

Abstract
Polyamines are essential aliphatic polycations that bind to nucleic acids and accordingly are involved in a variety of cellular processes. Polyamine function can be regulated by acetylation and deacetylation, just as histone function can be regulated by lysine acetylation and deacetylation. Acetylpolyamine amidohydrolase (APAH) from Mycoplana ramosa is a zinc-dependent polyamine deacetylase that shares approximately 20% amino acid sequence identity with human histone deacetylases. We now report the X-ray crystal structures of APAH–inhibitor complexes in a new and superior crystal form that diffracts to very high resolution (1.1–1.4 Å). Inhibitors include previously-synthesized analogues of N8-acetylspermidine bearing trifluoromethylketone, thiol, and hydroxamate zinc-binding groups [Decroos, C., Bowman, C. M., and Christianson, D. W. (2013) Bioorg. Med. Chem. 21, 4530], and newly synthesized hydroxamate analogues of shorter, monoacetylated diamines, the most potent of which is the hydroxamate analogue of N-acetylcadaverine (IC50 = 68 nM). The high resolution crystal structures of APAH–inhibitor complexes provide key inferences on the inhibition and catalytic mechanism of zinc-dependent deacetylases. For example, the trifluoromethylketone analogue of N8-acetylspermidine binds as a tetrahedral gem-diol that mimics the tetrahedral intermediate and its flanking transition states in catalysis. Surprisingly, this compound is also a potent inhibitor of human histone deacetylase 8 with an IC50 of 260 nM. Crystal structures of APAH– inhibitor complexes are determined at the highest resolution of any currently existing zinc deacetylase structure, and thus represent the most accurate references points for understanding structure-mechanism and structure-inhibition relationships in this critically important enzyme family.
INTRODUCTION
In all forms of life, the reversible covalent modification of proteins and other biologically important molecules is a critical chemical strategy for the regulation of molecular and cellular function. For example, proteins can undergo methylation, phosphorylation, acetylation, glycosylation, ADP ribosylation, farnesylation, myristoylation, or ubiquitination in the course of their biological functions.1 In particular, acetylation targets primary amino groups, as found in the side chain of lysine, the N-terminus of a protein, or polyamines such as spermidine and spermine. Lysine acetylation was first identified in histone proteins,2 where it plays a crucial role in transcriptional regulation.3 Histone acetylation can alter nucleosome structure, making it less condensed so as to facilitate transcription. Significantly, non-histone proteins also serve as targets for lysine acetylation.4 In total, the mammalian acetylome comprises over 3600 lysine acetylation sites that regulate myriad cellular functions.5,6
Acetyltransferases utilize acetyl-CoA to generate acetyllysine, whereas histone deacetylases (HDACs) catalyze the hydrolysis of acetyllysine to regenerate free lysine and acetate.7 The HDAC family contains 18 isoforms divided into four different classes. The class I (HDAC1, 2, 3, and 8), class IIa (HDAC4, 5, 7, and 9) and class IIb (HDAC6 and 10), and class IV (HDAC11) enzymes are metal-dependent deacetylases that share a common α/β fold.8 In contrast, the class III enzymes (sirtuins 1–7) are NAD-dependent deacetylases that are evolutionarily and mechanistically distinct.9 Notably, metal-dependent HDACs adopt the α/β fold originally observed for the binuclear manganese metallohydrolase arginase.10 The mechanism of HDAC catalysis has been established based on biochemical and structural studies of the histone deacetylase-like protein from Aquifex aeolicus and human HDAC8.11–17
In addition to proteins, small molecules are also subject to reversible acetylation in the course of their biological function. For example, polyamines such as putrescine, spermidine, and spermine are essential cationic metabolites involved in numerous cellular processes in all forms of life and are subject to acetylation (Figure 1).18 Enzymes of polyamine biosynthesis are tightly regulated,18,19 and dysregulation of polyamine metabolism is often associated with certain disease pathologies such as cancer.20,21 As with acetyllysine residues, the decreased overall charge of acetylpolyamines attenuates their affinity for negatively charged nucleic acids. In eukaryotes, two enzymes are responsible for polyamine acetylation: the cytosolic spermine/spermidine N1-acetyltransferase22 and the nuclear N8-acetyltransferase.23 Differences in selectivity and subcellular localization of these enzymes reflect different roles for the corresponding acetylated polyamines. For instance, N1-acetylspermidine, produced in the cytosol, is converted back to the shorter diamine putrescine through an oxidative deamination reaction catalyzed by polyamine oxidase.24 In contrast, N8-acetylation of spermidine in the nucleus enables the export of N8-acetylspermidine to the cytosol.25 Deacetylation of N8-acetylspermidine, but not N1-acetylspermidine, occurs in the cytosol to recycle spermidine, which either reenters the nucleus or serves as a substrate for spermine synthase.25,26 The mammalian polyamine deacetylase responsible for the specific deacetylation of N8-acetylspermidine remains unidentified, even though its activity has been detected in subcellular fractions and in cells.25–28
Figure 1.

Substrates and inhibitors of APAH from Mycoplana ramosa. Substrates include: acetylputrescine, acetylcadaverine, N1-acetylspermidine, N8-acetylspermidine, and N1-acetylspermine. Previously described33 inhibitors of APAH are analogues of the substrate N8-acetylspermidine bearing different metal-binding groups: trifluoromethylketone (1), thiol (2), or hydroxamic acid (3). New inhibitors reported in this study (4–7) are shorter hydroxamic acids with varying chain lengths.
Acetylpolyamine amidohydrolase (APAH) is a polyamine deacetylase identified in the Gram-negative bacterium Mycoplana ramosa.29 APAH exhibits broad substrate specificity in that it catalyzes the deacetylation of all short- and long-chain acetylpolyamines shown in Figure 1, although it exhibits a modest preference for short-chain polyamines.29,30 Like the metal-dependent HDACs, APAH is a zinc metallohydrolase that adopts the α/β arginase fold.31 However, APAHs are dimeric enzymes, as revealed by the crystal structures of APAH from M. ramosa and Burkholderia pseudomallei,31,32 whereas HDACs such as HDAC8 are monomers.12,13 Assembly of the APAH dimer results in a narrow “L”-shaped active site at the dimer interface that confers specifity for long, flexible, and positively charged acetylpolyamine substrates.
We recently reported the synthesis and evaluation of N8-acetylspermidine analogues as APAH inhibitors.33 These analogues differ by the nature of the functional group targeting Zn2+ coordination in the active site. The most potent inhibitors contain either a trifluoromethylketone or a hydroxamate Zn2+-binding group (Figure 1) and exhibit nanomolar inhibitory potency. Here, we report the synthesis and evaluation of new hydroxamate analogues of short-chain acetyldiamines, compounds 4–7 (Figure 1). These compounds exhibit increased inhibitory potency against APAH and also inhibit HDAC8. We additionally report high resolution X-ray crystal structures of APAH complexed with inhibitors 1–6 (Figure 1) in a new crystal form. These results provide new insight on catalysis and inhibition in the greater family of metal-dependent deacetylases.
METHODS
Chemistry
General Remarks
All reagents were of at least 95% purity, purchased from Fisher Scientific, Alfa Aesar, Sigma Aldrich or Bachem, and used without any further purification. All solvents were of HPLC grade and purchased from Fisher Scientific or Sigma Aldrich. For reactions requiring anhydrous conditions, solvents were purchased as anhydrous grade from Fisher Scientific except for CH2Cl2, which was freshly distilled under N2 from P2O5. Reactions were monitored by TLC with Sigma Aldrich aluminum plates (silica gel, 60 Å, 200 µm) and visualized by staining with ninhydrin solution. Flash column chromatography was performed using Fisher Scientific silica gel 60 (230–400 mesh). High-resolution mass spectrometry (HRMS) was performed on a Waters LC-TOF mass spectrometer (model LCT-XE Premier) using electrospray ionization (ESI) in positive mode. 1H and 13C spectra were recorded on a Bruker DRX 500 spectrometer operating at 500 MHz and 125.6 MHz for 1H and 13C NMR, respectively. 1H and 13C NMR chemical shifts (δ) are reported in ppm relative to the residual solvent peak. NMR coupling constants (J) are reported in Hz, and multiplicities are denoted as follows: s, singlet; d, doublet; t, triplet; q, quadruplet; m, multiplet; and dt, doublet of triplets.
The synthesis of compounds 1, 2, and 3 as dihydrochloride salts has already been described.33 The synthesis of compounds 4, 5, 6, and 7 is summarized in Scheme 1. Briefly, N-Boc-protected carboxylic acid starting materials were activated with carbonyldiimidazole (CDI) before reacting with unprotected hydroxylamine to form hydroxamic acids 8–11.33,34 Deprotection of compounds 8–11 was achieved under acidic conditions (anhydrous HCl (1 M) in ethyl acetate (AcOEt)) to yield inhibitors 4–7 as hydrochloride salts. Experimental details and characterization are given below for each new compound.
Scheme 1. Synthesis of new APAH inhibitors 4–7.

Reagents and conditions: (a) Carbonyldiimidazole (1.5 equivalents) in tetrahydrofuran, (rt, 1 h), then NH2OH·HCl (2 equivalents; room temperature, overnight); (b) Anhydrous HCl (1 M) in ethyl acetate, (room temperature, overnight).
5-(tert-Butoxycarbonylamino)-N-hydroxypentanamide (8)
To a solution of N-(tert-butoxycarbonyl)-5-aminovaleric acid (1.00 g, 4.60 mmol) under argon in dry tetrahydrofuran (30 mL) was added CDI (1.12 g, 6.91 mmol). After 1 hour at room temperature, hydroxylamine hydrochloride (640 mg, 9.21 mmol) was added to the solution. After stirring overnight, the reaction mixture was diluted with a 5% KHSO4 aqueous solution (100 mL) and extracted with AcOEt. Combined organic extracts were washed with brine and dried over Na2SO4, filtered, and concentrated in vacuo. The solid was further purified by flash chromatography on silica gel with hexanes/AcOEt followed by AcOEt/MeOH gradients to afford hydroxamic acid 8 as a white solid (953 mg, 89%). 1H NMR (500 MHz, DMSO-d6) δ: 10.32 (s, 1H), 868 (s, 1H), 6.77 (t, J = 5.0 Hz, 1H), 2.88 (apparent q (dt), J = 6.5 Hz), 1.92 (t, J = 7.3 Hz, 2H), 1.49–1.40 (m, 2H), 1.36 (s, 9H), 1.35–1.29 (m, 2H). 13C NMR (125.6 MHz, DMSO-d6) δ: 169.0, 155.6, 77.4, 39.6, 32.0, 29.2, 28.3 (3C), 22.6. HRMS (ESI) calcd for C10H20N2O4Na [M + Na]+ 255.1321, found 255.1327.
6-(tert-Butoxycarbonylamino)-N-hydroxyhexanamide (9)
Reaction of N-(tert-butoxycarbonyl)-6-aminocaproic acid (1.00 g, 4.32 mmol) with CDI (1.05 g, 6.48 mmol) and hydroxylamine hydrochloride (601 mg, 8.65 mmol) was performed under the same conditions as for 8, and afforded after purification hydroxamic acid 9 as a white solid (871 mg, 82%). 1H NMR (500 MHz, DMSO-d6) δ: 10.31 (s, 1H), 8.64 (s, 1H), 6.72 (t, J = 5.3 Hz, 1H), 2.87 (apparent q (dt), J = 6.3 Hz, 2H), 1.92 (t, J = 7.3 Hz, 2H), 1.48–1.40 (m, 2H), 1.39–1.32 (m, 2H), 1.36 (s, 9H), 1.23–1.16 (m, 2H). 13C NMR (125.6 MHz, DMSO-d6) δ: 169.1, 155.6, 77.3, 39.6, 32.3, 29.2, 28.3 (3C), 25.9, 24.9. HRMS (ESI) calcd for C11H22N2O4Na [M + Na]+ 269.1477, found 269.1476.
7-(tert-Butoxycarbonylamino)-N-hydroxyheptanamide (10)
Reaction of N-(tert-butoxycarbonyl)-7-aminoheptanoic acid (1.10 g, 4.48 mmol) with CDI (1.09 g, 6.72 mmol) and hydroxylamine hydrochloride (623 mg, 8.97 mmol) was performed under the same conditions as for 8, and afforded after purification hydroxamic acid 10 as a white solid (1.01 g, 86%). 1H NMR (500 MHz, DMSO-d6) δ: 10.32 (s, 1H), 8.64 (s, 1H), 6.74 (t, J = 5.3 Hz, 1H), 2.86 (apparent q (dt), J = 6.7 Hz, 2H), 1.92 (t, J = 7.5 Hz, 2H), 1.49–1.43 (m, 2H), 1.36 (s, 9H), 1.36–1.31 (m, 2H), 1.23–1.19 (m, 4H). 13C NMR (125.6 MHz, DMSO-d6) δ: 169.1, 155.6, 77.3, 39.7, 32.3, 29.4, 28.4, 28.3 (3C), 26.1, 25.1. HRMS (ESI) calcd for C13H24N2O4Na [M + Na]+ 283.1364, found 283.1364.
8-(tert-Butoxycarbonylamino)-N-hydroxyoctanamide (11)
Reaction of N-(tert-butoxycarbonyl)-8-aminooctanoic acid (1.10 g, 4.24 mmol) with CDI (1.03 g, 6.35 mmol) and hydroxylamine hydrochloride (589 mg, 8.48 mmol) was performed under the same conditions as for 8, and afforded after purification hydroxamic acid 11 as a white solid (910 mg, 78%). 1H NMR (500 MHz, DMSO-d6) δ: 10.32 (s, 1H), 8.65 (s, 1H), 6.74 (t, J = 5.3 Hz, 1H), 2.88 (apparent q (dt), J = 6.7 Hz, 2H), 1.92 (t, J = 7.5 Hz, 2H), 1.49–1.44 (m, 2H), 1.36 (s, 9H), 1.36– 1.32 (m, 2H), 1.25–11.19 (m, 6H). 13C NMR (125.6 MHz, DMSO-d6) δ: 169.2, 155.6, 77.3, 39.9, 32.3, 29.5, 28.6, 28.5, 28.3 (3C), 26.2, 25.1. HRMS (ESI) calcd for C14H26N2O4Na [M + Na]+ 297.1790, found 297.1790.
5-Amino-N-hydroxypentanamide hydrochloride salt (4)
To a solution of compound 8 (270 mg, 1.16 mmol) in 10 mL dry CH2Cl2 was added 30 mL of a 1 M anhydrous HCl solution in AcOEt. After stirring overnight under argon at room temperature, the precipitate was filtered, washed with dry Et2O, and dried in vacuo to afford hydroxamic acid 4 as an off-white powder (181 mg, 92%). 1H NMR (500 MHz, D2O) δ: 2.99 (t, J = 6.0 Hz, 2H), 2.22 (t, J = 6.8 Hz, 2H), 1.68–1.64 (m, 4H). 13C NMR (125.6 MHz, D2O) δ: 172.5, 39.0, 31.6, 26.0, 21.8. HRMS (ESI) calcd for C5H13N2O2 [M + H]+ 133.0977, found 133.0977.
6-Amino-N-hydroxyhexanamide hydrochloride salt (5)
Compound 9 (200 mg, 812 µmol) was deprotected under the same conditions as for 4 to afford after filtration hydroxamic acid 5 as an off-white powder (140 mg, 95%). 1H NMR (500 MHz, D2O) δ: 2.95 (t, J = 7.5 Hz, 2H), 2.15 (t, J = 7.3 Hz, 2H), 1.66–1.56 (m, 4H), 1.36–1.30 (m, 2H). 13C NMR (125.6 MHz, D2O) δ: 173.1, 39.2, 32.0, 26.3, 24.9, 24.3. HRMS (ESI) calcd for C6H15N2O2 [M + H]+ 147.1138, found 147.1138.
7-Amino-N-hydroxyheptanamide hydrochloride salt (6)
Compound 10 (300 mg, 1.15 mmol) was deprotected under the same conditions as for 4 to yield after filtration hydroxamic acid 6 as an off-white powder (207 mg, 91%). 1H NMR (500 MHz, D2O) δ: 3.01 (t, J = 7.5 Hz, 2H), 2.20 (t, J = 7.3 Hz, 2H), 1.71–1.66 (m, 2H), 1.65–1.60 (m, 2H), 1.44–1.33 (m, 4H). 13C NMR (125.6 MHz, D2O) δ: 173.4, 39.4, 32.2, 27.4, 26.5, 25.2, 24.6. HRMS (ESI) calcd for C7H17N2O2 [M + H]+ 161.1290, found 161.1298.
8-Amino-N-hydroxyoctanamide hydrochloride salt (7)
Compound 11 (200 mg, 729 µmol) was deprotected under the same conditions as for 4 to yield after filtration hydroxamic acid 7 as an off-white powder (143 mg, 93%). 1H NMR (500 MHz, D2O) δ: 2.95 (t, J = 7.5 Hz, 2H), 2.13 (t, J = 7.5 Hz, 2H), 1.64–1.58 (m, 2H), 1.58–1.53 (m, 2H), 1.37–1.26 (m, 6H). 13C NMR (125.6 MHz, D2O) δ: 173.5, 39.4, 32.2, 27.6, 26.5, 25.3, 24.7. HRMS (ESI) calcd for C8H19N2O2 [M + H]+ 175.1447, found 175.1441.
Expression and Purification of APAH and HDAC8
APAH was expressed from a pET-21b plasmid in Escherichia coli BL21(DE3) cells and purified as previously described.31 HDAC8 was expressed from a pHD2-Xa-His plasmid (modified pET-20b plasmid) in Escherichia coli BL21(DE3) cells and purified using previously described procedures.15,35
Inhibitory Activity Measurements
The inhibition of APAH by the newly-synthesized derivatives 4, 5, 6, and 7 was evaluated using a fluorimetric assay, as previously described.31,33 The IC50 values for compounds 1, 2, and 3 were reported previously.33 Activity was measured using the commercially available Fluor-de-Lys deacetylase fluorogenic substrate (BML-KI104, Enzo Life Sciences). Deacetylation of the acetyllysine-fluorophore substrate is followed by cleavage of the lysine-fluorophore amide bond by a protease developer, resulting in a fluorescence shift. In contrast with the shorter acetyllysine-fluorophore assay substrate used to assay APAH, the longer peptide fluorophore Ac-Arg-His-Lys(Ac)-Lys(Ac)-aminomethylcoumarin is a poor substrate for APAH, presumably due to the constricted APAH active site, as previously reported.31 Activity assays were run at 25°C and contained 250 nM APAH (∼50% Zn2+ occupancy), 150 µM substrate, 0–250 µM inhibitor in assay buffer (25 mM Tris (pH = 8.2), 137 mM NaCl, 2.7 mM KCl, and 1 mM MgCl2) in a final volume of 50 µL. Enzyme was first incubated with the inhibitor for 5 min before initiating the reaction with substrate; the most potent inhibitor, compound 6, was also evaluated after 30 and 60 min incubation times to assess the possibility of time-dependent inhibition. After 30 min, reactions were quenched by adding 100 µM M344 (Sigma Aldrich) and the appropriate Fluor-de-Lys developer (BML-KI105, Enzo Life Sciences, 50 µL). Since the developer is typically a serine protease, e.g., trypsin, we confirmed that trifluoromethylketone 1, even at millimolar concentrations, does not inhibit the developer enzyme (data not shown). Fluorescence was measured after 45 min using a Fluoroskan II plate reader (excitation = 355 nm, emission = 460 nm). Assays for each concentration of inhibitor were performed in triplicate in separate experiments. IC50 values for each compound were determined using the software Graphpad Prism (2008).
The inhibition of HDAC8 by compounds 1–7 was evaluated using a similar fluorimetric assay, as previously described.35 Activity assays were run at 25°C and contained 500 nM HDAC8 enzyme, 150 µM Ac-Arg-His-Lys(Ac)-Lys(Ac)-aminomethylcoumarin substrate (BML-KI178, Enzo Life Sciences), 0–10 mM inhibitor in assay buffer (25 mM Tris (pH = 8.2), 137 mM NaCl, 2.7 mM KCl, and 1 mM MgCl2; 250 µM tris-(2-carboxyethyl)phosphine was added for the assay of thiol compound 2) in a final volume of 50 µL. Enzyme was first incubated with inhibitor for 5 min before initiating the reaction with substrate; the most potent inhibitor, compound 1, was also evaluated after 30 and 60 min incubation times to assess the possibility of time-dependent inhibition. After 30 min, reactions were quenched by adding 100 µM M344 (Sigma Aldrich) and the appropriate Fluor-de-Lys developer (BML-KI176, Enzo Life Sciences, 50 µL). Fluorescence was measured after 45 min using a Fluoroskan II plate reader (excitation = 355 nm, emission = 460 nm). Assays for each concentration of inhibitor were performed in triplicate in separate experiments. IC50 values for each compound were determined using the software Graphpad Prism (2008).
Crystallization and Data Collection
Crystals of APAH–inhibitor complexes were prepared by cocrystallization at 4°C (except for the complex with 4, 21°C) in sitting drops using the vapor diffusion method. In general, a 500 nL drop containing 5 mg/mL (for 2 and 4) or 10 mg/mL (for 1, 3, 5, and 6) APAH, 20 mM Tris (pH = 8.0), 50 mM NaCl, 0.01% sodium azide, ZnCl2 (0.5 mM for 2, and 2 mM for 1, 3, 4, 5, and 6), and 2.5 mM inhibitor was added to a 500 nL drop of precipitant solution and equilibrated against a 100 µL reservoir of precipitant solution. Before mixing the drops, APAH was first incubated with ZnCl2 for 15 min on ice before adding the inhibitor. Enzyme solutions were filtered before crystallization due to the formation of a slight precipitate after addition of ZnCl2 and inhibitor. The precipitant solution for the cocrystallization of the APAH–1 and APAH–2 complexes consisted of 0.2 M LiNO3 and 20% (w/v) PEG 3,350. The precipitant solution for the cocrystallization of the APAH–3 complex consisted of 0.2 M ammonium chloride and 20% (w/v) PEG 3,350; after harvest, crystals were soaked for an additional two days in this precipitant solution supplemented with 2.5 mM 3. The precipitant solution for the cocrystallization of the APAH–4 complex consisted of 0.2 M KNO3 and 20% (w/v) PEG 3,350. The precipitant solution for the cocrystallization of the APAH–5 complex consisted of 0.2 M LiCl and 20% (w/v) PEG 3,350. The precipitant solution for the cocrystallization of the APAH–6 complex consisted of 0.2 M ammonium chloride and 20% (w/v) PEG 3,350; after harvest, crystals were soaked for an additional two days in this precipitant solution supplemented with 2.5 mM 6. We were unable to prepare suitable crystals of the APAH–7 complex.
Typically, crystals appeared within 2–7 days. Crystals were flash-cooled in liquid nitrogen after transfer to a cryoprotectant consisting of precipitant solution supplemented with 25–30% glycerol (v/v). X-ray diffraction data were collected on beamline X29 at the National Synchrotron Light Source (NSLS, Broohaven National Laboratory, New York) for APAH complexes with inhibitors 1, 2, 4, and 5; on beamline BL14-1 at the Stanford Synchrotron Radiation Lightsource (SSRL, Stanford Linear Accelerator Center, California); or on beamline NE-CAT 24-ID-E at the Advanced Photon Source (APS, Argonne National Laboratory, Illinois) for APAH in complex with 6. Data collection and reduction statistics are presented in Table 1. Diffraction data from crystals of all complexes except the APAH–6 complex were indexed, integrated, and scaled using HKL2000.36 Data collected from the APAH–6 complex were indexed and integrated with MOSFLM37 and scaled with SCALA (contained in the CCP4 suite of programs).38
Table 1.
Data collection and refinement statistics
| APAH–1 complex | APAH–2 complex | APAH–3 complex | APAH–4 complex | APAH–5 complex | APAH–6 complex | |
|---|---|---|---|---|---|---|
| Unit cell | ||||||
| space group symmetry |
P21 | P21 | P21 | P21 | P21 | P21 |
| a, b, c (Å) | 45.1, 121.4, 64.3 | 44.9, 121.0, 64.7 | 45.0, 121.1, 64.4 | 44.7, 120.9, 64.1 | 44.9, 120.5, 64.4 | 45.0, 120.7, 64.5 |
| α, β, γ (deg) | 90, 96.8, 90 | 90, 97.0, 90 | 90, 96.8, 90 | 90, 97.0, 90 | 90, 96.9, 90 | 90, 97.0, 90 |
| Data collection | ||||||
| wavelength (Å) | 1.075 | 1.075 | 1.069 | 1.075 | 1.075 | 0.979 |
| resolution limits (Å) | 38.9 – 1.42 | 41.8 – 1.40 | 38.9 – 1.33 | 38.7 – 1.42 | 41.8 – 1.22 | 22.8 – 1.13 |
| total/unique reflections |
644824/128460 | 955700/134698 | 1075040/155090 | 913682/126724 | 1391188/199748 | 1643256/248014 |
| Rmergea,b | 0.070 (0.328) | 0.088 (0.795) | 0.054 (0.272) | 0.070 (0.398) | 0.079 (0.525) | 0.077 (0.455) |
| I/σ(I)a | 20.4 (4.8) | 23.1 (2.4) | 23.4 (6.8) | 24.3 (5.0) | 21.5 (3.7) | 11.8 (3.9) |
| redundancy a | 5.0 (4.8) | 7.1 (5.7) | 6.9 (6.6) | 7.2 (6.8) | 7.0 (7.0) | 6.6 (6.1) |
| completeness (%)a | 99.9 (99.7) | 100 (100) | 99.3 (98.4) | 100 (100) | 99.4 (100) | 98.2 (94.4) |
| Refinement | ||||||
| reflections used in refinement/test set |
128411/6458 | 134649/6776 | 155046/7777 | 126680/6355 | 199693/10060 | 247921/12379 |
| Rcrystc | 0.133 | 0.146 | 0.133 | 0.131 | 0.137 | 0.114 |
| Rfreed | 0.153 | 0.164 | 0.147 | 0.152 | 0.152 | 0.136 |
| protein atomse | 5557 | 5444 | 5514 | 5406 | 5534 | 5481 |
| water moleculese | 953 | 856 | 1047 | 950 | 979 | 1094 |
| ligand moleculese | 2 | 2 | 2 | 2 | 2 | 2 |
| Zn2+ ionse | 2 | 4 | 2 | 4 | 4 | 2 |
| K+/Na+/NH4+ ionse | 2/2/0 | 2/2/0 | 2/0/2 | 4/0/0 | 2/2/0 | 2/0/2 |
| nitrate ionse | 4 | - | - | 4 | - | - |
| glycerol moleculese | 2 | 3 | 5 | 4 | 4 | 3 |
|
R.m.s. deviations from ideal geometry |
||||||
| bonds (Å) | 0.009 | 0.009 | 0.009 | 0.009 | 0.008 | 0.010 |
| angles (°) | 1.3 | 1.3 | 1.3 | 1.3 | 1.3 | 1.4 |
| dihedral angles (°) | 13 | 12 | 13 | 13 | 13 | 12 |
| Ramachandran plot (%)f | ||||||
| allowed | 92.6 | 91.2 | 92.4 | 90.8 | 92.4 | 92.0 |
| additionally allowed | 6.7 | 8.3 | 7.2 | 8.5 | 7.1 | 7.6 |
| generously allowed | 0.7 | 0.5 | - | 0.7 | 0.2 | - |
| disallowed | - | - | 0.4 | - | 0.4 | 0.4 |
| PDB accession code | 4ZUM | 4ZUN | 4ZUO | 4ZUP | 4ZUQ | 4ZUR |
Values in parentheses refer to the highest shell of data.
Rmerge = Σ|Ih〈I〉h|/ΣIh, where 〈I〉h is the average intensity calculated from replicate reflections.
Rcryst= Σ||Fo|-|Fc||/Σ|Fo| for reflections contained in the working set. |Fo| and |Fc| are the observed and calculated structure factor amplitudes, respectively.
Rfree = Σ||Fo|-|Fc||/Σ|Fo| for reflections contained in the test set held aside during refinement.
Per asymmetric unit.
Calculated with PROCHECK version 3.4.4.
Phasing, Model Building, and Structure Refinement
All structures belonged to space group P21 with two molecules (APAH dimer) in the asymmetric unit. Structures were solved by molecular replacement using PHENIX39 with the atomic coordinates of the APAH–M344 complex (PDB accession code 3Q9B) less inhibitor, ion, and solvent used as a search probe for rotation and translation function calculations. Each model was refined with iterative cycles of refinement in PHENIX and manual model rebuilding in COOT.40 Solvent molecules and inhibitors were added after several rounds of refinement for each structure. In the final stages, Translation Libration Screw (TLS) refinement was performed.41 TLS groups were automatically determined using PHENIX. For the APAH–6 complex (1.13 Å resolution), clear electron density was observed for many hydrogen atoms in late stages of refinement. Accordingly, hydrogen atoms were added to the model during the last stage of refinement, and anisotropic temperature factor refinement was performed for all atoms except hydrogens and water molecules. The protonation states of ionizable residues were determined by analysis of electron density as well as evaluation of their chemical environments. Refinement converged smoothly and detailed refinement statistics are recorded in Table 1.
In each structure, side chains of residues that were partially disordered were excluded from the final model as follows: APAH–1 complex, K187 (monomers A and B); APAH–2 complex, K84 and K187 (monomers A and B); APAH–3 complex, E106 (monomers A and B), K187 (monomer A); APAH–4 complex, K84 (monomers A and B) and K187 (monomer B); APAH–5 complex, R2 (monomers A and B), K79 (monomer B), K84 (monomers A and B), K187 (monomers A and B), and K267 (monomer B); and APAH–6 complex, K15 (monomer B), K84 (monomer A), K187 (monomer B), K237 (monomer A), and K267 (monomer B). For the APAH– 3, APAH–5, and APAH–6 complexes, H227 adopted a disallowed conformation that nonetheless was characterized by well-defined electron density.
Finally, in most of the structures reported, occasional ambiguous electron densities were observed either on the protein surface (e.g., near Y247 or F27) or in the protein interior (e.g., near M164). These electron densities were usually elongated and potentially corresponded to disordered PEG fragments or other molecules present in the buffer solution used for crystallization. However, since these electron density peaks were not confidently interpretable, they were left unmodeled.
RESULTS
Design, Synthesis, and Evaluation of New APAH Inhibitors
In our previous work, we designed analogues of N8-acetylspermidine as inhibitors of APAH.33 This led us to identify the trifluoromethylketone, hydroxamic acid, and thiol functional groups as the best ligands for coordination to the active site Zn2+ ion (Table 2). Given that shorter substrates such as acetylputrescine and acetylcadaverine bind to APAH with slightly higher affinity compared to larger substrates such as N1-acetylspermidine and N8-acetylspermidine (as reflected by Km values),30 we hypothesized that analogues of shorter acetylpolyamines might yield improved inhibitory potency. Consequently, we designed the four new hydroxamic acids 4–7 (Figure 1). These compounds were easily synthesized as hydrochloride salts in two steps from commercially available materials (Scheme 1; synthetic details are outlined in the Methods section).
Table 2.
Inhibitory potency of compounds 1–7 against against Mycoplana ramosa APAH and human HDAC8
| IC50 (µM) | ||
|---|---|---|
| Compound | APAH | HDAC8 |
| 1 | 0.27 ± 0.03a | 0.26 ± 0.02 |
| 38 ± 6a | ||
| 2 | 26 ± 3a | 12 ± 1 |
| 3 | 0.39 ± 0.03a | 14 ± 1 |
| 4 | 0.17 ± 0.01 | 120 ± 10 |
| 5 | 0.13 ± 0.01 | 19 ± 2 |
| 6 | 0.068 ± 0.006 | 5.3 ± 0.8 |
| 7 | 0.15 ± 0.02 | 2.2 ± 0.2 |
Values previously reported in ref. 33.
Compounds 4–7 were assayed for inhibitory activity against APAH, and results are summarized in Table 2. The IC50 plots are found in Figure S1, Supporting Information. Hydroxamate 6 (the hydroxamate analogue of acetylcadaverine) is the most potent APAH inhibitor of all compounds tested, with an IC50 of 68 nM (time-dependent inhibition is not observed). The other short-chain hydroxamates exhibit slightly diminished inhibitor potency with IC50 values of 130, 150, and 170 nM for compounds 5 (hydroxamate analogue of acetylputrescine), 7, and 4, respectively. Even so, all of these compounds exhibit increased potency relative to the authentic hydroxamate analogue of N8-acetylspermidine, 3, which has an IC50 value of 390 nM.33
Compounds 1–7 were additionally evaluated as inhibitors of the related deacetylase HDAC8 (Table 2). The IC50 plots are found in Figure S2, Supporting Information. Hydroxamates 3–7 are modest inhibitors of human HDAC8 with IC50 values in the mid- to low-micromolar range. In the short-chain analogue series, inhibitory potency increases with chain length, with IC50 values of 120, 19, 5.3, and 2.2 µM, for 4, 5, 6, and 7, respectively. Thiol 2 is also a modest inhibitor of HDAC8 with an IC50 of 12 µM, as is hydroxamate 3 (14 µM). Strikingly, among the compounds listed in Table 2, trifluromethylketone 1 is the most potent inhibitor of HDAC8 with IC50 = 260 nM (time-dependent inhibition is not observed).
Structure of APAH in a New Crystal Form
We previously reported the structure of APAH in a triclinic crystal form, in which the P1 unit cell contained two trimers of dimers.31 These crystals were exceedingly difficult to work with because they contained multiple lattices that grew with different orientations. Suitable X-ray diffraction intensities could be measured from a single crystal at 2.2–2.5 Å resolution only through the use of a microfocus X-ray beam centered on a crystal corner. Importantly, the APAH cocrystallization studies reported herein yielded a new monoclinic crystal form with superior diffraction properties, consistently yielding crystals diffracting to 1.1–1.4 Å resolution.
Although the overall dimer architecture of APAH in the new monoclinic crystal form is identical to that previously reported in the triclinic crystal form, dimer-dimer interactions differ between these two crystal forms.31 These differences give rise to alternative crystal symmetries. The coordination geometry of the active site Zn2+ ion is also similar in each crystal form. However, the conformation of catalytically important residue Y323 exhibits structural differences between the two crystal forms: whereas Y323 adopts both “inward” and “outward” conformations in triclinic structures, Y323 exclusively adopts the “inward” conformation in all monoclinic structures except for the structure of the APAH–2 complex, in which Y323 adopts a low occupancy (30%) intermediate conformation between the “inward” and “outward” conformations in monomer B only. As previously reported,31 the full-occupancy “outward” conformation of Y323 facilitates the binding of a third low-occupancy monovalent cation, but this additional cation is not observed in the structures reported herein. Additionally, the second monovalent cation site (comprised of F206, R209, V212, and T243) is observed to accommodate alternative cations in these structures: it is interpreted as Na+ in the APAH–1, APAH–2, and APAH–5 complexes; K+ in the APAH–4 complex; and NH4+ in the APAH–3 and APAH–6 complexes. The first monovalent cation site contains exclusively K+ in all structures.
Structure of the APAH-1 Complex
Trifluoromethylketones exists predominantly in the gem-diol form in aqueous solution due to the extreme electrophilicity of the carbonyl group adjacent to the trifluoromethyl moiety, and as such trifluoromethylketone derivatives are generally potent inhibitors of zinc metallohydrolases. The origin of tight binding is believed to result from the resemblance of the gem-diol to the tetrahedral intermediate and flanking transition states in the hydrolytic reaction catalyzed by a zinc metallohydrolase, as first demonstrated for a trifluoromethylketone inhibitor of carboxypeptidase A (Ki = 0.2 µM).42,43 More recently, the crystal structure of human HDAC4 complexed with a trifluoromethylketone inhibitor (IC50 = 0.367 µM) bound in the gem-diol form has been reported.44
As previously reported, trifluoromethylketone 1 inhibits APAH with IC50 = 0.27 ± 0.03 µM; a low affinity binding species is also observed in solution with IC50 = 38 ± 6 µM, which perhaps could correspond to the intact, substrate-like trifluoromethylketone instead of the tetrahedral gem-diol transition state mimic.33 However, in the crystal structure of the APAH–1 complex reported here at 1.42 Å resolution, only a single binding mode for the tetrahedral gem-diol is observed: one inhibitor binds in the active site of each APAH monomer, with the gem-diol moiety coordinating asymmetrically to the active site Zn2+ ion (Zn2+–O1 and Zn2+–O2 distances are 2.0 and 2.4 Å, respectively). Additionally, hydroxyl group O1 hydrogen bonds with Y323 (O---O separation = 2.6 Å) and hydroxyl group O2 hydrogen bonds with H158 and H159 (O---N separations = 2.7 Å). Overall metal coordination geometry is best described as trigonal bipyramidal, but distorted due to the bidentate coordination of the gem-diol moiety. This is generally comparable to Zn2+ coordination geometries observed in carboxypeptidase A complexes with inhibitory gem-diols.43,45 An electron density map of the HDAC8-1 complex is shown in Figure 2a.
Figure 2.

(a) Simulated annealing omit map showing trifluoromethylketone 1 bound as a gem-diol in the active site of APAH (monomer B, contoured at 3.5σ). Atomic color codes are as follows: C = wheat (protein, monomer B), brown (monomer A), or green (inhibitor), N = blue, O = red, F = light cyan, Zn2+ = magenta sphere. Water molecules are represented as red spheres. Metal coordination and selected hydrogen bond interactions are shown as solid black or dashed black lines, respectively. (b) Stereoview of the N8-acetylspermidine substrate bound in the active site of the inactive mutant APAH H159A (PDB accession code 3Q9C, monomer A). Atomic color codes are identical to those in (a); C atoms in brown correspond to adjacent monomer I of the APAH dimer. We note that in monomer J only of this structure, the secondary amine of N8-acetylspermidine donates a hydrogen bond to E117. Additionally, a water molecule makes a bridging interaction in some monomers between Y19 and the terminal amino group of the substrate. (c) Simulated annealing omit map of thiol 2 bound in the active site of APAH (monomer A, contoured at 3.0σ). Atomic color codes are identical as in (a), with S = gold.
The overall conformation of 1 is similar but not identical to that of N8-acetylspermidine in its complex with the inactive H159A APAH mutant (Figure 2b).31 As observed for N8-acetylspermidine, the primary amino group of 1 hydrogen bonds with E106 of the adjacent monomer of the dimer (N---O separation = 2.8 Å). However, in the present structure, E106 adopts two approximately equal-occupancy conformations, only one of which makes this hydrogen bond interaction. Although the secondary amino group of N8-acetylspermidine interacts with E117,31 E117 adopts an alternative conformation in the complex with 1 that precludes this interaction. Instead, the secondary amino group of 1 hydrogen bonds with water molecules that also interact with E17, E117, or T90 (from the adjacent monomer of the dimer). Additionally, the secondary amino group of N8-acetylspermidine makes an offset cation-π interaction with F225,31 and this interaction is conserved for the secondary amino group of 1.
Structure of the APAH-2 Complex
The thiol moiety of 2 comprises a good ligand for Zn2+ coordination, and a favorable Zn2+–S coordination interaction is proposed to account for moderate inhibitory potency (IC50 = 26 µM).33 Indeed, a favorable Zn2+–S coordination interaction accounts for the strong inhibitory potency of the marine depsipeptide inhibitor largazole against the histone deacetylases.46,47 The X-ray crystal structure of the HDAC8-largazole thiol complex (IC50 = 0.102 µM)48 reveals that the geometry of thiolate–Zn2+ coordination is nearly ideal49 with an average S–Zn2+ distance of 2.3 Å, a C–S–Zn2+ angle of 98°, and a C–C–S–Zn2+ dihedral angle of 92°; the Zn2+ ion is coordinated with nearly perfect tetrahedral geometry, with average ligand–Zn2+–ligand angles ranging 100°–115°, and Y306 donates a hydrogen bond to the Zn2+-bound thiolate anion.50 Comparable binding modes are observed in HDAC8 complexes with largazole analogues.51
Surprisingly, the 1.40 Å resolution crystal structure of the APAH-2 complex reveals the presence of two Zn2+ ions in the active site, as well as two mutually-exclusive positions for the catalytic Zn2+ ion separated by 0.7 Å. The catalytic Zn2+ ion is coordinated by D195, H197, and D284 in both positions, and the second Zn2+ ion (occupancy = 0.5) is coordinated by H158, H159, and a water molecule. The presence of this additional Zn2+ ion is due to the addition of ZnCl2 in the crystallization buffer. As a consequence, the thiol inhibitor 2 adopts two conformations. When the second Zn2+ ion is presumably absent, the structure reveals that the thiolate–Zn2+ coordination geometry (Figure 2c) is characterized by an average S–Zn2+ distance of 2.3 Å, a C–S–Zn2+ angle of 115°, and a C–C–S–Zn2+ dihedral angle of 51°; the Zn2+ ion is coordinated with distorted tetrahedral geometry, with average ligand–Zn2+–ligand angles ranging 86°–120°. The hydroxyl group of Y323 donates a hydrogen bond to the Zn2+-bound thiolate anion of 2. However, in monomer B, Y323 also partially adopts a conformation that is intermediate between the inward and outward conformation with 30% occupancy. When the second Zn2+ ion is present, the thiolate inhibitor adopts an alternative conformation in which the thiolate group bridges the two Zn2+ ions. For the catalytic Zn2+ ion, the average S–Zn2+ distance is 2.2 Å, the C–S–Zn2+ angle is 109°, and the C–C–S–Zn2+ dihedral angle is 173°; metal coordination geometry is distorted tetrahedral, with average ligand–Zn2+–ligand angles ranging 100°–116°. Similarly, the coordination geometry of the second Zn2+ ion is distorted tetrahedral, with an average S–Zn2+ distance of 2.3 Å, a C–S–Zn2+ angle of 98°, and a C–C–S–Zn2+ dihedral angle of −73°; average ligand–Zn2+–ligand angles range 85°–133°.
The rest of the inhibitor binds in the active site in a similar fashion to that observed for trifluoromethylketone 1. As observed for 1, the primary amino group of 2 hydrogen bonds with E106 of the adjacent monomer of the dimer (N---O separation = 2.7–3.1 Å); E106 adopts one and two conformations in monomers B and A, respectively. Similar to that observed for 1, the secondary amino group of 2 hydrogen bonds with water molecules that also interact with E17, E117, or T90 (from the adjacent monomer of the dimer).
Structures of the APAH Complexes with 3–6
The hydroxamate group generally comprises an excellent functional group for Zn2+ ion coordination in the active site of a metalloenzyme through the formation of a favorable five-membered ring chelate complex, as first observed in the crystal structure of thermolysin complexed with a hydroxamate inhibitor,52 although alternative binding modes are occasionally observed.53 The hydroxamate moieties of compounds 3, 4, 5, and 6 tend to form five-membered ring chelate complexes with the active site Zn2+ ion of APAH, as revealed in X-ray crystal structures determined at resolutions of 1.33 Å, 1.42 Å, 1.22 Å, and 1.13 Å, respectively. Electron density maps are shown in Figure 3 for APAH complexes with 3–6. Although the coordination mode of the hydroxamate OH group is identical in each enzyme-inhibitor complex (Zn2+–O separation = 1.9–2.0 Å in each monomer of each APAH–hydroxamate complex), the coordination mode of the hydroxamate carbonyl group is more variable and tends toward outer sphere interactions, with Zn2+–O separations ranging 2.3–3.1 Å for compounds 3–6. Additional interactions stabilize each bound hydroxamate moiety: Y323 donates a hydrogen bond to the hydroxamate carbonyl group; H158 donates a hydrogen bond to the anionic hydroxamate O; and H159 hydrogen bonds with the hydroxamate NH group. The Zn2+-bound hydroxamate is most likely ionized through the deprotonation of the hydroxamic acid OH group, as observed in other hydroxamate-metal ion complexes,54 and as observed here in the ultrahigh resolution structure of the APAH–6 complex.
Figure 3.

(a) Simulated annealing omit map of hydroxamate 4 bound in the active site of APAH (monomer A, contoured at 3.5σ). Atomic color codes are as follows: C = wheat (protein, monomer A), brown (monomer B), or green (inhibitor), N = blue, O = red, Zn2+ = magenta. Water molecules are represented as red spheres. Metal coordination and selected hydrogen bond interactions are shown as solid black or dashed black lines, respectively. (b) Simulated annealing omit map of hydroxamate 5 bound in two conformations in the active site of APAH (monomer B, contoured at 3.0σ). Atomic color codes are identical to those in (a). (c) Simulated annealing omit map of hydroxamate 6 bound in the active site of APAH (monomer A, contoured at 4.0σ). Atomic color codes are identical to those in (a). (d) Simulated annealing omit map of hydroxamate 3 bound in the active site of APAH (monomer B, contoured at 3.0σ). Atomic color codes are identical to those in (a).
Compounds 4, 5, and 6 differ by the chain length between the hydroxamate moiety and the terminal amine, with 4, 5, and 6 methylene groups, respectively; compound 3 is an analogue of N8-acetylspermidine. As a result, each binds differently in the active site of APAH. The 1.42 Å resolution structure of the APAH–4 complex reveals that hydroxamate 4 has the ideal chain length so that its its terminal amine directly interacts with the side chain of E117, which adopts two conformations in monomer B (with N---O separations of 2.5–3.1 Å in monomers A and B) (Figure 3a). The terminal amino group of 4 makes an offset cation-π interaction with F225. Additionally, the amino group of compound 4 hydrogen bonds with water molecules that also interact with the side chains of E17, Y168, or E106 (both from the adjacent monomer of the dimer), and various backbone amide groups of surrounding residues.
Compound 5 is one methylene longer than compound 4. The conformation of the inhibitor is bent such that the N–C–C–C torsion angle is synclinal (average value −57°), whereas it is antiperiplanar for compound 4 (average value 167°). This bent conformation allows the amino group of 5 to interact directly with one conformation of E117 (with occupancy = 0.6) in monomers A and B (N---O separations of 2.6 and 2.7 Å in monomers A and B, respectively) (Figure 3b). Additionally, the amino group hydrogen bonds with the hydroxyl group of Y168 (the N---O separation is 3.0 Å and 3.1 Å in monomers A and B, respectively). Interestingly, an alternative conformation is evident for compound 5 (occupancy = 0.3 and 0.4 in monomers B and A, respectively) such that the N–C–C–C torsion angle is antiperiplanar (average value −174°).
Compound 6 is one methylene group longer than compound 5. As such, even with a bent conformation, its terminal amine cannot interact with E117 (Figure 3c). As a result, the inhibitor conformation is extended such that the N-C-C-C torsion angle is antiperiplanar (average value −177°). As observed for hydroxamate 4, hydroxamates 5 and 6 also hydrogen bond with a network of water molecules interacting with side chains and backbone amide groups of surrounding residues.
Finally, compound 3 is an analogue of N8-acetylspermidine. Compared with the other hydroxamate inhibitors, 3 has the same number of methylene groups as 5, but possesses an extra 1,3-diaminopropane “tail”. From the hydroxamate moiety out to the secondary amino group, 3 adopts a conformation that is similar to that of 5 in its alternative antiperiplanar conformation. Surprisingly, the electron density for the 1,3-diaminopropane group of 3 is weak, indicating a higher degree of disorder that is also reflected in higher thermal B factors (Figure 3d). Contrary to 1, the 1,3-diaminopropane “tail” does not interact with E106, which is disordered in the present structure. Additionally, in contrast with N8-acetylspermidine and analogues 1 and 2, compound 3 makes no cation-π interaction with F225. The lack of this stabilizing interaction may account for the relative disorder of the 1,3-diaminopropane moiety. When the binding modes of compounds 3–6 are compared (Figure 4), it is clear that there are length-dependent effects that govern the conformation of the amino “tail” of each inhibitor while the hydroxamate-Zn2+ coordination geometry remains relatively constant.
Figure 4.

Superposition of APAH complexes with hydroxamate inhibitors 3–6 (monomer A for all). Atomic color codes are as follows: C = light grey (protein), teal (hydroxamate 3), orange (hydroxamate 4), yellow (hydroxamate 5), or green (hydroxamate 6), N = blue, O = red, F = light cyan, Zn2+ = magenta.
DISCUSSION
Compounds 1–3 are analogues of N8-acetylspermidine bearing different zinc-binding groups. The IC50 values recorded in Table 2 show that the trifluoromethylketone derivative 1 and the hydroxamate derivative 3 yield the most potent inhibition of APAH with IC50 values in the mid-nanomolar range.33 The thiol derivative 2 is relatively modest in comparison, which is surprising in view of the potent inhibition afforded by thiol zinc-binding groups of macrocyclic histone deacetylase inhibitors46–48 (the eukaryotic histone deacetylases are related to prokaryotic APAH by divergent evolution). Presumably, macrocycle interactions in the greater active site enhance the inhibitory potency of the thiol moiety in these systems.
While thiol derivative 2 is the least active of the three N8-acetylspermidine analogues, the structure of its complex with APAH reveals an unusual binding mode due to the presence of excess Zn2+ in the crystallization conditions. A second, half-occupancy Zn2+ ion binds in the APAH active site, and in the presence of two Zn2+ ions the ionized thiolate group of 2 bridges the binuclear metal cluster. The second Zn2+ ion is coordinated by the imidazole side chains of H158 and H159 as well as a water molecule. Interestingly, this binuclear zinc cluster is reminiscent of that formed in the active site of the unrelated bacterial deacetylase LpxC in the presence of excess zinc.55
Trifluoromethylketones are well known to exist as gem-diols in aqueous solution, and as such are usually excellent inhibitors of hydrolytic enzymes as first demonstrated by Gelb and colleagues.42 Accordingly, the gem-diol form of 1 binds to APAH as an analogue of the tetrahedral intermediate and its flanking transition states, and the structure of this complex provides critical inferences on the mechanism of deacetylation catalyzed by APAH. Specifically, this structure indicates that the developing negative charge of the tetrahedral oxyanion is stabilized by coordination to Zn2+ as well as a hydrogen bond with Y323. The imidazole side chains of H158 and H159 are presumed to hydrogen bond with the nucleophilic Zn2+-bound water molecule; by analogy with enzymological studies of HDAC8,16,56 H159 of APAH likely serves as a general base in a promoted-water mechanism in which both H159 and Zn2+ serve to activate the nucleophilic water molecule. Thus, the observed binding modes of trifluoromethylketone 1 as well as the substrate N8-acetylspermidine strongly support the mechanistic proposal for APAH recently presented by Lombardi and colleagues8 as summarized in Figure 5.
Figure 5.
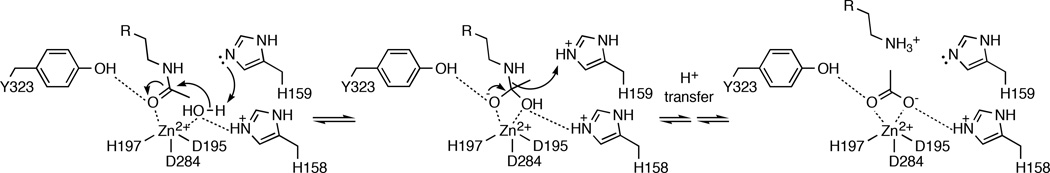
Proposed mechanism of APAH. The substrate binding mode is based on the complex of N8-acetylspermidine with the inactive mutant APAH H159A (PDB accession code 3Q9C), and the binding of the tetrahedral intermediate is mimicked by the binding of trifluoromethylketone 1 (Figure 2a).
Inhibitors 3–7 have in common a hydroxamate zinc-binding group that forms a favorable five-membered ring chelate complex with the catalytic Zn2+ ion in the APAH active site. Compounds 5 and 6 were designed as analogues of N-acetylputrescine and N-acetylcadaverine, respectively (Figure 1). These two acetylpolyamines are better substrates for APAH from Mycoplana ramosa than N8-acetylspermidine.30 The inhibitory potencies of the corresponding hydroxamate analogues follow a similar trend, since compounds 5 and 6 are more potent than compound 3 (Table 2). Compounds 4 and 7 are hydroxamate analogues of the mono N-acetylated derivatives of biological 1,3-diaminopropane and non-biological 1,6-diaminohexane. Compounds 4 and 7 exhibit comparable activity in the mid-nanomolar range (IC50 values of 170 and 150 nM, respectively). The structures of APAH complexed with 4, 5, and 6 reveal key interactions between the positively charged primary amino group of each inhibitor and E117 (for 4 and 5), and/or interactions with various surrounding residues through networks of hydrogen bonded water molecules. These results are consistent with enzymological measurements indicating that APAH is a promiscuous enzyme capable of accepting acetylpolyamines of various chain lengths as substrates.29,30
Finally, while compounds 1–7 were first designed as APAH inhibitors, it is interesting that compounds 2–7 are generally modest inhibitors of the related zinc deacetylase, human HDAC8; indeed, compounds 3–7 exhibit weaker inhibitory potency against HDAC8 compared with APAH (Table 2). Effective HDAC8 inhibitors generally contain a "capping" group of some sort capable of interacting with the outer active site cleft, such as the macrocycle moiety of Largazole.50,51 The lack of such a capping group in compounds 2–7 may account for generally modest inhibitory potency against HDAC8.
Strikingly, however, trifluoromethylketone 1 retains essentially identical nanomolar inhibitory potency against both APAH and HDAC8, even though it does not contain a capping group to capture additional affinity interactions in the outer active site cleft. Although we limited the current study to inhibitors that most closely resemble smaller polyamine substrates, it is interesting to consider that further derivatization of the primary amino group of 1 with a capping group might enable the capture of additional affinity interactions in the HDAC8 active site. Another capless inhibitor bearing a trifluoromethylketone zinc-binding group, a trifluoromethylketone analogue of L-arginine, was also reported to be a potent HDAC8 inhibitor.57 These results may suggest that the trifluoromethylketone moiety is an ideal functional group for coordination to the active site Zn2+ ion in the active sites of metal-dependent deacetylases, as also observed in an inhibitor complex with HDAC4.44 These results may also suggest that one of the enzymes currently annotated as a human histone deacetylase may additionally serve as a polyamine deacetylase. However, enzymological assays of HDAC8 conclusively demonstrate that it does not catalyze the deacetylation of N8-acetylspermidine: overnight incubation of N8-acetylspermidine with HDAC8 does not yield any measurable spermidine product (data not shown). Further results in the development of APAH inhibitors and the identification of the human enzyme responsible for catalyzing polyamine deacetylation will be reported in due course.
Supplementary Material
ACKNOWLEDGMENTS
We thank the NSLS for access to beamline X29, the APS for access to beamline NE-CAT 24-ID-E, and the SSRL for access to beamline BL14-1 for X-ray crystallographic data collection. Additionally, we thank Dr. Rakesh Kohli (University of Pennsylvania, Department of Chemistry) for recording the high resolution mass spectra.
FUNDING
We thank the NIH for grant GM49758 in support of this research.
ABBREVIATIONS
- APAH
acetylpolyamine amidohydrolase
- APS
Advanced Photon Source
- CDI
carbonyldiimidazole
- HDAC
histone deacetylase
- HRMS (ESI)
high-resolution mass spectrometry (electrospray ionization)
- M344
4-(dimethylamino)-N-[7-(hydroxyamino)-7-oxoheptyl]benzamide
- NSLS
National Synchrotron Light Source
- PEG
polyethylene glycol
- SSRL
Stanford Synchrotron Radiation Lightsource
- TLS
Translation Libration Screw
Footnotes
ASSOCIATED CONTENT
Supporting Information
IC50 plots showing inhibitory activity against APAH and HDAC8. This information is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The atomic coordinates and the crystallographic structure factors of APAH complexes with 1, 2, 3, 4, 5, and 6, have been deposited in the Protein Data Bank (www.rcsb.org) with accession codes 4ZUM, 4ZUN, 4ZUO, 4ZUP, 4ZUQ, and 4ZUR, respectively.
Notes
The authors declare no competing conflicts of interest.
REFERENCES
- 1.Walsh CT, Garneau-Tsodikova S, Gatto GJ., Jr Protein posttranslational modifications: the chemistry of proteome diversifications. Angew. Chem., Int. Ed. 2005;44:7342–7372. doi: 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- 2.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. U. S. A. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Workman JL, Kingston RE. Alteration of nucleosome as a mechanism of transcriptional regulation. Annu. Rev. Biochem. 1998;67:545–579. doi: 10.1146/annurev.biochem.67.1.545. [DOI] [PubMed] [Google Scholar]
- 4.Yang X-J, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol. Cell. 2008;31:449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 6.Philp A, Rowland T, Perez-Schindler J, Schenk S. Understanding the acetylome: translating targeted proteomics into meaningful physiology. Am. J. Physiol. Cell Physiol. 2014;307:C763–C773. doi: 10.1152/ajpcell.00399.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang X-J, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26:5310–5318. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- 8.Lombardi PM, Cole KE, Dowling DP, Christianson DW. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr. Opin. Struct. Biol. 2011;21:735–743. doi: 10.1016/j.sbi.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milne JC, Denu JM. The Sirtuin family: therapeutic targets to treat diseases of aging. Curr. Opin. Chem. Biol. 2008;12:11–17. doi: 10.1016/j.cbpa.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 10.Kanyo ZF, Scolnick LR, Ash DE, Christianson DW. Structure of a unique binuclear manganese cluster in arginase. Nature. 1996;383:554–557. doi: 10.1038/383554a0. [DOI] [PubMed] [Google Scholar]
- 11.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 12.Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang B-C, Verner E, Wynands R, Leahy EM, Dougan DR, Snell G, Navre M, Knuth MW, Swanson RV, McRee DE, Tari LW. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure. 2004;12:1325–1334. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 13.Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkühler C, Di Marco S. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2004;101:15064–15069. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gantt SL, Gattis SG, Fierke CA. Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry. 2006;45:6170–6178. doi: 10.1021/bi060212u. [DOI] [PubMed] [Google Scholar]
- 15.Dowling DP, Gantt SL, Gattis SG, Fierke CA, Christianson DW. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry. 2008;47:13554–13563. doi: 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gantt SL, Joseph CG, Fierke CA. Activation and inhibition of histone deacetylase 8 by monovalent cations. J. Biol. Chem. 2010;285:6036–6043. doi: 10.1074/jbc.M109.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dowling DP, Gattis SG, Fierke CA, Christianson DW. Structures of metal-substitutes human histone deacetylase 8 provide mechanistic inferences on biological function. Biochemistry. 2010;49:5048–5056. doi: 10.1021/bi1005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pegg AE, McCann PP. Polyamine metabolism and function. Am. J. Physiol. 1982;243:C212–C221. doi: 10.1152/ajpcell.1982.243.5.C212. [DOI] [PubMed] [Google Scholar]
- 19.Thomas T, Thomas TJ. Polyamines in cell growth and cell death: molecular mechanisms and therapeutic applications. Cell. Mol. Life Sci. 2001;58:244–258. doi: 10.1007/PL00000852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerner EW, Meyskens FL., Jr Polyamines and cancer: old molecules, new understanding. Nat. Rev. Cancer. 2004;4:781–792. doi: 10.1038/nrc1454. [DOI] [PubMed] [Google Scholar]
- 21.Casero RA, Jr, Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov. 2007;6:373–390. doi: 10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
- 22.Della Ragione F, Pegg AE. Purification and characterization of spermidine/spermine N1-acetyltransferase from rat liver. Biochemistry. 1982;21:6152–6158. doi: 10.1021/bi00267a020. [DOI] [PubMed] [Google Scholar]
- 23.Blankenship J, Walle T. Acetylation of spermidine and spermine by rat liver and kidney chromatin. Arch. Biochem. Biophys. 1977;179:235–242. doi: 10.1016/0003-9861(77)90108-4. [DOI] [PubMed] [Google Scholar]
- 24.Bolkenius FN, Seiler N. Acetylderivatives as intermediates in polyamine catabolism. Int. J. Biochem. 1981;13:287–292. doi: 10.1016/0020-711x(81)90080-x. [DOI] [PubMed] [Google Scholar]
- 25.Blankenship J. Deacetylation of N8-acetylspermidine by subcellular fractions of rat tissues. Arch. Biochem. Biophys. 1978;189:20–27. doi: 10.1016/0003-9861(78)90109-1. [DOI] [PubMed] [Google Scholar]
- 26.Marchant P, Manneh VA, Blankenship J. N1-acetylspermidine is not a substrate for N-acetylspermidine deacetylase. Biochim. Biophys. Acta. 1986;881:297–299. doi: 10.1016/0304-4165(86)90017-6. [DOI] [PubMed] [Google Scholar]
- 27.Wang Z, Fries D, Blankenship J. Effect of N8-acetylspermidine deacetylase inhibition on the growth of L1210 cells. Biochem. Pharmacol. 1999;57:1095–1103. doi: 10.1016/s0006-2952(99)00028-3. [DOI] [PubMed] [Google Scholar]
- 28.Mudumba S, Menezes A, Fries D, Blankenship J. Differentiation of PC12 cells induced by N8-acetylspermidine deacetylase inhibition. Biochem. Pharmacol. 2002;63:2011–2018. doi: 10.1016/s0006-2952(02)00991-7. [DOI] [PubMed] [Google Scholar]
- 29.Fujishiro K, Ando M, Uwajima T. Crystallization and some properties of acetylpolyamine amidohydrolase from Mycoplana bullata. Biochem. Biophys. Res. Commun. 1988;157:1169–1174. doi: 10.1016/s0006-291x(88)80997-5. [DOI] [PubMed] [Google Scholar]
- 30.Sakurada K, Ohta T, Fujishiro K, Hasegawa M, Aisaka K. Acetylpolyamine amidohydrolase from Mycoplana ramosa: gene cloning and characterization of the metal-substituted enzyme. J. Bacteriol. 1996;178:5781–5786. doi: 10.1128/jb.178.19.5781-5786.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lombardi PM, Angell HD, Whittington DA, Flynn EF, Rajashankar KR, Christianson DW. Structure of prokaryotic polyamine deacetylase reveals evolutionary functional relationships with eukaryotic histone deacetylases. Biochemistry. 2011;50:1808–1817. doi: 10.1021/bi101859k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abendroth J, Gardberg AS, Robinson JI, Christensen JS, Staker BL, Myler PJ, Stewart LJ, Edwards TE. SAD phasing using iodide ions in a high-throuhput structural genomics environment. J. Struct. Funct. Genomics. 2011;12:83–95. doi: 10.1007/s10969-011-9101-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Decroos C, Bowman CM, Christianson DW. Synthesis and evaluation of N8-acetylspermidine analogues as inhibitors of bacterial acetylpolyamine amidohydrolase. Bioorg. Med. Chem. 2013;21:4530–4540. doi: 10.1016/j.bmc.2013.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Usachova N, Leitis G, Jirgensons A, Kalvinsh I. Synthesis of hydroxamic acids by activation of carboxylic acids with N,N’-carbonyldiimidazole: exploring the efficiency of the method. Synth. Commun. 2010;40:927–935. [Google Scholar]
- 35.Decroos C, Bowman CM, Moser J-AS, Christianson KE, Deardorff MA, Christianson DW. Compromised structure and function of HDAC8 mutants identified in Cornelia de Lange Syndrome spectrum disorders. ACS Chem. Biol. 2014;9:2157–2164. doi: 10.1021/cb5003762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 37.Leslie AGW, Powell HR. Processing diffraction data with MOSFLM. In: Read RJ, Sussman JL, editors. Evolving Methods for Macromolecular Crystallography. Vol. 245. Berlin: Springer; 2007. pp. 41–51. [Google Scholar]
- 38.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr. 2011;D67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. 2001;D57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 42.Gelb MH, Svaren JP, Abeles RH. Fluoro ketone inhibitors of hydrolytic enzymes. Biochemistry. 1985;24:1813–1817. doi: 10.1021/bi00329a001. [DOI] [PubMed] [Google Scholar]
- 43.Christianson DW, Lipscomb WN. Complex between carboxypeptidase A and a possible transition-state analog: mechanistic inferences from high-resolution X-ray structures of enzyme-inhibitor complexes. J. Am. Chem. Soc. 1986;108:4998–5003. [Google Scholar]
- 44.Bottomley MJ, Lo Surdo P, Di Giovine P, Cirillo A, Scarpelli R, Ferrigno F, Jones P, Neddermann P, De Francesco R, Steinkühler C, Gallinari P, Carfi A. Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain. J. Biol. Chem. 2008;283:26694–26704. doi: 10.1074/jbc.M803514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Christianson DW, Lipscomb WN. Binding of a possible transition state analogue to the active site of carboxypeptidase A. Proc. Natl. Acad. Sci. USA. 1985;82:6840–6844. doi: 10.1073/pnas.82.20.6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taori K, Paul VJ, Luesch H. Structure and activity of largazole, a potent antiproliferative agent from the Floridian marine cyanobacterium Symploca sp. J. Am. Chem. Soc. 2008;130:1806–1807. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- 47.Ying Y, Taori K, Kim H, Hong J, Luesch H. Total synthesis and molecular target of largazole, a histone deacetylase inhibitor. J. Am. Chem. Soc. 2008;130:8455–8459. doi: 10.1021/ja8013727. [DOI] [PubMed] [Google Scholar]
- 48.Hong J, Luesch H. Largazole: from discovery to broad-spectrum therapy. Nat. Prod. Rep. 2012;29:449–456. doi: 10.1039/c2np00066k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chakrabarti P. Geometry of interactions with sulfur-containing ligands in protein structures. Biochemistry. 1989;28:6081–6085. doi: 10.1021/bi00440a052. [DOI] [PubMed] [Google Scholar]
- 50.Cole KE, Dowling DP, Boone MA, Phillips AJ, Christianson DW. Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. J. Am. Chem. Soc. 2011;133:12474–12477. doi: 10.1021/ja205972n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Decroos C, Clausen DJ, Haines BE, Wiest O, Williams RM, Christianson DW. Variable active site loop conformations accommodate the binding of macrocyclic largazole analogues to HDAC8. Biochemistry. 2015;54:2126–2135. doi: 10.1021/acs.biochem.5b00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holmes MA, Matthews BW. Binding of hydroxamic acid inhibitors to crystalline thermolysin suggests a pentacoordinate zinc intermediatein catalysis. Biochemistry. 1981;20:6912–6920. doi: 10.1021/bi00527a026. [DOI] [PubMed] [Google Scholar]
- 53.Scolnick LR, Clements AM, Liao J, Crenshaw L, Hellberg M, May J, Dean TR, Christianson DW. Novel binding mode of hydroxamate inhibitors to human carbonic anhydrase II. J. Am. Chem. Soc. 1997;119:850–851. [Google Scholar]
- 54.Zalkin A, Forrester JD, Templeton DH. Ferrichrome-A tetrahydrate. Determination of crystal and molecular structure. J. Am. Chem. Soc. 1966;88:1810–1814. doi: 10.1021/ja00960a040. [DOI] [PubMed] [Google Scholar]
- 55.Whittington DA, Rusche KM, Shin H, Fierke CA, Christianson DW. Crystal structure of LpxC, a zinc-dependent deacetylase essential for endotoxin biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 2003;100:8146–8150. doi: 10.1073/pnas.1432990100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wolfson NA, Pitcairn CA, Fierke CA. HDAC8 substrates: Histones and Beyond. Biopolymers. 2013;99:112–126. doi: 10.1002/bip.22135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ilies M, Dowling DP, Lombardi PM, Christianson DW. Synthesis of a new trifluoromethylketone analogue of L-arginine and contrasting inhibitory activity against human arginase I and histone deacetylase 8. Bioorg. Med. Chem. Lett. 2011;21:5854–5858. doi: 10.1016/j.bmcl.2011.07.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.