

Abstract
The human pregnane X receptor (hPXR), a member of the nuclear receptor superfamily, senses xenobiotics and controls the transcription of genes encoding drug-metabolizing enzymes and transporters. The regulation of hPXR’s transcriptional activation of its target genes is important for xenobiotic detoxification and endobiotic metabolism, and hPXR dysregulation can cause various adverse drug effects. Studies have implicated the putative phosphorylation site serine 350 (Ser350) in regulating hPXR transcriptional activity, but the mechanism of regulation remains elusive. Here we investigated the transactivation of hPXR target genes in vitro and in vivo by hPXR with a phosphomimetic mutation at Ser350 (hPXRS350D). The S350D phosphomimetic mutation reduced the endogenous expression of cytochrome P450 3A4 (an hPXR target gene) in HepG2 and LS180 cells. Biochemical assays and structural modeling revealed that Ser350 of hPXR is crucial for formation of the hPXR–retinoid X receptor alpha (RXRα) heterodimer. The S350D mutation abrogated heterodimerization in a ligand-independent manner, impairing hPXR-mediated transactivation. Further, in a novel humanized transgenic mouse model expressing the hPXRS350D transgene, we demonstrated that the S350D mutation alone is sufficient to impair hPXR transcriptional activity in mouse liver. This transgenic mouse model provides a unique tool to investigate the regulation and function of hPXR, including its non-genomic function, in vivo. Our finding that phosphorylation regulates hPXR activity has implications for development of novel hPXR antagonists and for safety evaluation during drug development.
Keywords: nuclear receptor, gene regulation, transcription regulation, receptor regulation, xenobiotic
Graphical Abstract
1. Introduction
The pregnane X receptor (PXR; NR1I2) is a member of the nuclear receptor (NR) superfamily, expressed predominantly in the liver and gastrointestinal tract. It has been well-characterized as a xenobiotic sensor that binds to structurally diverse chemicals, including numerous clinical drugs and endogenous substances [1]. As a xenobiotic sensor, human PXR (hPXR) functions mainly through its transcriptional activation of target genes encoding proteins involved in xenobiotic detoxification and endobiotic metabolism, such as drug-metabolizing enzymes and transporters [2]. Unwanted activation of hPXR by xenobiotics may lead to adverse drug-drug interactions (DDIs), cancer drug resistance, liver toxicity, and possible liability concerns affecting drug development and clinical therapy [3;4]. Therefore, a full understanding of hPXR regulation of transcriptional activation is crucial for understanding the metabolism of xenobiotics and circumventing the adverse effects of unwanted hPXR-mediated activation.
The transcriptional regulation of hPXR involves its 4 major structural domains: a sequence-specific DNA-binding domain (DBD), a flexible hinge, a ligand-binding domain (LBD), and an activation function-2 (AF-2) domain located in the LBD [5]. Ligand binding to the hPXR LBD is an important means of control of hPXR transcriptional activity. Upon ligand binding, a conformational change in the LBD and AF-2 domains of hPXR allows dissociation of the corepressor and recruitment of the coactivator; the hPXR-coactivator interaction facilitates DBD binding to site-specific DNA sequences of target genes, resulting in their transcriptional activation. Heterodimerization of hPXR with retinoid X receptor α (RXRα) is another step required for ligand-induced PXR activation [6]. Thus, ligand binding, hPXR-coactivator recruitment, and hPXR-RXRα heterodimerization are the three key steps in hPXR’s precise regulation of target gene expression.
Ligands binding of hPXR and coactivator recruitment have long been the main focus of the studies on regulation of transcriptional activity of hPXR. A number of hPXR ligands and hPXR-coactivators have been discovered [7]. However, mechanisms other than ligand binding and co-activator recruitment have been found involved in the regulation of other NR function, including post-translational modifications [7]. As a member of the NR superfamily, hPXR can be phosphorylated in biochemical and cell-based assays, and multiple putative phosphorylation sites have been proposed. Studies by our laboratory and others have shown that hPXR can be phosphorylated by cyclin-dependent kinase 2 (CDK2) and several other kinases including cAMP-dependent protein kinase A (PKA), protein kinase C (PKC), glycogen synthase kinase 3 (GSK3), casein kinase II (CK2), cyclin-dependent kinase 5 (CDK5), 70-kDa ribosomal S6 kinase (p70 S6K) [8–11]. To elucidate how phosphorylation affects the transcriptional activity of hPXR, phospho-mimetic mutations have been generated at different putative hPXR phosphorylation sites, and their transcriptional activity has been evaluated by a reporter-gene assay using the cytochrome P450 3A4 (CYP3A4) promoter. These assays have revealed increased (e.g., Thr248) or decreased (e.g., Ser8, Thr57, Ser114, Ser208, Ser350, Thr408, and Thr422) transcriptional activity associated with these residues [8;9;11;12].
In addition to post-translational modification, dimerization is also a key regulatory mechanism of NR, such as homodimerization of glucocorticoid receptors and hetero-dimerization of retinoic acid receptors (RARs) with RXRα [7]. The hPXR- RXRα hetero-dimerization has been established as a key step of hPXR activation, but how hPXR- RXRα dimerization regulates hPXR activation has not been well understood. A recent study of hPXR-RXRα LBD heterotetramer crystal structure identified 21 key amino acid residues of hPXR in the hPXR-RXRα interacting surface [13]. Among them, 15 (Lys325, Arg353, His359, Arg360, Asp363, Gln366, Glu367, Ile371, Lys374, Leu391, Met394, Glu399, Arg401, Ser402, Gln409) form either direct or water-meditated interaction with RXRα, while 6 (Lys332, Ser350, Glu378, Thr398, Gln406, Arg413), which are not involved in direct interaction with RXRα, can affect hPXR conformation and intramolecular interactions that potentially strengthen the dimer interface. However, it remains largely unknown whether and how these amino acid residues in hPXR affect the heterodimerization process and consequently hPXR’s function.
Ser350 residue of PXR is of particular interest in our study of the regulation of PXR function for a few reasons. First, Ser350 is present in a sequence (350SPDR353) that matches the consensus CDK phosphorylation motif [(S/T)PX(R/K)] [14], thus, it is a putative cyclin-dependent kinase 2 (CDK2) phosphorylation site. Since PXR can be phosphorylated by CDK2, and using a phospho-mimetic hPXR mutation of Ser350, S350D (a mutation of serine to a negatively charged aspartate), we and others have found that this mutation reduced hPXR-activated promoter transactivation of its target genes in cellular promoter-reporter assays, including phase I drug-metabolizing enzyme CYP3A4 [8;11;15] and phase II drug-metabolizing enzyme UDP-glucuronosyltransferase 1A1 [16]. Second, Ser350 is one of the 6 residues of PXR that can affect PXR conformation and intramolecular interactions, potentially leading to indirect effects on PXR-RXR dimerization [13]. The current knowledge suggests to us that Ser350 of hPXR could be crucial for hPXR’s function. A mechanistic understanding of the Ser350 would give us insights into the transcriptional regulation of hPXR at either post-translational modification or receptor dimerization levels.
In the present study, using a S350D mutation in hPXR, which allows constitutive mimic of phosphorylation of hPXR at Ser350 residue, we examined the functional effect of the S350D mutation on the hPXR transcriptional regulation of endogenous CYP3A4 gene expression in human liver-derived HepG2 and intestinal epithelia-derived LS180 cells, including its effects on the ligand binding, co-activator recruitment and hPXR-RXRα dimerization. We identified the molecular mechanism responsible for the impairment of transcriptional activity of the S350D mutant in these cells. Further, we demonstrated that the S350D mutation alone in hPXR is sufficient to impair hPXR activity in mouse liver in vivo by creating and functionally characterizing a novel humanized transgenic mouse model expressing the hPXRS350D transgene.
2. Materials and Methods
2.1 Chemicals and Plasmids
Rifampicin (RIF), SR12813 (SR), T0901317 (TO), hyperforin (Hyp) and 2,2,2-tribromoethanol were purchased from Sigma (St. Louis, MO). The plasmids pcDNA3-hPXR, pcDNA3-hPXRS350D, pcDNA3-hPXRS350A and pGL3-CYP3A4-luc were described previously [8].
2.2 Cell Culture
The human liver carcinoma cell line HepG2, the human intestinal epithelial cell line LS180 (derived from colorectal adenocarcinoma), and the 293T cell line were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and maintained in modified Eagle’s minimal essential medium (MEM) from ATCC with 10% FBS (Hyclone, Logan, UT), 2 mM L-glutamine (Invitrogen, Carlsbad, CA), and 100 U/ml penicillin/streptomycin (Invitrogen) at 37°C in a humidified 5% CO2 atmosphere.
2.3 hPXR Transactivation Assay
Cells were co-transfected with pGL3-CYP3A4-luc and with pcDNA3, pcDNA3-hPXR, pcDNA3-hPXRS350D, or pcDNA3-hPXRS350A by using FuGENE 6 (Roche Diagnostics, Indianapolis, IN). Twenty-four hours after transfection in growth medium, ~10,000 live cells were placed in each well of a 96-well culture plate (PerkinElmer, Waltham, MA) and grown for an additional 24 h in phenol red–free MEM (Invitrogen) supplemented with 1% charcoal/dextran-treated FBS (HyClone) and other additives, as described in the Cell Culture section. Forty-eight hours after transfection, a luciferase assay was performed using the Dual-Glo system (Promega, Madison, WI) and EnVision microplate reader (PerkinElmer).
2.4 RNA Isolation and Quantitative Real-Time PCR (qRT-PCR)
Total RNA was isolated from HepG2 cells, LS180 cells, or mouse liver tissue by using the Maxwell 16 LEV simplyRNA purification kit (Promega). qRT-PCR was performed according to the manufacturer’s protocol by using TaqMan gene expression assays (Applied Biosystems, Carlsbad, CA) specific for the CYP3A4, hPXR, and Cyp3a11 genes, with GAPDH as the reference gene in an ABI 7900HT system (Applied Biosystems). The comparative threshold (Ct) method was used for relative quantification of gene expression by the following formula: ΔCt = Ct (test gene) – Ct (GAPDH); ΔΔCt (test gene) = ΔCt (test gene in treatment group) – ΔCt (test gene in vehicle control group); the fold change of mRNA = 2−ΔΔCt, which indicates the mRNA level of the corresponding transcript in relation to that in the control samples.
2.5 Western Blot Analysis
Live mouse tissues were homogenized in 500 μL of cold RIPA lysis buffer (25 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, and 0.1% SDS) in a Bullet Blender Blue homogenizer (Next Advance, Averill Park, NY). The cells were rinsed once with cold phosphate-buffered saline and then lysed in RIPA lysis buffer. Whole lysates containing ~25 μg of total protein were loaded into Nupage 4% to 12% bis-Tris gels (Invitrogen) with Nupage MES SDS running buffer (Invitrogen). The proteins were then transferred to a nitrocellulose membrane by using the iBlot gel transfer system (Invitrogen) and iBlot gel transfer stacks (Invitrogen). For Western blotting, the membrane was blocked for 1 h with Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE), treated with mouse monoclonal antibodies to rat CYP3A11 (MAB10041, Millipore, Temecula, CA) [17]) and FALG M2 (Sigma) and a rabbit anti-RXRα antibody (D20, Santa Cruz Biotechnology) [18], and incubated with secondary goat anti-mouse antibody labeled with infrared dye (LI-COR Biosciences). Antigen-antibody interactions were visualized by using an Odyssey infrared imager (LI-COR Biosciences).
2.6 Mammalian Two-Hybrid Assay
The CheckMate mammalian two-hybrid system (Promega) was used to evaluate the hPXR-co-regulator interactions. LS180 cells were co-transfected with the pACT-hPXR, pACT-hPXRS350D, or pACT-hPXRS350A plasmids, pBIND-SRC-1(steroid receptor coactivator-1), pBIND-SMRTτ (silencing mediator for retinoid receptors-1τ, amino acids 2077 – 2471), pBIND-mNCoR (mouse nuclear receptor co-repressor, amino acids 1958 – 2401), or pBIND-RXRα in the presence of pG5-luc (a GAL4 luciferase reporter construct). The pBIND- plasmids constitutively expressed Renilla luciferase, which was used as an internal transfection control. The Dual-Glo Luciferase Assay (Promega) was used to measure luciferase activity. Expression of wild-type hPXR, hPXR mutants, SRC-1, SMRTτ, mNcoR, or RXRα was confirmed by Western blot analysis. The relative luciferase activity of pG5-luc was determined by normalizing firefly to Renilla luciferase activity.
2.7 Competitive Ligand-Binding Assay
A LanthaScreen TR-FRET PXR competitive binding assay was conducted according to the manufacturer’s protocol, as described previously [2;19]. Briefly, assays were performed in a volume of 20 μl in 384-well solid black plates containing serial dilutions of the PXR LBD fused to GST (GST-hPXR LBD, GST-hPXRS350D LBD, or GST-hPXRS350A LBD); 40 nM fluorescence-labeled hPXR ligand (Fluomore PXR Green, Invitrogen); 5 nM terbium-labeled anti-GST antibody; and different concentrations of test compound. The mixture was incubated at 25°C for 20 min, and the fluorescence emission (520 nm and 490 nm) of each well was then measured. The net TR-FRET ratio (520 nm/490 nm) of each well was calculated by subtracting the background TR-FRET ratio (obtained by adding 10 μM SR12813 to the reaction).
2.8 Electrophoretic Mobility Shift Assay (EMSA)
To measure the DNA-binding ability of hPXR and hPXRS350D, EMSA was performed as described previously [10]. FLAG-hPXR, FLAG-hPXRS350D, and RXRα proteins were synthesized in vitro by using the TNT rabbit reticulocyte lysate system (Promega), according to the manufacturer’s protocol. An equal amount of the in vitro–translated protein was added to each reaction. Competitive binding of the labeled oligonucleotides was assessed by using a 500-fold molar excess of unlabeled oligonucleotides. Each 20 μL reaction contained 10 mM Tris, pH 8.0, 40 mM KCl, 0.05% NP-40, 6% glycerol, 1 mM DTT, 0.2 μg of poly(dI-dC), 10 μM zinc chloride, and 2 μL of in vitro–translated protein. Oligonucleotides and synthesized proteins were added to the inner walls of microcentrifuge tubes, mixed by vortexing, and incubated on ice for 30 min. Complexes were separated by electrophoresis in a non-denaturing 4% polyacrylamide gel and analyzed with a Storm 860 PhosphoImager (GE Healthcare, Little Chalfont, Buckinghamshire, UK). The following double-stranded oligonucleotides representing the PXR DNA-binding sequence within the CYP3A4 promoter (CYP3A4-ER6, an everted repeat with a 6-base pair spacer) were used as 32P-labeled (radiolabeled) probes or unlabeled competitor probes as indicated: 5′-GATCAATATGAACTCAAAGGAGGTCAGTG-3′; or a mutant CYP3A4-ER6 5′-GATCAATATGCCATCAAAGGAATACAGTG-3′. The specific binding of hPXR to CYP3A4-ER6 has previously been validated [10].
2.9 Immunofluorescence
HepG2 cells were transfected with FLAG-hPXR, FLAG-hPXRS350D, or hPXRS350A construct and cultured in a 96-well view plate (PerkinElmer). After 24 h, cells were treated with either DMSO or the indicated concentration of SR for 12 h, fixed in 4% paraformaldehyde (EMS, Hatfield, PA, USA), permeabilized with 0.25% Triton X-100 in PBS, and incubated with the anti-FLAG M2 antibody overnight at 4 °C. After 1 h incubation with secondary antibody, cells were imaged in the IN Cell Analyzer 6000 system (GE Healthcare Life Sciences, Pittsburgh, PA). The percentage of transfected cells that showed nuclear staining of FLAG (i.e., a high nuclear-to-cytoplasmic ratio of pixel intensity) were tabulated for a Mann-Whitney nonparametric analysis using GraphPad Prism (GraphPad Software, La Jolla, CA, USA).
2.10 Animals and Drug Treatment
Pxr−/− (Pxr-null) mice and humanized PXR (hPXR-tg) mice were generated previously [20]. To create humanized hPXR mice carrying the S350D mutation (hPXRS350D-tg), transgenic mice were produced by microinjecting the hPXRS350D mutation transgene into the pronuclei of fertilized mouse eggs, as described previously [20–22]. The successful integration of the transgene was confirmed by Southern blot analysis. The hPXRS350D transgene was then backcrossed into the Pxr−/− background for over 5 generations, resulting in humanized hPXR S350D mice with a C57BL/6 genetic background. Mouse tail tips were genotyped to detect hPXR and hPXR with the S350D mutation. All animal experiments were performed in accordance with a protocol approved by St. Jude Children’s Research Hospital Institutional Animal Care and Use Committee. Male mice (8–16 weeks old) were housed in the St. Jude animal facility and used in all animal studies. Five mice in each group were dosed orally with vehicle control or 10 mg/kg RIF, every 24 h for three days. Eight hours after the last dose, the animals were euthanized by CO2 and liver tissues were harvested. A piece of each liver was preserved in RNAlater solution (Invitrogen) at 4 °C for mRNA isolation. The remaining tissue was instantly frozen in liquid nitrogen and stored at −80 °C for total protein extraction.
2.11 Loss of Righting Reflex (LORR) assay
Mice were intraperitoneally injected with 250 mg/kg of 2,2,2-tribromoethanol, which is metabolically cleared only via mouse CYP3A11 [23;24]. After the mice lost their righting reflex, they were placed on their backs under a heat lamp. The duration of LORR was measured as the time from the start of LORR to recovery (i.e., when mice could right themselves after being placed on their backs twice within 1 min). A baseline LORR duration was established for each mouse at the administered dose of 2,2,2-tribromoethanol. After a 1-wk washout period, each mouse was administered vehicle or RIF (10 mg/kg) by oral gavage once daily x 3, and the righting reflex experiment was repeated at least 8 h after the last treatment. The paired Student’s t-test was used to compare LORR duration between baseline and after treatment. A P value < 0.05 was considered to indicate a significant difference between compared groups.
3. Results
3.1 Reduced intracellular transactivation of endogenous hPXR target genes by hPXRS350D
We first evaluated the transcriptional activity of the hPXRS350D mutant in human HepG2 cells (liver cells) and LS180 cells (intestine cells), which are commonly used to investigate ligand-induced transactivation of hPXR target genes [25;26]. After treatment with RIF (a known potent hPXR agonist), endogenous CYP3A4 expression in HepG2 or LS180 cells transfected with empty vector, hPXR, hPXRS350D (phosphomimetic) mutant, or hPXRS350A (phospho-deficient) mutant was compared with that in the respective vehicle-treated cells. The expression level of hPXR was equivalent to that of the two mutant proteins by Western blot (Fig. 1, A–F, bottom panel). When hPXR was transfected, the induction of CYP3A4 mRNA was increased 4- to 14-fold in HepG2 cells and 9- to 30-fold in LS180 cells, in a dose-dependent manner (Fig. 1, A and B, top panel). In contrast, in cells with hPXRS350D transfection, induction of CYP3A4 mRNA was substantially reduced at all RIF concentrations (decreased to 2~3-fold in HepG2 cells and 3~12-fold in LS180 cells), to levels either similar to (in HepG2 cells) or less than (in LS180 cells) that in cells transfected with empty vector (Fig. 1, A and B). Conversely, RIF-induced CYP3A4 mRNA levels in hPXRS350A-transfected cells were similar to those in hPXR-transfected cells at all RIF concentrations (Fig. 1, A and B, top panel). Similar changes in CYP3A4 mRNA induction were observed in LS180 cells transfected with the three hPXR constructs and treated with TO, another potent hPXR agonist (Fig. 1C), suggesting that the reduced target-gene transactivation by hPXRS350D was not ligand-specific.
Figure 1. Attenuated transcriptional activation by hPXRS350D in human cells.

A–C) Human CYP3A4 mRNA level was quantified by real-time PCR in HepG2 liver cells and LS180 intestine cells transiently transfected for 24 h with pcDNA3 (empty vector, EV), pcDNA3-hPXR (hPXR), pcDNA3-hPXRS350D (hPXRS350D, or “D”), or pcDNA3-hPXRS350A (hPXRS350A, or “A”) and treated with 0.1% DMSO or increasing concentrations of rifampicin (RIF) or T0901317 (TO) as indicated for another 24 h. Results are presented as fold expression in DMSO-treated control cells. D-F) CYP3A4 promoter activity was determined in HepG2, 293T, and LS180 cells transiently co-transfected for 24 h with pGL3-CYP3A4-luc reporter and pRL-TK Renilla luciferase (Rluc, transfection control) and with empty vector, hPXR, hPXRS350D, or hPXRS350A plasmids and treated for another 24 h with 0.1% DMSO (“0” RIF), increasing concentrations of RIF (D and E), or different hPXR agonists (F). SR, SR12813, Hyp, hyperforin. CYP3A4 promoter activity is presented as relative luciferase units (RLU), normalized to Renilla luciferase. Data represent mean ± SEM from three independent experiments: *, P<0.05; **, P<0.01; ***, P<0.001 as compared by t test to cells expressing hPXRS350D mutant. The protein levels of hPXR, hPXRS350D (D) and hPXRS350A (A) in transfected cells were determined by Western blotting, and equal loading of lysates was verified by using Actin as control. The Western blots were placed under each bar graph (A, D, E and F); for B and C, only one Western blot showing the levels of PXR, S350D and S350A in LS180 cells was shown under the bar graphs.
To confirm that hPXRS350D transactivation of its target genes was reduced, we next tested hPXR-regulated CYP3A4-luc promoter activity by transiently co-transfecting HepG2 or 293T cells with a CYP3A4-luc reporter plasmid and with empty vector, hPXR, hPXRS350D, or hPXRS350A plasmids. Except for the empty vector-transfected cells, RIF treatment at all concentrations significantly but variably increased the promoter activity of CYP3A4-luc in the cells transfected with hPXR plasmids: the promoter activity of CYP3A4-luc in HepG2 and 293T cells transfected with hPXR was ~3–5 fold that in cells overexpressing hPXRS350D, but equivalent to that in cells transfected with hPXRS350A (Fig. 1, D and E), consistent with CYP3A4 mRNA induction by hPXR activation (Fig. 1, A and B) after RIF treatment. We next examined whether the impaired transcriptional regulation of CYP3A4-luc promoter by the hPXRS350D mutant is ligand-specific in LS180 cells. The cells were co-transfected with CYP3A4-luc and with different hPXR constructs, then treated with four commonly-used hPXR agonists with diverse chemical structures, at concentrations previously shown to induce the maximum hPXR-mediated CYP3A4-luc promoter activity [2;8]. After treatment with each of the agonists, cells overexpressing hPXRS350D showed only about half the promoter activity of CYP3A4-luc reporter in cells transfected with hPXR, and the promoter activity induced in cells transfected with hPXRS350A remained similar to that of hPXR (Fig. 1F). These results suggest that the transcriptional activity of hPXRS350D is attenuated in human cells in a manner not limited to specific hPXR ligands.
3.2 The interaction between hPXRS350D and hPXR co-regulators is intact
We next sought for mechanisms underlying the impaired transactivation capacity of hPXRS350D. Previous studies have shown that hPXR transcriptional activity is controlled by its interaction with hPXR co-regulators [27]. Phosphorylation of other NR proteins has been shown to regulate their ability to bind to their co-regulators (reviewed in [28]); therefore, we next used a mammalian two-hybrid system to examine whether the S350D, as a phosphomimetic hPXR mutation, can affect hPXR-co-regulator interactions. In this assay, expression vectors encoding GAL4DBD-coregulator (co-activator or co-repressor) fusion proteins and VP16-fused hPXR or mutant hPXR proteins were transiently expressed in LS180 cells; the specific GAL4-luc reporter gene can only be induced when VP16-hPXR interacts with the GAL4DBD-fusion protein, which binds to the GAL4 binding sites within the promoter of the GAL4-luc. After treatment with four commonly-used hPXR agonists, respectively, in the coactivator SRC-1-transfected cells, GAL4-luc reporter activity was not significantly different with the co-transfection of hPXR, hPXRS350D or hPXRS350A, indicating that ligand-induced association between SRC-1 coactivator and hPXR was not affected by either phosphomimetic (D) or phosphodeficient (A) mutation at the Ser350 position (Fig. 2A). Moreover, in cells transfected with the corepressor SMRTτ and mNCoR, reporter activity remained similar among cells co-expressing hPXR, hPXRS350D, or hPXRS350A (Fig. 2, B and C), suggesting that the interaction between hPXR and these two co-repressors was also not affected by the mutations.
Figure 2. The interaction between hPXRS350D and hPXR coregulators is intact.
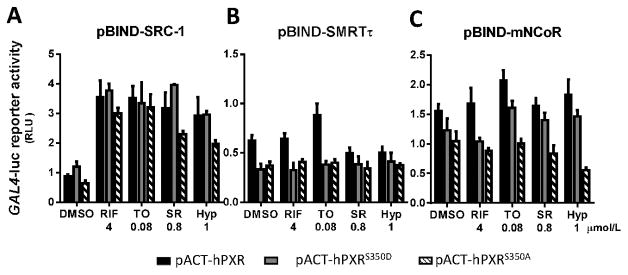
Mammalian two-hybrid assays measured the association of hPXR, hPXRS350D, and hPXRS350A with their coregulators A) SRC-1, B) SMRTτ, and C) mNCoR. In this assay, fusion proteins, VP16-hPXR, VP16-hPXRS350D or VP16-hPXRS350A mutants (full-length hPXR or the hPXRS350D or hPXRS350A mutants fused to Herpes simplex virus VP16 activation domain) and GAL4DBD-coregulator (GAL4-DNA binding domain fused to coregulator) were transiently expressed in LS180 cells in the absence or presence of the indicated hPXR agonists, and the interaction between the two fusion proteins was indicated by specific GAL4-luc reporter activity. GAL4-luc reporter activity is presented as RLU, normalized to Renilla luciferase internal control. Data represent mean ± SEM from three independent experiments.
3.3 The heterodimerization of hPXRS350D and RXRα is abolished
hPXR-RXRα heterodimerization is another important step required for hPXR transactivation of its target genes [6]. The dimerization interfaces of hPXR and RXRα are located within their LBDs [13;29]. Upon ligand binding, the hPXR-RXRα complex binds to specific DNA sequences of the hPXR-target gene promoters and activates gene expression. We tested whether the S350D mutation could affect hPXR-RXRα heterodimerization and thereby reduce transactivation of hPXR target genes. We first performed a mammalian two-hybrid assay in LS180 cells, and showed that VP16-hPXR or VP16-hPXRS350A induced higher GAL4-luc reporter activity than VP16-hPXRS350D did, in the absence of added hPXR agonist, suggesting that the S350D mutation may negatively affect the dimerization of hPXR and RXRα (Fig. 3A). After treatment with agonists, the GAL4-luc reporter signal was significantly increased in LS180 cells expressing VP16-hPXR or VP16-hPXRS350A protein, but not in cells expressing VP16-hPXRS350D mutant (Fig. 3A), further confirming the negative effect of S350D mutation. The agonist-mediated enhancement of GAL4-luc reporter activity in cells expressing VP16-hPXR or VP16-hPXRS350A might be caused by different mechanisms, such as enhanced interaction of hPXR with RXRα, or recruitment of additional co-activators. We then conducted a co-immunoprecipitation (co-IP) experiment to directly examine hPXR and RXRα dimerization. We transfected 293T cells with RXRα and FLAG-tagged hPXR plasmids, and used anti-FLAG antibodies to immunoprecipitate FLAG-hPXR and its binding partners. The RXRα and FLAG-hPXR and mutant proteins were expressed at equivalent levels in cells (Fig. 3B). With or without RIF treatment, RXRα was efficiently co-immunoprecipitated with FLAG-hPXR or FLAG-hPXRS350A proteins to a similar extent (Fig. 3B). However, RXRα was absent in the FLAG-hPXRS350D co-IP (Fig. 3B), indicating that RXRα could efficiently bind to hPXR or hPXRS350A but not hPXRS350D, regardless of agonist treatment. The basal level (untreated) of hPXR-RXRα interaction was then assessed by mammalian two-hybrid assay in the absence of hPXR agonist treatment. In this assay, the reporter signal in cells expressing PXRS350D was about 50% of that in cells expressing hPXR or PXRS350A (Fig. 3C), further suggesting that the S350D mutation reduced hPXR-RXRα interaction in cells in the absence of agonist activation. Taken together, these results indicate that the amino acid residue at position 350 in hPXR is crucial for hPXR-RXRα dimerization and subsequent hPXR activation of its target gene promoter; S350D, a phosphomimetic mutation, could prevent the dimerization of hPXR-RXRα and eliminate subsequent hPXR function. It is noted that a very low promoter activity of CYP3A4-luc reporter was detected in hPXRS350D expressing cells with hPXR agonist treatment (Figure 1D–E), suggesting a low residual transcriptional activity of hPXRS350D, which can only be detected in very sensitive assay system such as the reporter gene assay.
Figure 3. The heterodimerization of hPXRS350D and RXRα is impaired.

A) The interaction of RXRα with hPXR, hPXRS350D, and hPXRS350A in LS180 cells was determined by mammalian two-hybrid assay after treatment with the indicated hPXR agonists for 24 h. B) The heterodimerization of hPXRS350D mutant protein with RXRα was measured by co-immunoprecipitation assay in 293T cells without or with RIF treatment. 293T cells were transiently co-transfected with RXRα plasmids and FLAG-tagged hPXR, hPXRS350D, or hPXRS350A plasmids. Cell lysates were immunoprecipitated using FLAG-specific beads, resolved by SDS-PAGE, and probed with anti-FLAG (for hPXR and mutants) and anti-RXRα antibodies (top, immunoprecipitation: IP). Expression of hPXR and mutant hPXR proteins was analyzed by Western blot (bottom, lysates). Representative Western blots are shown from at least three independent experiments. C) Basal interaction between RXRα and hPXR or hPXR mutant proteins was measured in untreated LS180 cells by mammalian two-hybrid assay. GAL4-luc reporter activity indicating hPXR-RXRα interaction is presented as RLU normalized to Renilla luciferase. Data represent mean ± SEM from three independent experiments: *, P<0.05; **, P<0.01; ***, P<0.001, compared by t test to cells expressing hPXRS350D mutant protein. D) The S350D mutation in hPXR disrupts PXR-RXRα complex formation. The carbon atoms of PXR residues in direct contact with RXRα are depicted in green, R401 forms a water-mediated contact with RXRα in yellow, D352 and R353 in grey, and S350 in pink. Nitrogen and oxygen atoms are represented in blue and red, respectively. E) Interaction of RXRα-LBD (surface representation in brown) with PXR-LBD (cartoon representation in blue). The SRC-1 coactivator peptide is indicated in red. These images were created by using Protein Data Bank code PDB4J5W.
3.4 Possible mechanism for S350D disruption of the hPXR-RXRα complex formation
To further elucidate at a submolecular level how the S350D mutation impairs hPXR-RXRα dimerization, the 3D crystal structures of interaction between hPXRS350D and RXRα were computationally analyzed. A number of the 3D structures of PXR-LBD in the apo-form or with a bound agonist have been reported [30], indicating that Ser350 does not partake in direct contact with the ligand. However, Ser350 participates in a network of polar and electrostatic interactions that can be disrupted by the substitution of Ser350 with a charged residue such as glutamic acid (E) or aspartic acid (D), distorting the regional protein conformation. It is unlikely that these structural changes would affect the AF-2 domain or the recruitment of coactivators due to the extended distance between these regions and Ser350. A more discernible reason for the inactivation of the hPXRS350D mutant is its inability to heterodimerize with its partner protein RXRα, which can be explained by analyzing the recent crystal structure of the hPXR-RXRα complex [13]. There is a potential hydrogen bond between Ser350 and Gln366 of hPXR, and the disruption of this interaction can infringe on the optimum contact of Gln366 and Glu367 of hPXR with RXRα (Fig. 3D).
Ser350 also forms a potential hydrogen bond with Arg353, which in turn forms a salt bridge with Asp352. There is an electrostatic interaction between Asp352 and Arg401, the latter of which interacts with RXRα via a water molecule, according to the crystal structure [13]. Disturbances in Arg401 can also affect the interactions of neighboring residues, such as Glu399 and Ser402, within helix 10 of hPXR and RXRα (Fig. 3E). The disruption of this inter-residual network by substitution of the negatively charged residue aspartic acid (D) for Ser350 is consistent with the protein-destabilizing effects of the mutation predicted by the Site Directed Mutator method (SDM) [31], SortingIntolerant From Tolerant (SIFT) method [32], and Screening for Non-Acceptable Polymorphisms (SNAP) method [33]. In contrast, substitution of Ser350 by the uncharged alanine (A) showed no significant effect in biological assays (Fig. 3, A–C). Alanine, which is non-bulky while maintaining its secondary-structure integrity to some extent, would not dramatically interfere with the extended hydrogen-bond and electrostatic-bond network in the vicinity of Ser350. Accordingly, SDM, SIFT, and SNAP predicted that the S350A substitution would be tolerable in terms of structural stability. The computational analysis thus provides a possible mechanism for disruption of the PXR-RXRα complex formation by the S350D mutation.
3.5 The transcriptional activity of hPXRS350D is reduced independently of exogenous ligand binding
Because the hPXR-RXRα heterodimeric complex is necessary for transcriptional regulation by hPXR, we next tested whether the inefficient dimerization observed between PXRS350D and RXRα blunts the basal hPXR transactivation of CYP3A4 in the absence of any treatment (DMSO or agonist). We measured the CYP3A4 mRNA level in HepG2 cells and LS180 cells transfected with hPXR or with the two mutant constructs. The cellular expression levels of hPXR and the two mutant proteins were equivalent (Fig. 4, A and B). In HepG2 cells, compared to empty vector controls, transfection of hPXR or hPXRS350A led to more than 2 folds induction of endogenous CYP3A4 mRNA, while hPXRS350D overexpression did not alter the level of CYP3A4 mRNA (Fig. 4A), suggesting that the S350D substitution in hPXR negatively affects its transactivation of CYP3A4. Interestingly, in LS180 cells, compared to empty vector, transfection of hPXR or hPXRS350A had little impact on CYP3A4 mRNA level, but transfection of hPXRS350D resulted in ~50% reduction of endogenous CYP3A4 mRNA. These distinct change patterns of endogenous CYP3A4 mRNA levels in HepG2 and LS180 cells suggested that the activation of endogenous CYP3A4 promoter by endogenous hPXR was probably saturated in the LS180 cells (but not in the HepG2 cells) and that overexpression of hPXRS350D exerted a dominant negative-like effect on the intracellular regulation of endogenous CYP3A4 expression (Fig. 4B). In the reporter assay with HepG2 and LS180 cells expressing exogenous CYP3A4-Luc, hPXR-mediated CYP3A4-Luc promoter activity was significantly increased by hPXR or hPXRS350A transfection but was not changed by hPXRS350D co-transfection, further confirming the negative effect of S350D on hPXR transcriptional activity (Fig. 4, C and 4D).
Figure 4. Decreased transcriptional activation byPXRS350D is independent of exogenous ligand binding.

A and B) CYP3A4 mRNA expression was quantified by real-time PCR in A) HepG2 and B) LS180 cells transiently transfected for 24 h with empty vector pcDNA3 (EV), pcDNA3-hPXR (hPXR), pcDNA3-hPXRS350D (PXRS350D), or pcDNA3-hPXRS350A (hPXRS350A). Results are fold expression compared to control cells transfected with empty vector (EV). C and D) CYP3A4 promoter activity was determined in HepG2 and LS180 cells transiently co-transfected for 24 h with pGL3-CYP3A4-luc reporter and pRL-TK Renilla luciferase plasmids and with empty vector, hPXR, PXRS350D or hPXRS350A plasmids. CYP3A4 promoter activity is presented as relative luciferase units (RLU) normalized to Renilla luciferase. Data represent mean ± SEM from three independent experiments: *, P<0.05; **, P<0.01; ***, P<0.001, compared by t test with values in cells expressing hPXRS350D mutant protein. The protein levels of hPXR, hPXRS350D (D) and hPXRS350A (A) in transfected cells were determined by Western blotting, and equal loading of lysates was verified by using Actin as control.
3.6 Ligand binding, DNA binding, and cellular localization of hPXRS350D are unaffected
We next assessed whether the S350D substitution affected other molecular interactions of hPXR. First, we performed a competitive ligand binding assay to determine whether the mutation affected agonists’ binding to the LBD of hPXR. In a time-resolved fluorescence resonance transfer (TR-FRET) biochemical assay, a terbium-labeled anti-GST (Tb-anti-GST) antibody and a fluorescein-labeled hPXR ligand (a “tracer”) were incubated with purified GST-hPXR LBD, GST-hPXRS350D LBD, and GST-hPXRS350A LBD, respectively, at the indicated concentrations (Fig. 5A). The TR-FRET ratio indicated the binding of tracer to hPXR-LBD and increased in the presence of all three forms of hPXR-LBD, in a protein concentration-dependent manner, suggesting that hPXR-LBD ligand-binding was not affected by the S350D mutation. As full-length hPXR contains the DNA-binding domain, we next examined whether the S350D mutation affects its DNA-binding ability by EMSA. CYP3A4-ER6 DNA oligo, an hPXR-specific binding sequence within the CYP3A4 promoter, was used in this assay. The binding of 32P-labeled wild-type CYP3A4-ER6 DNA oligo to hPXR or hPXRS350D was indicated by the shift of bands in lanes 1 and 5 (Fig. 5B), which disappeared (lanes 2 and 6, Fig. 5B) when a high concentration of unlabeled CYP3A4-ER6 DNA oligo was added to compete with binding. These findings indicated that binding of the CYP3A4-ER6 DNA oligo to both versions of hPXR proteins was specific. When the radiolabeled mutant (mt) oligo, which had a mutation in the hPXR binding site, was used in the assay, no band shift appeared with either version of hPXR protein (lane 3, 4, 7 and 8, Fig. 5B), suggesting the failure of both versions of hPXR to bind to the mutant oligo and confirming their specific binding to the wild-type oligo. Taken together, our results suggested that the S350D mutation did not affect the DNA-binding ability of hPXR.
Figure 5. The ligand binding, DNA binding, and cellular localization of hPXRS350D is unaffected.

A) An in vitro time-resolved fluorescence resonance transfer (TR-FRET) assay measured the ligand binding of GST, GST-hPXR LBD (GST-hPXR), GST-hPXRS350D LBD (GST-hPXRS350D), or GST-hPXRS350A LBD (GST-hPXRS350A). Tb-anti-GST antibody and a fluorescein-labeled hPXR ligand (“tracer”) were incubated with the indicated GST-proteins. The TR-FRET ratio was calculated by dividing the emission signal at 520 nm (from acceptor fluorophore) by the emission signal at 490 nm (from donor terbium) to indicate the binding of tracer to GST-protein. An increase in the TR-FRET ratio compared to that of GST control indicated tracer binding to GST-fusion protein. B) Electrophoretic mobility shift assay with in vitro translated FLAG-hPXR and FLAG-hPXRS350D and 32P-labeled CYP3A4 PXR DNA binding sequence (3A4 ER6 oligo) as described in Materials and Methods. An equal amount of hRXR was added to all reactions. FLAG-hPXR and FLAG-hPXRS350D formed a complex with radiolabeled wild-type (wt) oligo (lanes 1 and 5), and this complex was efficiently out-competed by unlabeled wt oligo (lanes 2 and 6). Mutant (mt) oligo exhibited no binding to FLAG-hPXR and FLAG-hPXRS350D (lanes 3, 4, 7 and 8).
Because several studies suggested that hPXR localization is important for its function [34–36], we next examined whether the S350D mutation can alter the subcellular localization of hPXR. HepG2 cells expressing FLAG-tagged wild-type or mutant hPXR were treated with either DMSO or SR for 12 h; the subcellular localization of hPXR was visualized by fluorescence microscopy and quantified by the proportion of cells with nuclear FLAG staining. With or without SR12813 (a known potent hPXR agonist) treatment, hPXR, hPXRS350D, and hPXRS350A localized predominantly to the nucleus in a similar pattern, as previously observed [10;11], suggesting that the S350D mutation did not alter hPXR nuclear localization, with or without hPXR agonist.
3.7 Transcriptional activity of hPXRS350D is impaired in vivo in the livers of hPXRS350D-tg mice
The transcriptional activity of hPXR can be negatively or positively modulated by kinases, depending on its phosphorylation at different amino acid residues [11;37] and the availability and/or activity of kinases in the quiescent hepatocytes in the liver can be very different from that in the proliferative cultured cells. Thus, to further examine whether a single S350D mutation in hPXR was sufficient to impair its regulation of target gene expression in the liver, we first generated transgenic mice expressing phosphomimetic hPXRS350D in the mouse Pxr-null background, named humanized hPXRS350D-tg mice. The similar strategy was previously used to create wild-type hPXR humanized mice [20–22]. The integration and integrity of the transgene were confirmed by Southern blot analysis (Fig. 6A). The expression of the transgene was verified by Northern blot analysis (Fig. 6B). We first compared the change in expression of the Cyp3a11 gene, a human CYP3A4 homolog in mice regulated by hPXR [20], at both mRNA and protein levels, in the liver of Pxr−/−, hPXR-tg, and hPXRS350D-tg mice without and with RIF treatment. Compared to those in untreated mice, RIF treatment did not significantly alter Cyp3a11 mRNA levels in PXR−/− mice but tripled the induction of Cyp3a11 mRNA in hPXR-tg mice, indicating the activation of hPXR by RIF (Fig. 6C); however, the level of Cyp3a11 mRNA in hPXRS350D-tg mice was not altered by the same RIF treatment (Fig. 6C), suggesting that the phosphomimetic mutation at Ser350 impaired in vivo hPXR-mediated transactivation of Cyp3a11. At the protein level, the expression of hepatic CYP3A protein was doubled in hPXR-tg mice treated with RIF but was not significantly changed in either Pxr−/− mice or hPXRS350D-tg mice treated with RIF, consistent with the change of Cyp3a11 mRNA level by RIF treatment (Fig. 6D).
Figure 6. Reduced transcriptional activation by hPXRS350D in vivo.
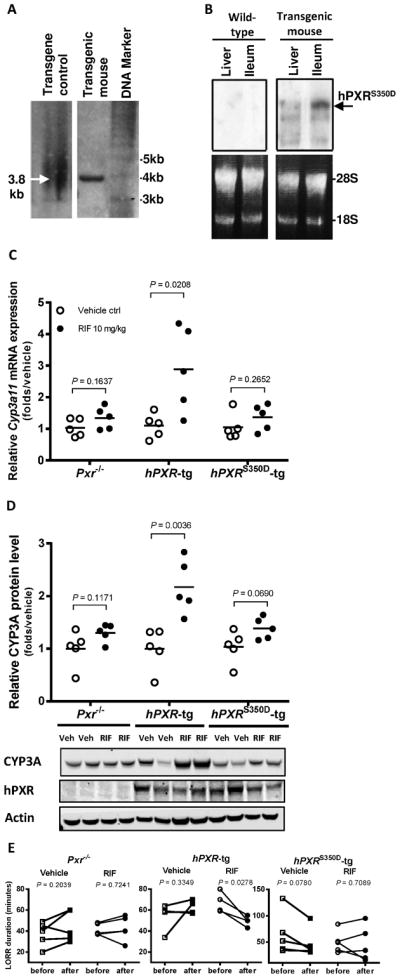
A) Southern blot analysis on the integration and integrity of the transgene. B) Northern blot analysis on the expression of the transgene in mouse liver and intestinal tracts. C) Mouse liver Cyp3a11 mRNA was analyzed by real-time PCR in Pxr−/−, hPXR-tg, and hPXRS350D-tg mice treated with vehicle control (Vehicle Ctrl) or RIF (10 mg/kg) for 72 h. E) liver CYP3A protein levels in the same mice as determined by Western blotting. Each data point represents level of Cyp3a11 mRNA (C) or protein (D) in an individual mouse; lines indicate the mean value for 5 mice per group. Representative Western blots from 2 mice in each group are shown. E) Loss of righting reflex (LORR) duration, recorded as described in Materials and Methods to measure metabolism of the anesthetic (2,2,2-tribromoethanolamine) in Pxr−/−, hPXR-tg, and hPXRS350D-tg mice before and after vehicle or RIF treatment. Each data point represents LORR duration in an individual mouse; lines indicate LORR duration change in individual mouse before and after treatment. P values indicate comparison of values between two groups by unpaired (C and D) and paired t test (E).
Because a relation has been shown between hepatic Cyp3a11 gene expression and enzymatic activity [38], we compared CYP3A11 enzyme activity before vs. after RIF administration in the three groups of mice by using a “loss of righting reflex” (LORR) assay. In this assay, Mice were injected intraperitoneally with the anesthetic drug 2,2,2-tribromoethanol, a CYP3A11-specific substrate whose duration of LORR effect is inversely related to CYP3A11 enzymatic activity, which hepatically metabolizes injected 2,2,2-tribromoethanol [20;23;24]. Compared to the LORR times before the treatment, RIF administration significantly decreased LORR duration in hPXR-tg mice (Fig. 6E), indicating RIF elevation of CYP3A11 enzymatic activity, but did not significantly alter the duration of LORR in PXR−/− or hPXRS350D-tg mice (Fig. 6E), indicating unaltered CYP3A11 enzymatic activity by RIF in them. Together, these results show consistent changes in Cyp3a11 levels of mRNA, protein, and enzyme activity in response to RIF treatment, suggesting that the hPXR agonist RIF could not effectively induce transcriptional activation by mutant hPXRS350D in mouse liver, and, thus the in vivo function of hPXR in mouse liver was impaired by the phosphomimetic S350D mutation.
4. Discussion
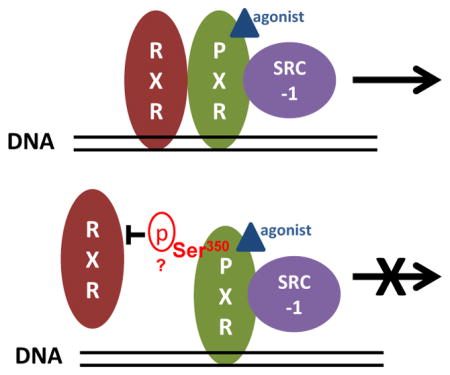
To our knowledge, the present study is the first to demonstrate that Ser350 of hPXR is crucial for heterodimerization between hPXR and RXRα and the subsequent transcriptional activation of hPXR target genes. As shown schematically in Fig. 7, this conclusion was proved by establishing, through different in vitro and in vivo approaches, that a phosphomimetic mutation of Ser350 prevents hPXR-RXRα dimerization, which is a necessary step in hPXR-mediated transcriptional activation (Fig. 7).
Figure 7. A proposed mechanistic model of the role of Ser350 in hPXR function.

A) Agonist binding, hPXR-RXRα heterodimerization and hPXR-coactivator recruitment are the three key steps for the precise regulation of hPXR on target gene expression (top panel). B) The putative phosphorylation of Ser350 of PXR halts transcriptional activation of target genes by preventing hPXR heterodimerization with RXRα (bottom panel).
Although hPXR-RXRα heterodimerization is known to be required for hPXR-mediated transcriptional activation, control of phosphorylation-mediated heterodimerization with RXRα is a novel model for the regulation of hPXR transcriptional activity. Although NR phosphorylation is known to alter its transcriptional activation function by changing its ligand binding affinity (reviewed by [7]), our study suggested a model in which phosphorylation modulates NR activity through its effect on NR-RXRα interaction. Studies of other NRs support this model. For example, human retinoic acid receptor alpha (RARα) showed decreased capacity to heterodimerize with hRXRα, and a reduced transcriptional activation function, when phosphorylated at Ser157 [39]. Similarly, peroxisome proliferator-activated receptor alpha (PPARα) carrying an S179A mutation showed impaired dimerization with RXRα [40] due to its resistance to phosphorylation by PKC, resulting in decreased ligand-induced transactivation activity [41]. The present study revealed that Ser350 is crucial for hPXR-RXRα dimerization and that the phosphomimetic S350D mutation impairs dimerization, impairing hPXR’s transcriptional activity. Therefore, phosphorylation of Ser350 can be a regulatory mechanism for hPXR activity. However, although several kinases appear to phosphorylate hPXR, the actual prevalence and magnitude of hPXR Ser350 phosphorylation in different cells and tissues, influenced by different hemostatic and xenobiotic factors, remain to be defined. Further studies using mass spectrometry and biochemical tools are warranted.
Our finding that phosphorylation of hPXR can prevent its interaction with RXRα independently of ligand binding has implications for clinical drug development. Ser350 of hPXR is followed by a proline within a consensus site of proline-dependent kinases, such as CDKs. Our studies suggested that CDKs may regulate hPXR function in the absence of ligand by phosphorylating it, thus modulating the hPXR-RXRα interaction; therefore, kinase inhibitor drugs, such as CDK inhibitors, could undesirably affect PXR function. CDK inhibitors, such as pan-CDKs inhibitors and CDK2 inhibitors [42], have been widely developed for cancer therapy and have shown promising clinical efficacy [42]. It remains to be determined whether and how recently developed CDK inhibitors affect hPXR functions. Our results in this study suggested that CDK inhibitors can indirectly affect hPXR activity in a ligand-independent manner by mediating the phosphorylation of hPXR, thereby disrupting the heterodimerization of hPXR-RXRα. CDK inhibitors are usually recommended for use in combination with other clinical anticancer drugs. Considering that unwanted activation of hPXR can cause adverse drug-drug interactions in combination therapy, the effects of these CDK inhibitors on hPXR function warrant further investigation in vitro and in vivo.
Our findings also suggest that specific disruption of hPXR-RXRα interaction offers an alternative approach for development of hPXR antagonists. Studies in humans and animal models have shown that unwanted activation of hPXR has multiple clinical ramifications, such as hPXR-mediated adverse drug-drug interactions, cancer drug resistance, and liver toxicity [3;4;43]; therefore, significant efforts have been made to develop hPXR antagonists to tackle these adverse effects. It has been suggested that developing PXR antagonists fitting into the LBD of hPXR can be difficult because of the highly flexible and promiscuous property of its structure which can change its shape to allow various ligands to bind [43;44]. Therefore, targeting the hPXR-coactivators interaction becomes an appealing approach to develop modulators of hPXR activity. The recent development of azole compounds to inhibit agonist-induced hPXR activation by disrupting hPXR-SRC1 interaction offers a successful example [45;46]. Our finding that a single phosphomimetic mutation that disrupts hPXR-RXRα interaction is sufficient to impair the transcriptional activity of hPXR indicates that the domain of hPXR that interacts with RXRα is crucial for its function. Thus, compounds or peptides that specifically target the hPXR-RXRα interaction may exert hPXR-antagonistic effects, and screening for such compounds may be an alternative approach for the discovery of hPXR antagonists.
In the hPXRS350D-tg mouse model, the mutant hPXR fails to activate the transcription of its target genes in vivo; therefore, this model may prove a useful tool for segregating the genomic (i.e. transcriptional) and non-genomic (i.e., non-transcriptional) functions of hPXR, as the transcriptional function of hPXRS350D is defective despite ligand binding. This model may also provide new avenues to further investigate some remaining questions about hPXR. For example, there are discrepancies about different roles of hPXR observed in colon cancer cells. In cellular models, genetically or pharmacologically activated hPXR showed an antiapoptotic function that facilitates the progression of colon cancer [22;47]; however, in the absence of agonists, the overexpressed hPXR displayed a proapoptotic effect that inhibits the proliferation and tumorigenicity of HT-29 colon cancer cells [48]. One possible explanation is that hPXR can be anti-apoptotic as a result of its ligand-induced transcriptional activity, while it can be proapoptotic as the result of a non-genomic function acting on proteins in the apoptosis pathway. If this explanation applies, then under certain experimental conditions, the apoptosis phenotype might differ in colon cells of Pxr−/−, hPXR-tg, and hPXRS350D-tg mice, with the hPXRS350D-tg mice showing mainly a phenotype reflecting the proapoptotic process due to hPXR’s non-genomic effects in this mouse model. Therefore, the hPXRS350D-tg mouse model presents a tool for studies of the potential non-genomic effects of hPXR in vivo. Further definition of the non-genomic effects of hPXR and characterization of the hPXRS350D-tg mouse model are needed.
In summary, this study demonstrated that: 1) Ser350 of hPXR, a putative phosphorylation site, is crucial for hPXR heterodimerization with RXRα and the resulting transcriptional activation of hPXR target genes and 2) in humanized transgenic mice, the hPXRS350D mutant displays impaired function in regulating its target genes upon agonist activation. Our findings suggest new avenues for safety evaluation during drug development (e.g., CDK inhibitors) and a novel strategy for the development of hPXR antagonists to prevent and manage hPXR-induced adverse DDIs. Our hPXRtgS350D mouse model provides a potentially unique tool to elucidate hPXR’s non-genomic functions.
Acknowledgments
We thank the St. Jude Animal Resources Center for technical assistance, Dr. Martin Privalsky for kindly providing the SMRTτ construct, other members of the Chen research laboratory for valuable discussions, and Sharon Naron (St. Jude Department of Scientific Editing) for editing the manuscript. This work was supported by the American Lebanese Syrian Associated Charities (ALSAC), St. Jude Children’s Research Hospital, and the National Institutes of Health [Grants RO1GM086415, RO1GM110034, & P30-CA21765].
Footnotes
Conflict of interest
The authors have no conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Willson TM, Kliewer SA. PXR CAR and drug metabolism. Nat Rev Drug Discov. 2002;1:259–66. doi: 10.1038/nrd753. [DOI] [PubMed] [Google Scholar]
- 2.Wang YM, Lin W, Chai SC, Wu J, Ong SS, Schuetz EG, Chen T. Piperine activates human pregnane X receptor to induce the expression of cytochrome P450 3A4 and multidrug resistance protein 1. Toxicol Appl Pharmacol. 2013;272:96–107. doi: 10.1016/j.taap.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sinz M, Kim S, Zhu Z, Chen T, Anthony M, Dickinson K, Rodrigues AD. Evaluation of 170 xenobiotics as transactivators of human pregnane X receptor (hPXR) and correlation to known CYP3A4 drug interactions. Curr Drug Metab. 2006;7:375–88. doi: 10.2174/138920006776873535. [DOI] [PubMed] [Google Scholar]
- 4.Sinz MW. Evaluation of pregnane X receptor (PXR)-mediated CYP3A4 drug-drug interactions in drug development. Drug Metab Rev. 2013;45:3–14. doi: 10.3109/03602532.2012.743560. [DOI] [PubMed] [Google Scholar]
- 5.Chen T. Nuclear receptor drug discovery. Curr Opin Chem Biol. 2008;12:418–26. doi: 10.1016/j.cbpa.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 6.Evans RM, Mangelsdorf DJ. Nuclear Receptors, RXR, and the Big Bang. Cell. 2014;157:255–66. doi: 10.1016/j.cell.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Germain P, Staels B, Dacquet C, Spedding M, Laudet V. Overview of nomenclature of nuclear receptors. Pharmacol Rev. 2006;58:685–704. doi: 10.1124/pr.58.4.2. [DOI] [PubMed] [Google Scholar]
- 8.Lin W, Wu J, Dong H, Bouck D, Zeng FY, Chen T. Cyclin-dependent kinase 2 negatively regulates human pregnane X receptor-mediated CYP3A4 gene expression in HepG2 liver carcinoma cells. J Biol Chem. 2008;283:30650–7. doi: 10.1074/jbc.M806132200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lichti-Kaiser K, Brobst D, Xu C, Staudinger JL. A systematic analysis of predicted phosphorylation sites within the human pregnane X receptor protein. J Pharmacol Exp Ther. 2009;331:65–76. doi: 10.1124/jpet.109.157180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pondugula SR, Brimer-Cline C, Wu J, Schuetz EG, Tyagi RK, Chen T. A phosphomimetic mutation at threonine-57 abolishes transactivation activity and alters nuclear localization pattern of human pregnane x receptor. Drug Metab Dispos. 2009;37:719–30. doi: 10.1124/dmd.108.024695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elias A, High AA, Mishra A, Ong SS, Wu J, Peng J, Chen T. Identification and characterization of phosphorylation sites within the pregnane X receptor protein. Biochem Pharmacol. 2014;87:360–70. doi: 10.1016/j.bcp.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doricakova A, Novotna A, Vrzal R, Pavek P, Dvorak Z. The role of residues T248, Y249 and T422 in the function of human pregnane X receptor. Arch Toxicol. 2013;87:291–301. doi: 10.1007/s00204-012-0937-9. [DOI] [PubMed] [Google Scholar]
- 13.Wallace BD, Betts L, Talmage G, Pollet RM, Holman NS, Redinbo MR. Structural and functional analysis of the human nuclear xenobiotic receptor PXR in complex with RXRalpha. J Mol Biol. 2013;425:2561–77. doi: 10.1016/j.jmb.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ubersax JA, Ferrell JE., Jr Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. 2007;8:530–41. doi: 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]
- 15.Sivertsson L, Edebert I, Palmertz MP, Ingelman-Sundberg M, Neve EP. Induced CYP3A4 expression in confluent Huh7 hepatoma cells as a result of decreased cell proliferation and subsequent pregnane X receptor activation. Mol Pharmacol. 2013;83:659–70. doi: 10.1124/mol.112.082305. [DOI] [PubMed] [Google Scholar]
- 16.Sugatani J, Uchida T, Kurosawa M, Yamaguchi M, Yamazaki Y, Ikari A, Miwa M. Regulation of pregnane X receptor (PXR) function and UGT1A1 gene expression by posttranslational modification of PXR protein. Drug Metab Dispos. 2012;40:2031–40. doi: 10.1124/dmd.112.046748. [DOI] [PubMed] [Google Scholar]
- 17.Ma X, Shah Y, Cheung C, Guo GL, Feigenbaum L, Krausz KW, Idle JR, Gonzalez FJ. The PREgnane X receptor gene-humanized mouse: a model for investigating drug-drug interactions mediated by cytochromes P450 3A. Drug Metab Dispos. 2007;35:194–200. doi: 10.1124/dmd.106.012831. [DOI] [PubMed] [Google Scholar]
- 18.Zhou H, Liu W, Su Y, Wei Z, Liu J, Kolluri SK, Wu H, Cao Y, Chen J, Wu Y, Yan T, Cao X, Gao W, Molotkov A, Jiang F, Li WG, Lin B, Zhang HP, Yu J, Luo SP, Zeng JZ, Duester G, Huang PQ, Zhang XK. NSAID sulindac and its analog bind RXRalpha and inhibit RXRalpha-dependent AKT signaling. Cancer Cell. 2010;17:560–73. doi: 10.1016/j.ccr.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin W, Chen T. A vinblastine fluorescent probe for pregnane X receptor in a time-resolved fluorescence resonance energy transfer assay. Anal Biochem. 2013;443:252–60. doi: 10.1016/j.ab.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie W, Barwick JL, Downes M, Blumberg B, Simon CM, Nelson MC, Neuschwander-Tetri BA, Brunt EM, Guzelian PS, Evans RM. Humanized xenobiotic response in mice expressing nuclear receptor SXR. Nature. 2000;406:435–9. doi: 10.1038/35019116. [DOI] [PubMed] [Google Scholar]
- 21.Gong H, Singh SV, Singh SP, Mu Y, Lee JH, Saini SP, Toma D, Ren S, Kagan VE, Day BW, Zimniak P, Xie W. Orphan nuclear receptor pregnane X receptor sensitizes oxidative stress responses in transgenic mice and cancerous cells. Mol Endocrinol. 2006;20:279–90. doi: 10.1210/me.2005-0205. [DOI] [PubMed] [Google Scholar]
- 22.Zhou J, Liu M, Zhai Y, Xie W. The antiapoptotic role of pregnane X receptor in human colon cancer cells. Mol Endocrinol. 2008;22:868–80. doi: 10.1210/me.2007-0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poulton EJ, Levy L, Lampe JW, Shen DD, Tracy J, Shuhart MC, Thummel KE, Eaton DL. Sulforaphane is not an effective antagonist of the human pregnane X-receptor in vivo. Toxicol Appl Pharmacol. 2013;266:122–31. doi: 10.1016/j.taap.2012.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mani S, Huang H, Sundarababu S, Liu W, Kalpana G, Smith AB, Horwitz SB. Activation of the steroid and xenobiotic receptor (human pregnane X receptor) by nontaxane microtubule-stabilizing agents. Clin Cancer Res. 2005;11:6359–69. doi: 10.1158/1078-0432.CCR-05-0252. [DOI] [PubMed] [Google Scholar]
- 25.Harmsen S, Koster AS, Beijnen JH, Schellens JH, Meijerman I. Comparison of two immortalized human cell lines to study nuclear receptor-mediated CYP3A4 induction. Drug Metab Dispos. 2008;36:1166–71. doi: 10.1124/dmd.107.017335. [DOI] [PubMed] [Google Scholar]
- 26.Gupta A, Mugundu GM, Desai PB, Thummel KE, Unadkat JD. Intestinal human colon adenocarcinoma cell line LS180 is an excellent model to study pregnane X receptor, but not constitutive androstane receptor mediated CYP3A4 and multidrug resistance transporter 1 induction: studies with anti-human immunodeficiency virus protease inhibitors. Drug Metab Dispos. 2008;36:1172–80. doi: 10.1124/dmd.107.018689. [DOI] [PubMed] [Google Scholar]
- 27.Helsley RN, Sui Y, Ai N, Park SH, Welsh WJ, Zhou C. Pregnane X receptor mediates dyslipidemia induced by the HIV protease inhibitor amprenavir in mice. Mol Pharmacol. 2013;83:1190–9. doi: 10.1124/mol.113.085753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Staudinger JL, Lichti K. Cell signaling and nuclear receptors: new opportunities for molecular pharmaceuticals in liver disease. Mol Pharm. 2008;5:17–34. doi: 10.1021/mp700098c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teotico DG, Frazier ML, Ding F, Dokholyan NV, Temple BR, Redinbo MR. Active nuclear receptors exhibit highly correlated AF-2 domain motions. PLoS Comput Biol. 2008;4:e1000111. doi: 10.1371/journal.pcbi.1000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang YM, Ong SS, Chai SC, Chen T. Role of CAR. PXR in xenobiotic sensing and metabolism. Expert Opin Drug Metab Toxicol. 2012;8:803–17. doi: 10.1517/17425255.2012.685237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Worth CL, Preissner R, Blundell TL. SDM--a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011;39:W215–W222. doi: 10.1093/nar/gkr363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40:W452–W457. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35:3823–35. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matic M, Nakhel S, Lehnert AM, Polly P, Clarke SJ, Robertson GR. A novel approach to investigate the subcellular distribution of nuclear receptors in vivo. Nucl Recept Signal. 2009;7:e004. doi: 10.1621/nrs.07004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Squires EJ, Sueyoshi T, Negishi M. Cytoplasmic localization of pregnane X receptor and ligand-dependent nuclear translocation in mouse liver. J Biol Chem. 2004;279:49307–14. doi: 10.1074/jbc.M407281200. [DOI] [PubMed] [Google Scholar]
- 36.Kawana K, Ikuta T, Kobayashi Y, Gotoh O, Takeda K, Kawajiri K. Molecular mechanism of nuclear translocation of an orphan nuclear receptor, SXR. Mol Pharmacol. 2003;63:524–31. doi: 10.1124/mol.63.3.524. [DOI] [PubMed] [Google Scholar]
- 37.Smutny T, Mani S, Pavek P. Post-translational and post-transcriptional modifications of pregnane X receptor (PXR) in regulation of the cytochrome P450 superfamily. Curr Drug Metab. 2013;14:1059–69. doi: 10.2174/1389200214666131211153307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raybon JJ, Pray D, Morgan DG, Zoeckler M, Zheng M, Sinz M, Kim S. Pharmacokinetic-pharmacodynamic modeling of rifampicin-mediated Cyp3a11 induction in steroid and xenobiotic X receptor humanized mice. J Pharmacol Exp Ther. 2011;337:75–82. doi: 10.1124/jpet.110.176677. [DOI] [PubMed] [Google Scholar]
- 39.Delmotte MH, Tahayato A, Formstecher P, Lefebvre P. Serine 157, a retinoic acid receptor alpha residue phosphorylated by protein kinase C in vitro is involved in RXR.RARalpha heterodimerization and transcriptional activity. J Biol Chem. 1999;274:38225–31. doi: 10.1074/jbc.274.53.38225. [DOI] [PubMed] [Google Scholar]
- 40.Gray JP, Burns KA, Leas TL, Perdew GH, Vanden Heuvel JP. Regulation of peroxisome proliferator-activated receptor alpha by protein kinase C. Biochemistry. 2005;44:10313–21. doi: 10.1021/bi050721g. [DOI] [PubMed] [Google Scholar]
- 41.Blanquart C, Mansouri R, Paumelle R, Fruchart JC, Staels B, Glineur C. The protein kinase C signaling pathway regulates a molecular switch between transactivation and transrepression activity of the peroxisome proliferator-activated receptor alpha. Mol Endocrinol. 2004;18:1906–18. doi: 10.1210/me.2003-0327. [DOI] [PubMed] [Google Scholar]
- 42.Guha M. Cyclin-dependent kinase inhibitors move into Phase III. Nat Rev Drug Discov. 2012;11:892–4. doi: 10.1038/nrd3908. [DOI] [PubMed] [Google Scholar]
- 43.Chen T. Overcoming drug resistance by regulating nuclear receptors. Adv Drug Deliv Rev. 2010;62:1257–64. doi: 10.1016/j.addr.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mani S, Dou W, Redinbo MR. PXR antagonists and implication in drug metabolism. Drug Metab Rev. 2013;45:60–72. doi: 10.3109/03602532.2012.746363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Venkatesh M, Wang H, Cayer J, Leroux M, Salvail D, Das B, Wrobel JE, Mani S. In vivo and in vitro characterization of a first-in-class novel azole analog that targets pregnane X receptor activation. Mol Pharmacol. 2011;80:124–35. doi: 10.1124/mol.111.071787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H, Redinbo MR, Venkatesh M, Ekins S, Chaudhry A, Bloch N, Negassa A, Mukherjee P, Kalpana G, Mani S. Novel yeast-based strategy unveils antagonist binding regions on the nuclear xenobiotic receptor PXR. J Biol Chem. 2013;288:13655–68. doi: 10.1074/jbc.M113.455485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang H, Venkatesh M, Li H, Goetz R, Mukherjee S, Biswas A, Zhu L, Kaubisch A, Wang L, Pullman J, Whitney K, Kuro-o M, Roig AI, Shay JW, Mohammadi M, Mani S. Pregnane X receptor activation induces FGF19-dependent tumor aggressiveness in humans and mice. J Clin Invest. 2011;121:3220–32. doi: 10.1172/JCI41514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ouyang N, Ke S, Eagleton N, Xie Y, Chen G, Laffins B, Yao H, Zhou B, Tian Y. Pregnane X receptor suppresses proliferation and tumourigenicity of colon cancer cells. Br J Cancer. 2010;102:1753–61. doi: 10.1038/sj.bjc.6605677. [DOI] [PMC free article] [PubMed] [Google Scholar]