of PDH activation (32). We chose a concentration of 10mM DCA based on previous work (e.g. Sanchez et al. (39)) and first confirmed that, at this concentration, DCA did not significantly inhibit cell growth in any of our cell lines (Fig. S2). We then assessed target modulation by measuring PDH activity. Our results (Fig. 6A and 6B) indicate that DCA induced activation of PDH most effectively in IDH1 mutant cells. PDH activity increased by 730±219% (p<0.005) in U87IDHmut, by 41±26% (p<0.05) in U87IDHwt (Fig. 6A), by 578±222% (p<0.05) in NHAIDHmut cells and not detectably (8±23% p=0.6) in NHAIDHwt cells (Fig. 6B).
Figure 6. DCA inhibits clonogenicity of IDH1 mutant cells.

Effect of DCA on PDH activity in U87 (A) and NHA (B) models. Effect of DCA on hyperpolarized 5-13C-glutamate production in U87 (C) and NHA (D) models. Effect of DCA on clonogenicity in U87 (E) and NHA (F) models.
Next, we confirmed the effect of DCA on metabolism using hyperpolarized 13C-MRS. Hyperpolarized 5-13C-glutamate increased by 142±76% (p<0.05) in U87IDHmut and by 43±30% (p<0.05) in U87IDHwt (Fig. 6C). It increased by 586±146% (p<0.001) in NHAIDHmut cells and not significantly by 23±6% (p=0.1) in NHAIDHwt (Fig. 6D).
Finally, we tested the effect of DCA on clonogenicity. In the U87 model, both IDH1 wild-type and mutant cells readily formed colonies on soft agar and DCA inhibited clonogenicity equally in U87IDHwt (46±10%, p<0.01) and U87IDHmut (44±8%, p<0.05), likely reflecting the GBM background of this model and pointing to a limited growth advantage associated with PDH down-regulation in these cells (Fig. 6E). In the NHA model, consistent with its effect on PDH activity, DCA had a clear inhibitory effect on NHAIDHmut cells with a very significant drop of 86±2% (p<0.005) in the number of colonies (Fig. 6F). In the case of NHAIDHwt the number of colonies produced was also smaller but the effect barely significant (57±11%, p=0.05). Furthermore, we noted that in the absence of DCA, NHAIDHmut cells showed a significant 834±187% (p<0.005) increase in colony formation on soft agar compared to NHAIDHwt (Fig. 6F). However, DCA completely abrogated the effect of the IDH1 mutation, resulting in equivalent colony numbers for untreated NHAIDHwt and DCA-treated NHAIDHmut cells (8±2 versus 11±3, p=0.2; Fig. 6F). These results highlighted the importance of PDH down-regulation for IDH1 mutation-induced tumorigenesis in the NHA model.
PDH is essential for proliferation of patient-derived mutant IDH1 neurospheres
In order to confirm our findings in clinically relevant models, we extended our studies to the BT54 and BT142 patient-derived mutant IDH1 neurosphere models (26,27). In a small study we assessed the contribution of glucose to the total glutamate and 2-HG pools. As in the case of our genetically engineered models, we used 1-13C-glucose labeling followed by metabolite extraction and 13C-MRS to determine the contribution of glucose to glutamate and 2-HG production. 1H-MRS was used to measure the total glutamate and 2-HG pools. We found that glucose contributed 0.18 fmol/cell out of a total glutamate pool of 0.6 fmol/cell (n=1) in the BT54 model. In the BT142 model, 0.2 fmol/cell of glutamate came from glucose out of a total of 0.43 fmol/cell (n=1). In the case of 2-HG we found that in BT54 cells, 0.08 fmol/cell out of a total 2-HG pool of 0.22 fmol/cell was derived from glucose (n=1). In BT142 cells 2-HG levels were too low to be accurately quantified by MRS, consistent with previous studies (27).
We then treated with DCA and found that it activated PDH by 28±10% (p<0.05) in BT54 and by 49±20% (p<0.05) in BT142 cells (Fig. 7A). We then used hyperpolarized 2-13C-pyruvate to study metabolism in the relatively faster growing BT142 model and confirmed that increased PDH activity resulted in elevated hyperpolarized 5-13C-glutamate production by 170±72% (p<0.001, Fig. 7B). Finally we determined that DCA significantly inhibited neurosphere proliferation by 50±5% (p<0.05) and 37±14% (p<0.05) in BT54 and BT142 cells respectively (Fig. 7C). Collectively, our findings in the patient-derived mutant IDH1 models confirmed our observations in genetically engineered IDH1 mutant cells.
Figure 7. DCA inhibits proliferation of patient-derived mutant IDH1 models.
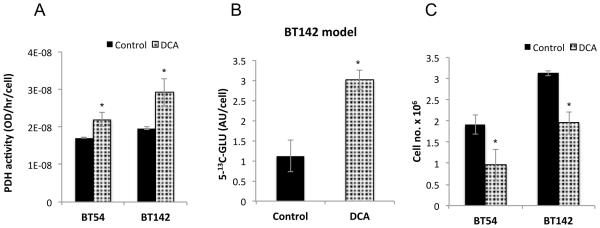
Effect of DCA on PDH activity (A), glutamate production (B) and cell proliferation (C) in BT54 and BT142 models.
DISCUSSION
Targeting metabolic reprogramming in cancer cells is increasingly being considered as a therapeutic option (3,40). In the context of the IDH1 mutation, several studies have cataloged the metabolic changes associated with mutant IDH (10-17), but the biological significance and therapeutic potential of these changes is not fully understood. Here, we focused on the enzyme PDH and found that PDH activity and glucose flux towards glutamate were reduced in genetically engineered IDH1 mutant cells compared to wild-type. Furthermore, we found that activation of PDH, both in our genetically engineered and in patient-derived mutant IDH1 models, significantly reduced proliferation and clonogenicity, pointing to an essential role for PDH.
Reduced PDH activity was likely mediated in our cells by elevated levels of HIF-1α, which led to downstream activation of PDK3 expression. The PDKs are highly specific regulators of PDH activity (32). While several glycolytic and TCA cycle enzymes often moonlight as transcriptional or apoptotic regulators, no such activity has been reported for the PDKs. Therefore, the increased expression of PDK3 observed in our studies is likely responsible for the increased inhibitory phosphorylation of PDH resulting in reduced PDH activity.
Previous work indicates that mutant IDH1 tumors reprogram their metabolism differently from wild-type IDH1 glioblastoma (11,15). Nonetheless, our findings regarding PDH are consistent with observations in most cancer cells, wherein HIF-1α up-regulates expression of PDK1 and PDK3, which mediate inhibitory phosphorylation of PDH (33). Importantly, these can be directly linked to mutant IDH. Mutant IDH1 via 2-HG production can stabilize HIF-1α levels by inhibiting α-KG-dependent prolyl hydroxylases (PHD) (34). Koivunen et al. (41) showed that introduction of the IDH1 mutation or addition of 2-HG to cell lines resulted in diminished HIF-1α levels and reduced expression of HIF-1α target genes. However, in contrast, and in line with our findings, two other studies showed that expression of mutant IDH1 or 2-HG treatment in U87 cells elevated HIF-1α levels and increased expression of HIF-1α target genes, and this effect was abrogated by treatment with an IDH1 inhibitor (34,42). Furthermore, and also consistent with our observations, Sasaki et al. observed that HIF-1α levels and target genes were up-regulated in brain tissue from IDH1 R132H knock-in mice (43).
In most cancer cells, up-regulated HIF-1α expression results in increased LDHA expression (33). Therefore, U87 and NHA IDH1 mutant cells might be expected to show increased LDHA expression relative to wild-type cells. However, Chesnelong et al. (15) demonstrated that the LDHA gene in IDH1 mutant glioma cells is silenced by methylation of CpG sites within the promoter. Therefore, elevated HIF-1α expression in IDH1 mutant cells might not translate to increased LDHA expression. Indeed, expression of LDHA was not significantly altered in our NHAIDHmut cells compared to NHAIDHwt cells (15), nor have we observed any alteration in LDHA expression in our U87 model.
Our data indicates that PDH activity is ~10 fold higher in NHA (akin to low-grade gliomas) and BT142 models compared to U87 GBM (Table S1). This is consistent with a recent study demonstrating a 10-fold reduction in spare respiratory capacity in high-grade GBMs compared to SV-40 immortalized human astrocytes, associated with modulation of PDH activity (44). Thus, while the IDH1 mutation leads to reduced PDH activity and flux to the TCA cycle, this flux remains higher in the NHA and BT models than in high-grade GBMs and points to a greater dependence on the TCA cycle in mutant IDH1 low-grade glioma compared to GBM. Our findings are thus consistent with observations showing that HIF-1α target genes are under-expressed in mutant IDH1 patient-derived models (including BT54 and BT142) compared to primary GBM models wild-type for IDH1 (15) and concur with observations of reduced PDH activity in engineered IDH1 mutant colorectal cancer cells (16). Reduced flux through the TCA cycle would allow IDH1 mutant cells to divert carbon precursors towards macromolecular biosynthesis, even if these cells proliferate slower than GBM (6).
In our studies we used the classic PDK inhibitor, DCA, to understand the biological significance of reduced PDH activity in IDH1 mutant cells. DCA is a pyruvate mimetic and its crystal structures with PDK1, PDK2 and PDK3 show binding at the enzymatic active site. Moreover, siRNA-mediated inhibition of some PDKs mimics the effect of DCA, further confirming its specificity (45). Finally, although DCA is known to induce DNA hypomethylation in mouse liver, its general lack of toxicity in patients suggests a fairly selective mechanism of action (45).
Following DCA treatment, we found that clonogenicity was inhibited equally for U87IDHwt and U87IDHmut cells. This finding concurs with the reported efficacy of DCA against GBM (46) and with the fact that lower baseline PDH activity in the U87 GBM background allows for a response even in IDH1 wild-type cells. It is also consistent with the fact that introduction of the IDH1 mutation did not increase the clonogenicity of the cells and thus a differential effect of DCA on clonogenicity would not be expected. In contrast, in the NHA model, introduction of the IDH1 mutation significantly increased clonogenicity, and, importantly, this effect was completely abrogated by DCA treatment. We also confirmed that DCA inhibited growth of patient-derived neurospheres. Our data thus suggest that the reprogramming of PDH activity by the IDH1 mutation is crucial for tumorigenesis and to the best of our knowledge, this is the first report of the efficacy of DCA against low-grade mutant IDH1 glioma cells.
Our studies also shed light on the different factors contributing to reduced glutamate levels in IDH1 mutant gliomas (10-13,19). A drop in BCAT-1, GDH and AST activity contribute to this reduction (13). However, our studies show that the drop in PDH flux into the TCA cycle and reduced contribution of glucose to glutamate synthesis also contribute to the lower levels of glutamate in IDH1 mutant cells.
Our data also indicate that some 2-HG can be derived from glucose, in addition to its production from glutamine previously reported (38). Reducing 2-HG production by inhibiting glutaminolysis is being explored as a therapeutic option (47). In this context recognizing that glucose can also serve as a source of 2-HG is important.
Finally, our study highlights the value of MRS in probing metabolic reprogramming in IDH1 mutant cells as well as their response to therapies that target metabolism. 1H-MRS is a non-invasive method used to monitor metabolite levels in preclinical models and human patients (48). It has been used to probe for elevated 2-HG levels (49-52). It was also used to detect reduced glutamate levels in patients with mutant IDH1 tumors (12), and our study mechanistically validates reduced glutamate as a potential complementary metabolic biomarker of mutant IDH1 tumors. In addition, hyperpolarized 13C-MRS can be used to monitor metabolic fluxes (13,22-24). We have previously used hyperpolarized 13C-MRS to image 2-HG production in tumors expressing mutant IDH1 (24). We have also shown that we can detect a reduction in α-KG to glutamate conversion that occurs as part of the metabolic reprogramming of mutant IDH1 tumors (13). Our current findings point to the value of hyperpolarized 2-13C-pyruvate and its conversion to glutamate as another potential complementary imaging approach for mutant IDH1 tumors. Importantly, hyperpolarized pyruvate has been used clinically (23) and the effect of DCA on hyperpolarized pyruvate metabolism has been detected in rat brain (53).
In summary, our study demonstrates that reprogramming of PDH activity is essential for mutant IDH1 cell proliferation, that MRS can be used to detect this reprogramming and that modulation of PDH activity could potentially serve as a therapeutic approach for mutant IDH1 tumors.
Supplementary Material
Acknowledgments
Financial support: This work was supported by NIH R01CA172845 (SMR), NIH R01CA154915 (SMR), NIH R21CA161545 (SMR) and the Terry Fox Research Institute and Foundation (JGC).
Footnotes
Conflict of interest: None
REFERENCES (53/50)
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell. 2008;13(6):472–82. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 4.Warburg O, Posener K, Negelein E. Ueber den Stoffwechsel der Tumoren. Biochemische Zeitschrift. 1924;152:319–44. [Google Scholar]
- 5.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4(11):891–9. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 6.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang H, Ye D, Guan KL, Xiong Y. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res. 2012;18(20):5562–71. doi: 10.1158/1078-0432.CCR-12-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17(3):225–34. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reitman ZJ, Jin G, Karoly ED, Spasojevic I, Yang J, Kinzler KW, et al. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc Natl Acad Sci U S A. 2011;108(8):3270–5. doi: 10.1073/pnas.1019393108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohka F, Ito M, Ranjit M, Senga T, Motomura A, Motomura K, et al. Quantitative metabolome analysis profiles activation of glutaminolysis in glioma with IDH1 mutation. Tumour Biol. 2014;35(6):5911–20. doi: 10.1007/s13277-014-1784-5. [DOI] [PubMed] [Google Scholar]
- 12.Choi C, Ganji S, Hulsey K, Madan A, Kovacs Z, Dimitrov I, et al. A comparative study of short- and long-TE (1)H MRS at 3 T for in vivo detection of 2-hydroxyglutarate in brain tumors. NMR Biomed. 2013;26(10):1242–50. doi: 10.1002/nbm.2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaumeil MM, Larson PE, Woods SM, Cai L, Eriksson P, Robinson AE, et al. Hyperpolarized [1-13C] glutamate: a metabolic imaging biomarker of IDH1 mutational status in glioma. Cancer Res. 2014;74(16):4247–57. doi: 10.1158/0008-5472.CAN-14-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Esmaeili M, Hamans BC, Navis AC, van Horssen R, Bathen TF, Gribbestad IS, et al. IDH1 R132H mutation generates a distinct phospholipid metabolite profile in glioma. Cancer Res. 2014;74(17):4898–907. doi: 10.1158/0008-5472.CAN-14-0008. [DOI] [PubMed] [Google Scholar]
- 15.Chesnelong C, Chaumeil MM, Blough MD, Al-Najjar M, Stechishin OD, Chan JA, et al. Lactate dehydrogenase A silencing in IDH mutant gliomas. Neuro Oncol. 2014;16(5):686–95. doi: 10.1093/neuonc/not243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grassian AR, Parker SJ, Davidson SM, Divakaruni AS, Green CR, Zhang X, et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014;74(12):3317–31. doi: 10.1158/0008-5472.CAN-14-0772-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reitman ZJ, Duncan CG, Poteet E, Winters A, Yan LJ, Gooden DM, et al. Cancer-associated isocitrate dehydrogenase 1 (IDH1) R132H mutation and d-2-hydroxyglutarate stimulate glutamine metabolism under hypoxia. J Biol Chem. 2014;289(34):23318–28. doi: 10.1074/jbc.M114.575183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glunde K, Bhujwalla ZM, Ronen SM. Choline metabolism in malignant transformation. Nat Rev Cancer. 2011;11(12):835–48. doi: 10.1038/nrc3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Izquierdo-Garcia JL, Viswanath P, Eriksson P, Chaumeil MM, Pieper RO, Phillips JJ, et al. Metabolic Reprogramming in Mutant IDH1 Glioma Cells. PLoS One. 2015;10(2):e0118781. doi: 10.1371/journal.pone.0118781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ardenkjaer-Larsen JH, Fridlund B, Gram A, Hansson G, Hansson L, Lerche MH, et al. Increase in signal-to-noise ratio of > 10,000 times in liquid-state NMR. Proc Natl Acad Sci U S A. 2003;100(18):10158–63. doi: 10.1073/pnas.1733835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Golman K, Zandt R, Thaning M. Real-time metabolic imaging. Proc Natl Acad Sci U S A. 2006;103(30):11270–5. doi: 10.1073/pnas.0601319103. in 't. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurhanewicz J, Vigneron DB, Brindle K, Chekmenev EY, Comment A, Cunningham CH, et al. Analysis of cancer metabolism by imaging hyperpolarized nuclei: prospects for translation to clinical research. Neoplasia. 2011;13(2):81–97. doi: 10.1593/neo.101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nelson SJ, Kurhanewicz J, Vigneron DB, Larson PE, Harzstark AL, Ferrone M, et al. Metabolic imaging of patients with prostate cancer using hyperpolarized [1-(1)(3)C]pyruvate. Sci Transl Med. 2013;5(198):198ra08. doi: 10.1126/scitranslmed.3006070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaumeil MM, Larson PE, Yoshihara HA, Danforth OM, Vigneron DB, Nelson SJ, et al. Non-invasive in vivo assessment of IDH1 mutational status in glioma. Nat Commun. 2013;4:2429. doi: 10.1038/ncomms3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Izquierdo-Garcia JL, Cai LM, Chaumeil MM, Eriksson P, Robinson AE, Pieper RO, et al. Glioma cells with the IDH1 mutation modulate metabolic fractional flux through pyruvate carboxylase. PLoS One. 2014;9(9):e108289. doi: 10.1371/journal.pone.0108289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelly JJ, Blough MD, Stechishin OD, Chan JA, Beauchamp D, Perizzolo M, et al. Oligodendroglioma cell lines containing t(1;19)(q10;p10) Neuro Oncol. 2010;12(7):745–55. doi: 10.1093/neuonc/noq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luchman HA, Stechishin OD, Dang NH, Blough MD, Chesnelong C, Kelly JJ, et al. An in vivo patient-derived model of endogenous IDH1-mutant glioma. Neuro Oncol. 2012;14(2):184–91. doi: 10.1093/neuonc/nor207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hamburger AW, Salmon SE. Primary bioassay of human tumor stem cells. Science. 1977;197(4302):461–3. doi: 10.1126/science.560061. [DOI] [PubMed] [Google Scholar]
- 29.Ronen S, Degani H. Studies of the metabolism of human breast cancer spheroids by NMR. Magnetic Resonance in Medicine. 1989;12(2):274–81. doi: 10.1002/mrm.1910120217. [DOI] [PubMed] [Google Scholar]
- 30.Lodi A, Woods SM, Ronen SM. Treatment with the MEK inhibitor U0126 induces decreased hyperpolarized pyruvate to lactate conversion in breast, but not prostate, cancer cells. NMR Biomed. 2013;26(3):299–306. doi: 10.1002/nbm.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ward CS, Venkatesh HS, Chaumeil MM, Brandes AH, Vancriekinge M, Dafni H, et al. Noninvasive detection of target modulation following phosphatidylinositol 3-kinase inhibition using hyperpolarized 13C magnetic resonance spectroscopy. Cancer Res. 2010;70(4):1296–305. doi: 10.1158/0008-5472.CAN-09-2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roche TE, Hiromasa Y. Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell Mol Life Sci. 2007;64(7-8):830–49. doi: 10.1007/s00018-007-6380-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. The Journal of Clinical Investigation. 2013;123(9):3664–71. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bralten LB, Kloosterhof NK, Balvers R, Sacchetti A, Lapre L, Lamfers M, et al. IDH1 R132H decreases proliferation of glioma cell lines in vitro and in vivo. Ann Neurol. 2011;69(3):455–63. doi: 10.1002/ana.22390. [DOI] [PubMed] [Google Scholar]
- 36.Schroeder MA, Atherton HJ, Ball DR, Cole MA, Heather LC, Griffin JL, et al. Real-time assessment of Krebs cycle metabolism using hyperpolarized 13C magnetic resonance spectroscopy. FASEB J. 2009;23(8):2529–38. doi: 10.1096/fj.09-129171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen R, Nishimura MC, Kharbanda S, Peale F, Deng Y, Daemen A, et al. Hominoid-specific enzyme GLUD2 promotes growth of IDH1R132H glioma. Proceedings of the National Academy of Sciences. 2014;111(39):14217–22. doi: 10.1073/pnas.1409653111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–44. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanchez WY, McGee SL, Connor T, Mottram B, Wilkinson A, Whitehead JP, et al. Dichloroacetate inhibits aerobic glycolysis in multiple myeloma cells and increases sensitivity to bortezomib. Br J Cancer. 2013;108(8):1624–33. doi: 10.1038/bjc.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tennant DA, Duran RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010;10(4):267–77. doi: 10.1038/nrc2817. [DOI] [PubMed] [Google Scholar]
- 41.Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483(7390):484–8. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324(5924):261–5. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio II, et al. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012;26(18):2038–49. doi: 10.1101/gad.198200.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prabhu A, Sarcar B, Miller CR, Kim S-H, Nakano I, Forsyth P, et al. Ras-mediated modulation of pyruvate dehydrogenase activity regulates mitochondrial reserve capacity and contributes to glioblastoma tumorigenesis. Neuro-Oncology. 2015 doi: 10.1093/neuonc/nou369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sutendra G, Michelakis ED. Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front Oncol. 2013;3:38. doi: 10.3389/fonc.2013.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic Modulation of Glioblastoma with Dichloroacetate. Science Translational Medicine. 2010;2(31):31ra34. doi: 10.1126/scitranslmed.3000677. [DOI] [PubMed] [Google Scholar]
- 47.Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70(22):8981–7. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nelson SJ. Assessment of therapeutic response and treatment planning for brain tumors using metabolic and physiological MRI. NMR Biomed. 2011;24(6):734–49. doi: 10.1002/nbm.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Elkhaled A, Jalbert LE, Phillips JJ, Yoshihara HA, Parvataneni R, Srinivasan R, et al. Magnetic resonance of 2-hydroxyglutarate in IDH1-mutated low-grade gliomas. Sci Transl Med. 2012;4(116):116ra5. doi: 10.1126/scitranslmed.3002796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andronesi OC, Kim GS, Gerstner E, Batchelor T, Tzika AA, Fantin VR, et al. Detection of 2-hydroxyglutarate in IDH-mutated glioma patients by in vivo spectral-editing and 2D correlation magnetic resonance spectroscopy. Sci Transl Med. 2012;4(116):116ra4. doi: 10.1126/scitranslmed.3002693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Choi C, Ganji SK, DeBerardinis RJ, Hatanpaa KJ, Rakheja D, Kovacs Z, et al. 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat Med. 2012;18(4):624–9. doi: 10.1038/nm.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pope WB, Prins RM, Albert Thomas M, Nagarajan R, Yen KE, Bittinger MA, et al. Non-invasive detection of 2-hydroxyglutarate and other metabolites in IDH1 mutant glioma patients using magnetic resonance spectroscopy. J Neurooncol. 2012;107(1):197–205. doi: 10.1007/s11060-011-0737-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park JM, Josan S, Grafendorfer T, Yen Y-F, Hurd RE, Spielman DM, et al. Measuring mitochondrial metabolism in rat brain in vivo using MR Spectroscopy of hyperpolarized [2-13C]pyruvate. NMR in Biomedicine. 2013;26(10):1197–203. doi: 10.1002/nbm.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.