

Abstract
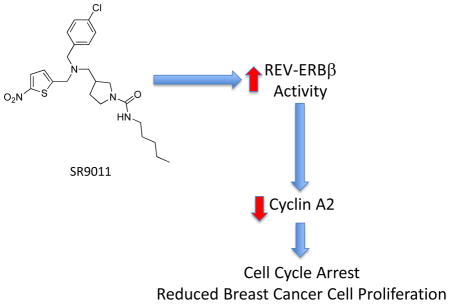
REV-ERBα and REV-ERBβ are nuclear receptors that are ligand-dependent transcriptional repressors. Heme is the natural ligand for these receptors, but several synthetic agonists and antagonists have been designed recently. The gene that encodes REV-ERBα, NR1D1, is closely associated with ERBB2, the gene that encodes the HER2 oncogene, which is amplified in HER2+ breast cancers. We examined the effect of a synthetic REV-ERB agonist, SR9011, on a range of estrogen receptor positive (ER+), ER−, HER2+, HER2− and triple negative breast cancer cell lines. We found that SR9011 suppressed proliferation of the breast cancer cell lines regardless of their ER or HER2 status. SR9011 had no effect on MCF10A cell proliferation. SR9011 appears to pause the cell cycle of the breast cancer cells prior to M phase. Cyclin A (CCNA2) was identified as a direct target gene of REV-ERB suggesting that suppression of expression of this cyclin by SR9011 may mediate the cell cycle arrest. These data indicate that synthetic REV-ERB ligands may hold utility in treatment of diseases associated with uncontrolled cellular proliferation such as cancer.
Keywords: Nuclear receptor, circadian rhythm, steroid receptor, heme, cell cycle
Graphical Abstract
1. Introduction
REV-ERBα and REV-ERBβ are members of the nuclear receptor superfamily and serve as receptors for heme 1–3. These two receptors respond to heme binding by altering the conformation of the ligand binding domain (LBD) leading to recruitment of the corepressor NCoR and subsequent repression of expression of their target genes 1–3. REV-ERBs play an important role in a range of physiological processes including regulation of glucose and lipid metabolism as well as regulation of the circadian rhythm 3–5. We and others have designed a number of synthetic ligands that target the REV-ERBs 6–13. In fact, we recently demonstrated that the REV-ERB agonists, SR9011 and SR9009, display a range of activity in vivo, including modulation of behavior and metabolism 10, 12, 14
Interestingly, the REV-ERBα gene (NR1D1) resides on the ERBB2 (HER2)-containing 17q12–21 amplicon and has been suggested to play a role in the viability of HER2+ breast cancers. Additionally, REV-ERBα has been suggested to be a survival factor for HER2+ breast cancers 15. Based on these reports, we sought to examine the effect of a synthetic REV-ERB agonist, SR9011, on a range of breast cancer cells including estrogen receptor positive and negative cells, HER2 positive and negative cells as well as triple negative cells.
2. Materials and Methods
2.1 Plasmids
The CCNA2 promoter (−957 to +17) was amplified from genomic DNA of HepG2 cells and cloned into pTAL-Luc luciferase report vector (Clontech, Mountain View, CA) to make the pTAL-CCNA2 reporter construct. The CCNA2 promoter mutant construct was made using QuikChange II site-directed mutagenesis kit (Stratagene, Santa Clara, CA) according to the manufacturer’s instructions. The REVRE (−802 to −791) was mutated from TAATAAGGTCAT to TAATAAGGCCAT. The muted primers targeting REV-ERB binding site are: CTTCTGAAAGGAACATAATTATATCTAGGCCACTAGAACGTCATTGTG (forward) and CACAATGACGTTCTAGTGgCCTAGATATAATTATGTTCCTTTCAGAAG (reverse). The pGL4.73, pG5luc, pGL3-mBmal1, Gal4-REV-ERBα and Gal4-REV-ERBβ were previously described. pcDNA-REV-ERBα and pcDNA-REV-ERBβ were cloned in our lab.
2.2 Luciferase assay
HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum at 37 °C under 5% CO2. Cells were plated in 96-well plates at a density of 15 X 103 cells/well 24 h before transfection. Eight hour post-transfection, the cells were treated with SR9011 or DMSO. Twenty-four hours post-treatment, the luciferase activity was measured using the Dual-GloTM luciferase assay system (Promega, Madison, WI). The values indicated represent the means ± S.E. from three independently transfected wells. The experiments were repeated three times, and representative experiments are shown.
2.3 Cell culture, compound treatment, overexpression and knockdown
MCF10A, MDA-MB-231, MCF-7, MDA-MB-361, SKBR3, BT474 are from ATCC. Cells were plated in 6-well plates one day before treatment. The cells were treated with SR9011 or DMSO for 24 hr and harvested for RNA isolation or western blot. For over expression, the cells were infected with adenovirus for 24 hours and then switched to regular growth media. Twenty-four hours later, the cells were harvested to isolate total RNA. For knockdown assay, the control siRNA, human REV-ERBβ siRNA (Thermo Scientific) were transfected with LipofectamineTM RNAiMAX (Invitrogen, Carlsbad, CA) by using reverse transfection following the manufacture’s instruction. After 24 hours, cells were harvested to perform quantitative PCR assay.
2.4 MTT assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell proliferation assays (Invitrogen)16 were performed according to the manufacturer’s manual. Briefly, 3 × 103 to 5 × 103 cells per well were plated in 96-well plates. Twenty-four hours later, cells were treated with SR9011 or DMSO. Seventy-two hours after treatment, the cells were labeled with 1.2 mM MTT and incubated for 4 hours. DMSO was then added and readings were taken on a plate reader at 540 nm.
2.5 Cell Cycle and BrdU assays
The SKBR3 cells were incubated in growth media without serum for 72 hours and re-stimulated with normal growth media to synchronize the phase of the cell cycle. Every four hours after synchronization, the cells harvested for isolation of mRNA and assessment of CCNA2 expression by QPCR. In MDA-MD231 cells, a BrdU assay17 was used to determine their phase within the cell cycle. The cells were incubated with Brdu (Invitrogen)) for 30 min and then trypsinised and fixed with 70% ice-cold ethanol on ice for at least 30 minutes. The cells were then incubated with 2 M hydrochloric acid for 20 minutes, washed and stained with an anti-BrdU antibody (Invitrogen), FITC-labeled goat anti-mouse IgG (eBiosciences, San Diego, CA) and propidium iodide (Sigma, St. Louis, MO) subsequently. Data were collected in a LSRII flow cytometer and analyzed using FloJo software.
2.6 cDNA synthesis and quantitative PCR
Total RNA extraction and cDNA synthesis were performed as described before. The quantitative PCR was performed using ABI Prism 7900 HT detection system (Applied Biosystems, Foster City, CA). The primers for quantitative PCR are: CYPB, GCAAATTCCATCGTGTAATCAAG (forward) and CGTAGATGCTCTTTCCTCCTG (reverse); REV-ERBα, TTCCGCTTCGGTGGAGCAGC (forward) and CCGGTTCTTCAGCACCAGAG (reverse); REV-ERBβ, GAACAGACAGCCTTGCCAGC (forward) and AGCTCTGGTCACCATGCCAA (reverse); CCNA2, CGGGACAAAGCTGGCCTGAA (forward) and GTTGTGCATGCTGTGGTGCT (reverse). The expression of target gene was normalized to housekeeping gene CYPB.
2.7 Western blot
Antibodies against human cyclin A were obtained from Santa Cruz (Dallas, TX). Tubulin is from Sigma. Western blot were performed according to the standard protocols. Briefly, cells were washed once with phosphate-buffered saline and then incubated for 10 min at 4 °C in 100 μl of TNT lysis buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, and 1% Triton X-100) and a complete miniprotease inhibitor mixture (Roche Applied Science, Indianapolis, IN). Samples were then scraped and harvested into 1.5-ml microcentrifuge tubes, vortexed, and then centrifuged. Protein levels in the supernatants were determined using a Coomassie protein assay kit (Bio-Rad, Hercules, CA), and 20 μg of protein from each sample was separated by SDSPAGE (BioRad - 10%) and then transferred to a polyvinylidene difluoride membrane (Millipore, Milford, MA) and immunoblotted with primary antibodies and horseradish peroxidase-conjugated secondary antibodies (Jackson Immunoresearch, West Grove, PA). Detection of the bound antibody by enhanced chemiluminescence was performed according to the manufacturer’s instructions (Santa Cruz).
2.8 ChIP/Microarray and ChIP assay
The REV-ERBα Chromatin Immunoprecipitation (ChIP)/Microarray experiment was previously described18. Standard ChIP assays were performed according to the manufacturer’s instructions (Active Motif, Carlsbad, CA) as we have previously described19. Generally, the cells were grown to 70–80% confluency, fixed with 1% formaldehyde and sheared by sonication. Sheared chromatin was immunoprecipitated with IgG (Abcam, Cambridge MA), anti-HIstone H3 (Abcam) and anti-REV-ERBα (Cell Signalling, Danvers, MA) overnight. The eluted chromatin was reverse cross-linked at 95°C for 15 min and treated with Proteinase K at 37°C for 1hr. The primers for the PCR reaction are: TGTTAAAGGCACGTATAGTTAAGAGAGT (forward) and AGATGGCACCTTGAACTACTGTTG (reverse).
3. Results
3.1 SR9011 Inhibits Breast Cancer Cell Proliferation
In order to examine the effect of SR9011 (Fig. 1a) on breast cancer cell proliferation, we assessed the ability of this compound to alter cell viability using a MTT assay. We began using two cell lines. In the non-tumorigenic breast epithelial MCF-10A cell line had no effect on cell viability; however, in the tumorigenic HER2+ (estrogen receptor (ER) negative and progesterone receptor (PR) negative) SKBR3 cell line there was a dose-dependent decrease in viability of the cells (Fig. 1b). MCF-10A cells are commonly used as normal controls in breast cancer studies 20 and it is interesting to note that the SR9011 effects were specific to the tumorigenic breast cancer cell line. When we examined the effects of SR9011 on additional breast cancer cell lines including ER+ PR+ HER2− (MCF-7), ER+ PR+ HER2+ (BT-474 and MDA-MB361), and triple negative (MDA-MB231) cell lines we observed that the compound also caused a dose-dependent reduction in cell viability (Fig. 1c). Thus, the effect of SR9011 on the breast cancer cells appeared to be independent of ER, PR or HER2 status. As shown in Fig 2, treatment with SR9011 resulted in an increase in cells in the G0/G1 phase and a decrease of cells in S and G2/M phase suggesting that activation of REV-ERB may be resulting in decreased transition from G1 to S phase and/or from S to G2/M phase. There was no evidence of induction of apoptosis or cell death as indicated by the absence of a sub-G1 phase.
Figure 1.

Activation of REV-ERB suppresses breast cancer cell viability (MTT assay). a. Chemical structure of the REV-ERB agonist SR9011. b. Effect of SR9011 on viability of SKBR3 and MCF10A cells. c. Effect of SR9011 on a range of breast cancer cell viability. See results for description of each cell line in terms of ER and HER2 expression.
Figure 2.

SR9011 treatment of breast cancer cells (MDA-MD231) results in cell cycle arrest. Results from flow cytometry analysis of MDA-MD231 cells stained with BrdU and propidium idodide (vehicle or SR9011 treatment (10 μM)) are shown. The percentage of cells identified in various stages of the cell cycle are illustrated. *, indicates p<0.05 using Student’s t-test.
3.2 Cyclin A is a REV-ERB target gene
Given the specific effects we observed on the cell cycle, we initially searched for putative REVERB target genes that could be responsible for accumulation of cells in the G0/G1 phase. Assessment of ChIP/Microarry data revealed that the cyclin A gene (CCNA2) contained a putative REV-ERB response element within its promoter (Fig. 3a) and the function of cyclin A in regulation of progression of the cell cycle into the S and M phases via interaction with CDK2 and CDK1, respectively 21–23. A subsequent ChIP assay revealed REV-ERBα occupancy of the promoter region of CCNA2 (Fig. 3b). Adenoviral overexpression of REV-ERBα in SKBR3 cells led to significant repression of CCNA2 gene expression, as one would expect if this transcriptional repressor directly targeted this gene (Fig. 3c). A putative REV-ERB response element (REVRE) that displayed considerably conservation between human and mouse was identified in the promoter (Fig. 3d). We constructed luciferase reporter constructs containing the promoter of the wild type CCNA2 REVRE or a mutant CCNA2 REVRE (Fig. 3d bottom) and assessed the ability of either REV-ERBα or REV-ERBβ to regulate this reporter in a cotransfection assay in HEK293 cells. As shown in Fig. 3e, when either REV-ERBα or REV-ERBβ were cotransfected into cells along with the wild type reporter significant repression of transcription was noted. If the REV-ERB agonist SR9011 was added, additional suppression of transcription was observed. However, if the reporter contained the mutant REVRE instead of the wild type the repressive activity of either of the REV-ERBs was lost (Fig. 3e). Additionally, and as one would expect, the response to SR9011 was lost when examining the mutant reporter (Fig. 3e)
Figure 3.

The CCNA2 promoter contains a functional RORE site and is responsive to REV-ERB. a. Illustration of REV-ERBα occupancy of the CCNA2 gene. b. Results from a chromatin immunoprecipitation assay examining RNA polymerase II occupancy (positive control) or REV-ERBα occupancy of the CCNA2 promoter. c. Overexpression of REV-ERBα with an adenovirus suppresses CCNA2 expression. d. Illustration of the putative REV-ERB response element (REVRE) in the CCNA2 promoter. The human sequence is compared with the mouse and the mutation is indicated at bottom. e. Mutation of the REVRE (CCNA2mt:luc) reduces CCNA2 expression in the presence of REV-ERBα or REV-ERBβ. Cells were cotransfected with wt or a mutant (REVRE mutated) CCNA2 promoter-reporter with or without REV-ERBα or REV-ERBβ expression vectors. SR9011 treatment at 10 μM. *, indicates p<0.05 vs. untreated control and # indicates p<0.05 vs. SR9011 treated control.
In order to perform REV-ERB loss of function studies, we first examined the expression of the REV-ERB subtypes in the various breast cancer cell lines. We found that REV-ERBβ was the predominate form of REV-ERB expressed in all of the breast cancer cells lines with the exception of the MCF10A line. REV-ERBβ was expressed to a level approximately 9- to 35-fold greater than REV-ERBα in these cell lines (Fig 4). In MCF-10A cells REV-ERBα and REV-ERBβ expression levels were similar (Fig. 4). Given that REV-ERBβ is the dominant REV-ERB in the cells, we performed loss of function studies targeting REV-ERBβ expression with siRNA. Suppression of REV-ERBβ expression in MDA-MB231 cells resulted in loss of the ability of SR9011 to reduce cell viability as detected in a MTT assay as shown in Figure 5a. Furthermore, overexpression of REV-ERBβ in the resistant MCF-10A cells resulted in gain of ability of SR9011 to reduce cell viability in the MTT assay (Fig. 5b). These data suggest that expression of REVERBβ is critical for the anti-proliferative effects of SR9011 in breast cancer cells.
Figure 4.

REV-ERBβ mRNA is highly expressed in breast cancer cell lines. REV-ERB mRNA expression was measured by QPCR and normalized to cyclophilin. REV-ERBβ is overexpressed relative to REV-ERBα in breast cancer cells. Most tissues we have examined with the exception of the breast cancer cell lines display a ratio near 1 (data not shown). *, indicates p<0.05 relative to MCF10A cell expression using Student’s t-test.
Figure 5.

REV-ERBβ is critical for the effects of SR9011 on breast cancer cells. A) Suppression of REV-ERBβ expression in MDA-MBA231 cells renders the cells less sensitive to the actions of SR9011 as observed in a MTT cell viability assay. B) Overexpression of REV-ERBβ in MCF10A cells renders these cells sensitive to the action of SR9011 in terms of suppression of cell viability. *, indicates p<0.05 relative to control cells using Student’s t-test.
We also examined the effect of suppression of REV-ERBβ on CCNA2 gene expression in three cell lines, MCF7, MDA-MBA-231 and SKBR3. Transfection of these cells with REV-ERBβ siRNA led to substantial decrease in REV-ERBβ expression in all three cases ranging from 70–90% (Fig. 6). In all three cases, suppression of REV-ERBβ expression led to an increase in CCNA2 gene expression (Fig. 6). These data clearly indicate that the cyclin A gene is a direct REV-ERB target gene.
Figure 6.

CCNA2 expression is tonically suppressed by REV-ERBβ. When REV-ERBβ expression is suppressed by siRNA treatment, CCNA2 mRNA expression increases in the cell lines examined above. Expression was normalized to cyclophilin and control cells received scrambled siRNA. *, indicates p<0.05 using Student’s t-test.
3.4 SR9011 suppresses cyclin A expression in breast cancer cells
Since we had observed a clear effect of the REV-ERB agonist SR9011 on cell cycle that led us to hypothesize that CCNA2 was a direct REV-ERB target gene, we next examined whether SR9011 would regulate CCNA2 expression. First, we examined this possibility using the CCNA2 promoter luciferase reporter transfected along with either REV-ERBα or REV-ERBβ in HEK293 cells. As shown in Fig. 7a, in cells transfected with the CCNA2 reporter only SR9011 treatment resulted in suppression of transcription consistent with SR9011 enhancing the transcriptional repressor activity of endogenously expressed REVERB. Overexpression of either REV-ERBα or REV-ERBβ enhanced the effect of SR9011, as one would expect (Fig. 7a). Treatment of a range of breast cancer cell lines with SR9011 for 24h led to suppression of CCNA2 gene expression in all cases (Fig. 7b). Interestingly, the most sensitive cell line in terms of SR9011-dependent suppression of proliferation (SKBR3) was also most sensitive in terms of maximal suppression of CCNA2 gene expression (~90% suppression) while the least sensitive cell line for proliferation response (MCF10A) was also the least sensitive to SR9011 in terms of CCNA2 response (~15%) (Fig. 7b). We also examined CCNA2 expression in SKBR3 cells where we synchronized the cells for the phase of cell cycle and found that 20h after synchronization CCNA2 expression was suppressed in SR9011 treated cells vs. vehicle treated cells (Fig. 7c). This suppression was sustained for the duration of the experiment that concluded at 44h post synchronization. When we examined Cyclin A2 protein expression in three of the cell lines (MDA-MB231, MDA-MB361, and BT-474) in response to SR9011 treatment we also observed a decrease consistent with the decrease in gene expression (Fig. 7d).
Figure 7.

The REV-ERB agonist, SR9011, suppresses the expression of CCNA2. a. SR9011 suppresses luciferase expression driven by the CCNA2 promoter. In HEK293 cells, treatment with SR9011 (10 μM) suppresses CCNA2 expression and coexpression of either REV-ERBα or REV-ERBβ enhances the SR9011-mediated effect. b. SR9011 treatment suppresses the expression of CCNA2 in multiple breast cancer cell lines. Expression was normalized to cyclophilin. c. SR9011 treatment suppresses CCNA2 expression in synchronized SKBR3 cells. d. SR9011 treatment suppresses the expression of CYCLIN A protein in multiple cancer cell lines. *, indicates p<0.05 using Student’s t-test.
4. Discussion
Two subfamilies of nuclear receptors, REV-ERBs (REV-ERBα and β) and retinoic acid receptor-related orphan receptors (RORα, β, γ) play an important role in modulation of the circadian clock by directly regulation the expression of core circadian clock genes such as BMAL1, NPAS2, and CLOCK 18, 24–27. Circadian (~24h) rhythms in biological processes are essential for normal physiological function in nearly all living organisms. These 24h recurrent patterns of behavior and physiology are controlled by a feedback loop at the cellular level. Circadian rhythms play an essential role in aspects of physiology and behavior including the sleep-wake cycle, body temperature, blood pressure, and renal function, and are generated by feedback loops in gene expression where heterodimers of BMAL1 and CLOCK (the positive limb) activate the expression of the Cryptochrome and Period genes (the negative limb). Once CRY and PER have reached a critical level of expression they are able to block the stimulatory effect of the CLOCK/BMAL1 complex on their own genes completing the loop. Epidemiological data indicates that disruption of circadian rhythmicity is associated with an increased risk of development of breast cancer 28–31. Based on these data, the World Health Organization has classified shift-work associated with a disrupted circadian rhythm as a probable carcinogen 32. The observation that disruption of the circadian rhythm leads to increased risk of cancer has been replicated in animal models where rodents experience an increased rate of tumor progression if their rhythms are disrupted 33–36. The molecular mechanism underlying the link between circadian rhythm disruption in humans and the increased risk of breast cancer is not clear, but several studies have demonstrated links between abnormal regulation of “clock” genes (genes that regulate the core circadian rhythm) and tumorigenesis 37–40.
These studies led us to consider that our efforts to pharmacologically target components of the clock machinery may hold utility in treatment of cancer. We were particularly intrigued by recent studies implicating REV-ERB in regulation of proliferation and cancer 15, 41, 42. Our data indicate that activation of REV-ERB with a synthetic agonist leads to decreased proliferation of a range of breast cancer cells independent of their ER or HER2 status. It appears that this may be due to direct targeting of cyclin A2 expression. A previous study linked REV-ERBα to breast cancer based on the localization of the NR1D1 gene within the same chromosomal region of the ERBB2 gene (HER2) that is amplified in many breast cancers Interestingly, the REV-ERBα gene (NR1D1) resides on the ERBB2 (HER2)-containing 17q12–21 amplicon and has been suggested to play a role in the viability of HER2+ breast cancers. 15. REV-ERBα expression was associated with increased lipogenesis that improved BT474 cell survival 15. Based on this, we might expect that a REV-ERB antagonist would be beneficial, which is in contrast to our current work indicating that activation of REV-ERB leads to decreased breast cancer cell proliferation in the BT474 cell line as well as a range of other breast cancer cells lines. In a more recent study, suppression of REV-ERBα expression did not alter BT474 cell viability 42. However, this same study found that although a REVERBβ antagonist did not alter BT474 viability alone, it did enhance the cytotoxicity of the autophagy inhibitor chloroquine 42.
Thus, there is some controversy surrounding whether REV-ERB is a “survival” factor in HER2+ breast cancer cells based on REV-ERBα knock-down experiments performed in BT474 cells, but our data clearly demonstrate that pharmacological activation of REV-ERB leads to reduced proliferation of a range of breast cancer cells independent of their ER or HER2 status. Overexpression of REV-ERBβ relative to REV-ERBα appears to be critical for the efficacy of SR9011 in the breast cancer cells since suppression of REV-ERBβ expression reduces that activity of SR9011 in MDA-MB231 cells while overexpression of REV-ERBβ in the “resistant” MCF10A cells renders them sensitive to the drug. Overall, we have not observed cytotoxicity of SR9011 or other REV-ERB agonists in “normal” non-cancerous cells as well as in a number of in vivo studies indicating that these compounds are not overtly toxic10, 12, 14, 43. These data suggest that targeting REV-ERB may be an effective method for treating breast cancers clinically.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (R01MH093429).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Raghuram S, Stayrook KR, Huang P, Rogers PM, Nosie AK, McClure DB, Burris LL, Khorasanizadeh S, Burris TP, Rastinejad F. Identification of heme as the ligand for the orphan nuclear receptors REV-ERBalpha and REV-ERBbeta. Nat Struct Mol Biol. 2007;14:1207–1213. doi: 10.1038/nsmb1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yin L, Wu N, Curtin JC, Qatanani M, Szwergold NR, Reid RA, Waitt GM, Parks DJ, Pearce KH, Wisely GB, Lazar MA. Rev-erb{alpha}, a Heme Sensor That Coordinates Metabolic and Circadian Pathways. Science. 2007;318:1786–1789. doi: 10.1126/science.1150179. [DOI] [PubMed] [Google Scholar]
- 3.Burris TP. Nuclear hormone receptors for heme: REV-ERBalpha and REV-ERBbeta are ligand-regulated components of the mammalian clock. Mol Endocrinol. 2008;22:1509–1520. doi: 10.1210/me.2007-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yin L, Wu N, Lazar MA. Nuclear receptor Rev-erbalpha: a heme receptor that coordinates circadian rhythm and metabolism. Nucl Recept Signal. 2010;8:e001. doi: 10.1621/nrs.08001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duez H, Staels B. Rev-erb-alpha: an integrator of circadian rhythms and metabolism. Journal of applied physiology. 2009;107:1972–1980. doi: 10.1152/japplphysiol.00570.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meng QJ, McMaster A, Beesley S, Lu WQ, Gibbs J, Parks D, Collins J, Farrow S, Donn R, Ray D, Loudon A. Ligand modulation of REV-ERB{alpha} function resets the peripheral circadian clock in a phasic manner. J Cell Sci. 2008;121:3629–3635. doi: 10.1242/jcs.035048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grant D, Yin L, Collins JL, Parks DJ, Orband-Miller LA, Wisely GB, Joshi S, Lazar MA, Willson TM, Zuercher WJ. GSK4112, a Small Molecule Chemical Probe for the Cell Biology of the Nuclear Heme Receptor Rev-erbα. ACS Chem Biol. 2010;5:925–932. doi: 10.1021/cb100141y. [DOI] [PubMed] [Google Scholar]
- 8.Kojetin D, Wang Y, Kamenecka TM, Burris TP. Identification of SR8278, a Synthetic Antagonist of the Nuclear Heme Receptor REV-ERB. ACS Chem Biol. 2011;6:131–134. doi: 10.1021/cb1002575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar N, Solt LA, Wang Y, Rogers PM, Bhattacharyya G, Kamenecka TM, Stayrook KR, Crumbley C, Floyd ZE, Gimble JM, Griffin PR, Burris TP. Regulation of Adipogenesis by Natural and Synthetic REV-ERB Ligands. Endocrinology. 2010;151:3015–3025. doi: 10.1210/en.2009-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Solt LA, Wang Y, Banerjee S, Hughes T, Kojetin DJ, Lundasen T, Shin Y, Liu J, Cameron MD, Noel R, Yoo S-H, Takahashi JS, Butler AA, Kamenecka TM, Burris TP. Regulation of Circadian Behavior and Metabolism by Synthetic REV-ERB Agonists. Nature. 2012;485:62–68. doi: 10.1038/nature11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noel R, Song X, Shin Y, Banerjee S, Kojetin D, Lin L, Ruiz CH, Cameron MD, Burris TP, Kamenecka TM. Synthesis and SAR of tetrahydroisoquinolines as Rev-erbalpha agonists. Bioorganic & medicinal chemistry letters. 2012;22:3739–3742. doi: 10.1016/j.bmcl.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banerjee S, Wang Y, Solt LA, Griffet K, Kazantizis M, Amador A, El-Gendy BM, Huitron-Resendiz S, Roberts AJ, Shin Y, Kamenecka TM, Burris TP. Pharmacological Targeting of the Mammalian Clock Regulates Sleep Architecture and Emotional Behavior. Nature Communications. 2014;5:575. doi: 10.1038/ncomms6759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shin Y, Noel R, Banerjee S, Kojetin D, Song X, He Y, Lin L, Cameron MD, Burris TP, Kamenecka TM. Small molecule tertiary amines as agonists of the nuclear hormone receptor Rev-erbalpha. Bioorganic & medicinal chemistry letters. 2012;22:4413–4417. doi: 10.1016/j.bmcl.2012.04.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woldt E, Sebti Y, Solt LA, Duhem C, Lancel S, Eeckhoute J, Hesselink MKC, Paquet C, Delhaye S, Shin Y, Kamenecka TM, Schaart G, Lefebvre P, Neviere R, Burris TP, Schrauwen P, Staels B, Duez H. Rev-erba modulates skeletal muscle oxidative capacity by regulated mitochondrial biogenesis and autophagy. Nature Medicine. 2013;19:1039–1046. doi: 10.1038/nm.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kourtidis A, Jain R, Carkner RD, Eifert C, Brosnan MJ, Conklin DS. An RNA Interference Screen Identifies Metabolic Regulators NR1D1 and PBP as Novel Survival Factors for Breast Cancer Cells with the ERBB2 Signature. Cancer Research. 2010;70:1783–1792. doi: 10.1158/0008-5472.CAN-09-1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berridge MV, Herst PM, Tan AS. Tetrazolium dyes as tools in cell biology: new insights into their cellular reduction. Biotechnology annual review. 2005;11:127–152. doi: 10.1016/S1387-2656(05)11004-7. [DOI] [PubMed] [Google Scholar]
- 17.Leif RC, Stein JH, Zucker RM. A short history of the initial application of anti-5-BrdU to the detection and measurement of S phase. Cytometry Part A : the journal of the International Society for Analytical Cytology. 2004;58:45–52. doi: 10.1002/cyto.a.20012. [DOI] [PubMed] [Google Scholar]
- 18.Crumbley C, Wang Y, Kojetin DJ, Burris TP. Characterization of the core mammalian clock component, NPAS2, as a REV-ERBalpha/RORalpha target gene. Journal of Biological Chemistry. 2010;285:35386–35392. doi: 10.1074/jbc.M110.129288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stayrook KR, Rogers PM, Savkur RS, Wang Y, Su C, Varga G, Bu X, Wei T, Nagpal S, Liu XS, Burris TP. Regulation of human 3alpha-hydroxysteroid dehydrogenase (AKR1C4) expression by the liver X receptor alpha. Mol Pharmacol. 2008;73:607–612. doi: 10.1124/mol.107.039099. [DOI] [PubMed] [Google Scholar]
- 20.Zientek-Targosz H, Kunnev D, Hawthorn L, Venkov M, Matsui SI, Cheney RT, Ionov Y. Transformation of MCF-10A cells by random mutagenesis with frameshift mutagen ICR191: A model for identifying candidate breast-tumor suppressors. Mol Cancer. 2008;7 doi: 10.1186/1476-4598-7-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massague J, Pavletich NP. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature. 1995;376:313–320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- 22.Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin A is required at two points in the human cell cycle. The EMBO journal. 1992;11:961–971. doi: 10.1002/j.1460-2075.1992.tb05135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coverley D, Laman H, Laskey RA. Distinct roles for cyclins E and A during DNA replication complex assembly and activation. Nat Cell Biol. 2002;4:523–528. doi: 10.1038/ncb813. [DOI] [PubMed] [Google Scholar]
- 24.Guillaumond F, Dardente H, Giguere V, Cermakian N. Differential control of Bmal1 circadian transcription by REV-ERB and ROR nuclear receptors. Journal of biological rhythms. 2005;20:391–403. doi: 10.1177/0748730405277232. [DOI] [PubMed] [Google Scholar]
- 25.Akashi M, Takumi T. The orphan nuclear receptor ROR alpha regulates circadian transcription of the mammalian core-clock Bmal1. Nature Structural & Molecular Biology. 2005;12:441–448. doi: 10.1038/nsmb925. [DOI] [PubMed] [Google Scholar]
- 26.Preitner N, Damiola F, Molina LL, Zakany J, Duboule D, Albrecht U, Schibler U. The orphan nuclear receptor REV-ERB alpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002;110:251–260. doi: 10.1016/s0092-8674(02)00825-5. [DOI] [PubMed] [Google Scholar]
- 27.Crumbley C, Burris TP. Direct regulation of CLOCK expression by REV-ERB. PLoS One. 2011;6:e17290. doi: 10.1371/journal.pone.0017290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schernhammer ES, Laden F, Speizer FE, Willett WC, Hunter DJ, Kawachi I, Colditz GA. Rotating night shifts and risk of breast cancer in women participating in the nurses’ health study. J Natl Cancer Inst. 2001;93:1563–1568. doi: 10.1093/jnci/93.20.1563. [DOI] [PubMed] [Google Scholar]
- 29.Schernhammer ES, Laden F, Speizer FE, Willett WC, Hunter DJ, Kawachi I, Fuchs CS, Colditz GA. Night-shift work and risk of colorectal cancer in the Nurses’ Health Study. J Natl Cancer Inst. 2003;95:825–828. doi: 10.1093/jnci/95.11.825. [DOI] [PubMed] [Google Scholar]
- 30.Megdal SP, Kroenke CH, Laden F, Pukkala E, Schernhammer ES. Night work and breast cancer risk: A systematic review and meta-analysis. Eur J Cancer. 2005;41:2023–2032. doi: 10.1016/j.ejca.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 31.Hansen J. Increased breast cancer risk among women who work predominantly at night. Epidemiology. 2001;12:74–77. doi: 10.1097/00001648-200101000-00013. [DOI] [PubMed] [Google Scholar]
- 32.Straif K, Baan R, Grosse Y, Secretan B, El Ghissassi F, Bouvard V, Altieri A, Benbrahim-Tallaa L, Cogliano V, Monograph WHOIARC. Carcinogenicity of shift-work, painting, and fire-fighting. Lancet Oncology. 2007;8:1065–1066. doi: 10.1016/S1470-2045(07)70373-X. [DOI] [PubMed] [Google Scholar]
- 33.Filipski E, Delaunay F, King VM, Wu MW, Claustrat B, Grechez-Cassiau A, Guettier C, Hastings MH, Francis L. Effects of chronic jet lag on tumor progression in mice. Cancer Research. 2004;64:7879–7885. doi: 10.1158/0008-5472.CAN-04-0674. [DOI] [PubMed] [Google Scholar]
- 34.Shah PN, Mhatre MC, Kothari LS. EFFECT OF MELATONIN ON MAMMARY CARCINOGENESIS IN INTACT AND PINEALECTOMIZED RATS IN VARYING PHOTOPERIODS. Cancer Research. 1984;44:3403–3407. [PubMed] [Google Scholar]
- 35.Filipski E, King VM, Li XM, Granda TG, Mormont MC, Liu XH, Claustrat B, Hastings MH, Levi F. Host circadian clock as a control point in tumor progression. J Natl Cancer Inst. 2002;94:690–697. doi: 10.1093/jnci/94.9.690. [DOI] [PubMed] [Google Scholar]
- 36.van den Heiligenberg S, Depres-Brummer P, Barbason H, Claustrat B, Reynes M, Levi F. The tumor promoting effect of constant light exposure on diethylnitrosamine-induced hepatocarcinogenesis in rats. Life Sciences. 1999;64:2523–2534. doi: 10.1016/s0024-3205(99)00210-6. [DOI] [PubMed] [Google Scholar]
- 37.Lin YM, Chang JH, Yeh KT, Yang MY, Li TC, Lin SF, Su WW, Chang JG. Disturbance of Circadian Gene Expression in Hepatocellular Carcinoma. Mol Carcinog. 2008;47:925–933. doi: 10.1002/mc.20446. [DOI] [PubMed] [Google Scholar]
- 38.Kuo SJ, Chen ST, Yeh KT, Hou MF, Chang YS, Hsu NC, Chang JG. Disturbance of circadian gene expression in breast cancer. Virchows Archiv. 2009;454:467–474. doi: 10.1007/s00428-009-0761-7. [DOI] [PubMed] [Google Scholar]
- 39.Chen ST, Choo KB, Hou MF, Yeh KT, Kuo SJ, Chang JG. Deregulated expression of the PER1, PER2 and PER3 genes in breast cancers. Carcinogenesis. 2005;26:1241–1246. doi: 10.1093/carcin/bgi075. [DOI] [PubMed] [Google Scholar]
- 40.Yang XM, Wood PA, Oh EY, Du-Quiton J, Ansell CM, Hrushesky WJM. Down regulation of circadian clock gene Period 2 accelerates breast cancer growth by altering its daily growth rhythm. Breast Cancer Research and Treatment. 2009;117:423–431. doi: 10.1007/s10549-008-0133-z. [DOI] [PubMed] [Google Scholar]
- 41.Wang J, Lazar MA. Bifunctional Role of Rev-erb{alpha} in Adipocyte Differentiation. Mol Cell Biol. 2008;28:2213–2220. doi: 10.1128/MCB.01608-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Mei C, Ercolani L, Parodi C, Veronesi M, Vecchio CL, Bottegoni G, Torrente E, Scarpelli R, Marotta R, Ruffili R, Mattioli M, Reggiani A, Wade M, Grimaldi B. Dual inhibition of REV-ERBbeta and autophagy as a novel pharmacological approach to induce cytotoxicity in cancer cells. Oncogene. 2014 doi: 10.1038/onc.2014.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sitaula S, Billon C, Kamenecka TM, Solt LA, Burris TP. Suppression of Atherosclerosis by Synthetic REV-ERB Agonist. Biochem Biophys Res Commun. 2015 doi: 10.1016/j.bbrc.2015.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]