

Summary
Neurotrophins are important for neuronal health and function. Here, statins, inhibitors of HMG-CoA reductase and cholesterol lowering drugs, were found to stimulate expression of neurotrophins in brain cells independent of the mevalonate pathway. Time-resolved fluorescence resonance energy transfer (FRET) analyses, computer-derived simulation, site-directed mutagenesis, thermal shift assay, and de novo binding followed by electrospray ionization tandem mass spectrometry (ESI-MS) demonstrates that statins serve as ligands of PPARα and that Leu331 and Tyr 334 residues of PPARα are important for statin binding. Upon binding, statins upregulate neurotrophins via PPARα-mediated transcriptional activation of cAMP-response element binding protein (CREB). Accordingly, simvastatin increases CREB and brain-derived neurotrophic factor (BDNF) in the hippocampus of Ppara null mice receiving full-length lentiviral PPARα, but not L331M/Y334D statin-binding domain-mutated lentiviral PPARα. This study identifies statins as ligands of PPARα, describes neurotrophic function of statins via the PPARα-CREB pathway, and analyzes the importance of PPARα in the therapeutic success of simvastatin in an animal model of Alzheimer's disease.
Graphical abstract
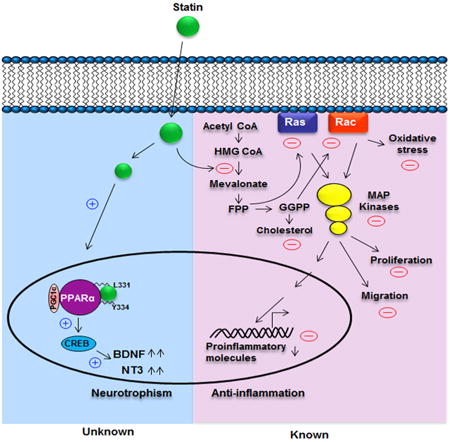
Introduction
Brain-derived neurotrophic factor (BDNF) and NT-3 are neurotrophins belonging to the NGF family of neuronal growth factors. BDNF infusion in the substantia nigra of rodent brain protects dopaminergic neurons, suggesting therapeutic prospects for Parkinson's disease. Moreover, the injection of BDNF in hippocampus and amygdaloid regions leads to the protection of cholinergic neurons (Morse et al., 1993) and improvement in cognitive impairment (Nagahara et al., 2009), implicating BDNF in Alzheimer's disease. BDNF is also capable of repairing spiny striatal interneurons in Huntington's disease (Kells et al., 2004) and remyelinating the lower motor neurons (Stadelmann et al., 2002) in multiple sclerosis. Therefore, BDNF promotes both structural and functional integrity of neurons to alleviate different neurological disorders. Similarly, NT-3 protects damaged neurons, stimulates neurogenesis and restores neuronal functions in the CNS affected by neurodegenerative diseases (Abe, 2000; Cheng and Mattson, 1994). However, the mechanisms by which the production of these neurotrophins could be therapeutically increased in the CNS are poorly understood.
Although statins are cholesterol-lowering drugs, lovastatin inhibits the activation of NF-κB and the expression of proinflammatory molecules in brain cells via modulation of the mevalonate pathway, thus prompting investigation of the efficacy of statins as an anti-inflammatory and neuroprotective drug (Pahan et al., 1997). Therefore, in addition to cholesterol-lowering, statins are currently known to control inflammation, attenuate cell proliferation and cell migration, favor vasodilation, modulate adaptive immunity, and suppress oxidative stress via modulation of the mevalonate pathway (Pahan, 2006; Roy and Pahan, 2011). Here, we report that statins also exhibit a neurotrophic effect. Different statins upregulate BDNF and NT-3 in neurons, microglia, and astrocytes. Although most of the biological functions of statins depend on their ability to inhibit the mevalonate-cholesterol pathway, statins stimulate the expression of neurotrophins independently of this pathway. Interestingly, we have found that statins directly interact with two critical residues Leu331 and Tyr334 located in the ligand-binding domain of PPARα to regulate the transcription of CREB, leading to expression of neurotrophic molecules. Finally, we demonstrate that simvastatin increases BDNF and improves memory and learning in an animal model of Alzheimer's disease (AD) via PPARα.
Results
Different Statins Induce the Expression of BDNF and NT-3 in Primary Glia and Neurons
Astrocytes are the predominant cell type in the CNS and we first investigated the effect of time-dependent effect of simvastatin on BDNF expression in astrocytes. Although simvastatin-mediated increase in BDNF mRNA was not visible at 2 hr, marked upregulation was observed at 5 hr of stimulation (Figure S1A). Accordingly, mevastatin, simvastatin, and pravastatin dose-dependently upregulated the mRNA expression of BDNF and NT-3 in mouse primary astrocytes (Figures S1B and S1C) and microglia (Figures S1D and S1E). Consistently, our protein results show upregulation of BDNF and NT-3 in both astrocytes and microglia by ELISA (Figures S1F and S1G) and immunocytochemistry (Figures S1H and S1I). Similar to mouse astrocytes, mevastatin dose-dependently increased the mRNA (Figures S1J and S1K) and protein (Figure S1L) expression of BDNF in primary human astrocytes. Immunofluorescence analyses revealed that both astrocytes (Figure S1M) and microglia (Figure S1N) in the cortex of simvastatin-fed mice expressed higher levels of BDNF than the brain of saline-fed mice. These results demonstrate that statins are capable of stimulating BDNF and NT-3 in glial cells in culture as well as in vivo in the brain. Earlier, Wu et al. (2008) showed increase in BDNF in vivo in the brain by simvastatin.
While astrocytes and microglia produce neurotrophic factors under neurodegenerative conditions (Kerschensteiner et al., 1999; Roy et al., 2007), neurons are major contributors of neurotrophic factors in normal brain (Maisonpierre et al., 1990). Similar to glia, the different statins stimulated mRNA (Figures S1O and S1P) and protein expression (Figures S1Q and S1R) of neurotrophins in mouse cortical neurons. To further confirm, we knocked down the BDNF gene by small interfering (siRNA) (Figures S1S and S1T) and found that siRNA knock down of BDNF decreased the production of BDNF protein in simvastatin-treated cortical neurons (Figure S1U). Furthermore, glutamate significantly induced the production of BDNF in neurons (Figure S1U). Similar to mouse cells, different statins also dose-dependently stimulated the expression of neurotrophins in primary human neurons (Figures S1V and S1W). Accordingly, oral administration of simvastatin upregulated the neuronal expression of BDNF in the cortex compared with vehicle-fed mice (Figure S1X). Therefore, statins are also able to stimulate the expression of neurotrophic factors in neurons.
Statin-Induced Expression of BDNF and NT-3 Is Independent of the Mevalonate Pathway
Next, we investigated the mechanism by which statins might up-regulate the expression of neurotrophins in glia. Since statins selectively inhibit HMG-CoA reductase, most of the known biological functions of statins depend on their ability to modulate the mevalonate pathway (Endo et al., 1976; Pahan et al., 1997). However, pretreatment with increasing doses of mevalonate and farnesyl pyrophosphate (FPP) failed to abrogate the statin-induced mRNA expression of neurotrophins in astrocytes (Figures 1A and 1B), suggesting that statins upregulate the expression of neurotrophic factors independently of mevalonate metabolites. By contrast, mevalonate, FPP, and geranylgeranyl pyrophosphate (GPP) dose-dependently abrogated simvastatin-mediated inhibition of iNOS in IL-1β-stimulated astrocytes (Figures 1C and 1D). Moreover, the selective inhibition of small G proteins by dominant-negative mutants of p21ras (ΔRas) and p21rac (ΔRac) failed to stimulate the mRNA expression of BDNF (Figure 1E). On the other hand, both ΔRas and ΔRac inhibited the IL-1β-induced mRNA expression of iNOS in astrocytes (Figure 1F). Consistent with this finding, while the siRNA-mediated knockdown of p21ras failed to increase the expression of BDNF in astrocytes (Figure 1G), it downregulated iNOS protein (Figures 1H–1I) expression. Farnesyl pyrophosphate transferase (FPT) transfers the farnesyl group to p21ras, resulting in membrane attachment of p21ras and its activation. It was previously reported that FPT inhibitor (FPTi) inhibited the downstream activation of various inflammatory pathways such as NF-κB (Pahan et al., 1997). Interestingly, FPTi had no stimulatory effect on the mRNA expression of BDNF in astrocytes (Figure 1J), suggesting that the farnesylation-p21ras pathway is not involved in the upregulation of neurotrophic factors. Similarly, geranylgeranyl transferase transfers the geranylgeranyl group to p21rac for its membrane attachment and activation. Similar to FPTi, geranyl-geranyl transferase inhibitor (GGTi) also failed to induce the mRNA expression of BDNF (Figure 1J), indicating that the geranylgeranylation-p21rac pathway is also not involved in the stimulation of neurotrophic factors. However, as expected, both FPTi and GGTi suppressed the IL-1β-induced expression of iNOS in astrocytes (Figures 1K and 1L). Taken together, these data suggest that statins employ two independent signaling pathways for their neurotrophic and anti-inflammatory effects.
Figure 1. Anti-inflammatory, but Not Neurotrophic, Activity of Simvastatin Depends on Mevalonate Metabolites and Isoprenylation of p21ras and p21rac.
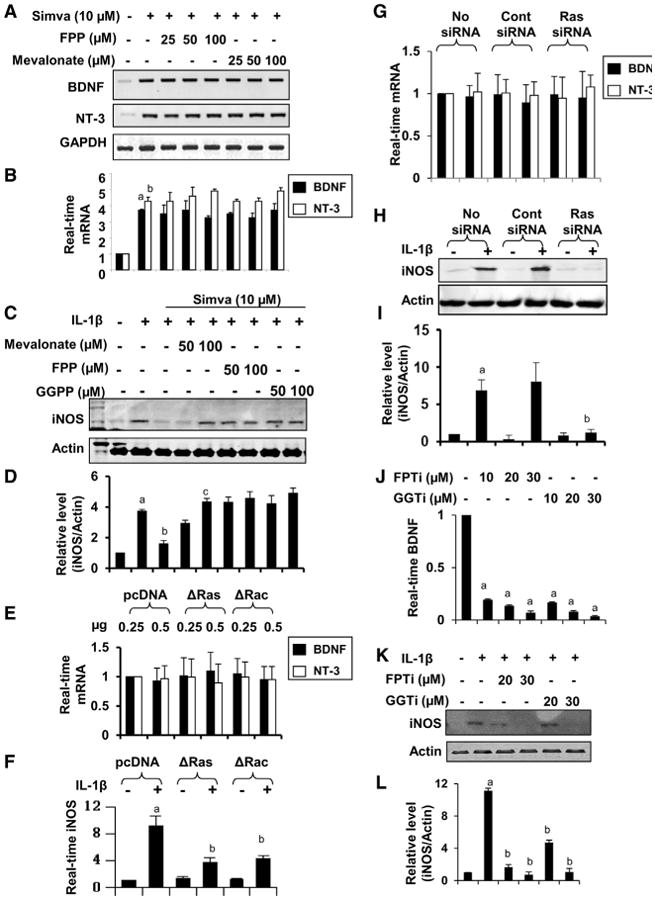
(A and B) Mouse astrocytes pre-treated with different doses of farnesyl pyrophosphate (FPP) and mevalonate for 1 hr were stimulated with 10 μM simvastatin for another 5 hr under serum-free condition followed by mRNA analysis of neurotrophins by semi-quantitative RT-PCR (A) and real-time PCR (B). Results represent three independent analyses. ap < 0.001 versus control-BDNF; bp < 0.001 versus control-NT3.
(C) Astrocytes pre-treated with mevalonate, farnesyl pyrophosphate (FPP), and geranylgeranyl pyrophosphate (GGPP) in the presence of 10 μM simvastatin for 6 hr were stimulated with IL-1β (20 ng/ml) for another 12 hr followed by analysis of iNOS by western blot.
(D) Bands were scanned and presented as relative to control. ap < 0.001 versus control; bp < 0.001 versus IL-1β; cp < 0.001 versus IL-1β + Simva.
(E) Astrocytes were transfected with pcDNA3 (empty vector) and dominant-negative mutants of p21ras (ΔRas) and p21rac (ΔRac). After a 24-hr transfection, cells were incubated with serum-free media for 6 hr followed by analysis of mRNA expression of BDNF and NT-3 by real-time PCR.
(F) Similarly, 24 hr after transfection, cells were stimulated with IL-1β (20 ng/ml) for 6 hr followed by monitoring the expression of iNOS mRNA by real-time PCR.
(G) After transfection with control and p21rassiRNA, cells were incubated with serum-free media for 6 hr followed by monitoring Bdnf and Nt3 mRNAs by real-time PCR.
(H) Similarly, after transfection, cells were stimulated with IL-1β for 12 hr followed by analysis of iNOS protein by western blot.
(I) Bands were scanned and presented as relative to control. ap < 0.001 versus control; bp < 0.001 versus control-siRNA-IL-1β.
(J) Astrocytes were incubated with farnesyl phosphotransferase inhibitor (FPTi) and geranylgeranyl phosphotransferase inhibitor (GGTi) for 6hr followed by analysis of Bdnf mRNA by realtime PCR.
(K) Astrocytes pretreated with FPTi and GGTi for 1 hr were stimulated with IL-1β followed by monitoring iNOS protein after a 12-hr incubation.
(L) Bands were scanned and presented as relative to control. ap < 0.001 versus control; bp < 0.001 versus IL-1β. Results represent three independent analyses. See also Figure S1.
The Role of PPARα in Statin-Induced Expression of Neurotrophic Factors in Glia
Simvastatin inhibits IL-1β-induced mRNA (Figures 2A and 2B) and protein (Figures 2C and 2D) expression of iNOS equivalently in wild-type (WT) and Ppara null astrocytes, suggesting that simvastatin does not require PPARα to suppress iNOS. We next examined whether PPARα was involved in statin-mediated expression of neurotrophic factors in glia. Interestingly, simvastatin dose-dependently stimulated the mRNA expression of BDNF and NT-3 in WT, but not in Ppara null, microglia (Figures 2E and 2F) and astrocytes (Figures 2G and 2H). Similarly, simvastatin and pravastatin stimulated the protein expression of BDNF in WT, but not Ppara null, astrocytes (Figures 2I–2K) and neurons (Figures S2A and S2B). Together, these results suggest that PPARα is involved in the statin-mediated upregulation of neurotrophic factors in glia and neurons.
Figure 2. PPARα Is Involved in Neurotrophic, but Not Anti-inflammatory, Properties of Statins.
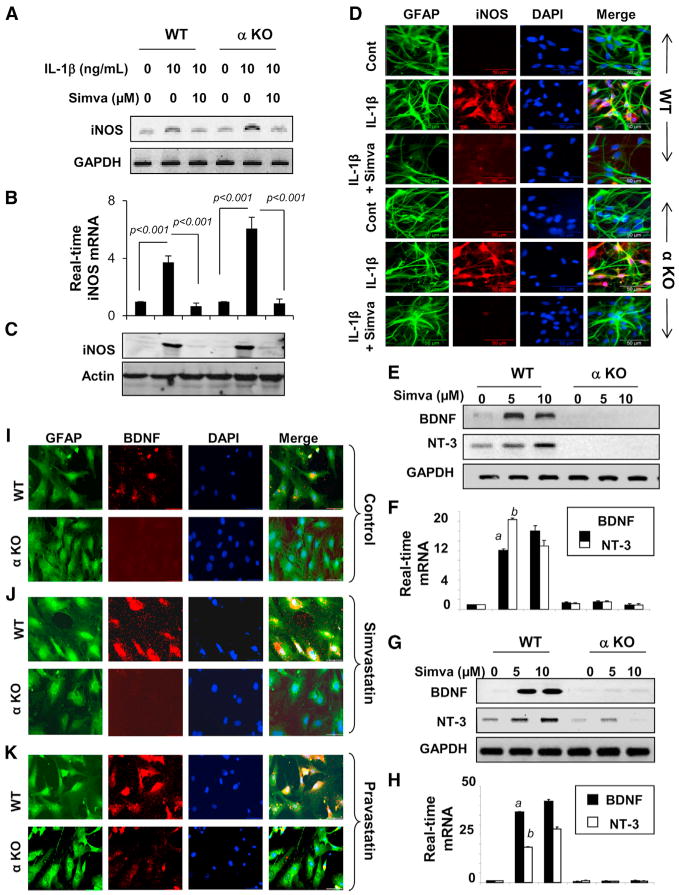
(A–H) Astrocytes isolated from WT and Ppara null (α KO) mice were treated with simvastatin for 6 hr followed by stimulation with IL-1β. After 6 hr, the mRNA expression of iNOS was monitored by RT-PCR (A) and real-time PCR (B). Results are mean ± SD of three independent experiments. After 24 hr, the protein level of iNOS was monitored by western blot (C) and immunofluorescence (D). Microglia (E and F) and astrocytes (G and H) isolated from WT and α KO mice were treated with simvastatin. After 6 hr, cells were analyzed for mRNA encoding BDNF and NT-3 by RT-PCR (E and G) and real-time PCR (F and H). ap < 0.001 versus control-BDNF; bp < 0.001 versus control-NT-3.
(I–K) WT and α KO primary astrocytes were incubated with 10 μM simvastatin (J) and pravastatin (K) under serum-free conditions. After 24 hr, the BDNF level was analyzed by double-label immunofluorescence. Results represent three independent experiments.
See also Figures S2 and S3.
PPARβ and PPARγ Are Not Involved in Statin-Induced Expression of Neurotrophins
Simvastatin was found to stimulate the expression of PPARα, PPARβ, and PPARγ in astrocytes (Figure S2C). Therefore, we next examined the role of PPARβ and PPARγ in statin-induced expression of BDNF and NT-3. Although activation of PPARγ has been shown to induce the expression of BDNF (Jin et al., 2013; Kariharan et al., 2015), antisense knockdown of PPARγ and PPARβ failed to inhibit simvastatin-stimulated expression of BDNF and NT-3 in astrocytes (Figure S2D), suggesting that neither PPARβ nor PPARγ is involved in simvastatin-mediated upregulation of neurotrophic factors. However, it is PPARα, but not PPARβ, that is involved in statin-mediated upregulation of neurotrophic factors, as simvastatin stimulated BDNF expression in Pparb null astrocytes (Figure S2E). The direct involvement of PPARα in the statin-stimulated expression of neurotrophins was further confirmed when simvastatin stimulated the expression of BDNF in PPARα cDNA-, but not empty vector-transfected Ppara null astrocytes (Figures S2F and S2G). These data suggest that statins require only PPARα for the induction of BDNF and NT-3 in glial cells. Consistent to the role of PPARα in the induction of neurotrophins, prototype ligands of PPARα (WY14643 and gemfibrozil) induced the expression of BDNF and NT-3 in primary astrocytes (Figure S2H). Similar to simvastatin, WY14643 and gemfibrozil were also unable to induce the expression of BDNF and NT-3 in Ppara null astrocytes and PPARα overexpression restored the ability of these ligands to induce CREB and neurotrophins in Ppara null astrocytes (Figure S2I).
Statins Stimulate the Expression of BDNF and NT-3 In Vivo in Mouse Brain via PPARα
Next, we examined whether statins required PPARα (Figure S3A) for the upregulation of neurotrophins in vivo in the brain. As evident from RT-PCR (Figure S3B) real-time PCR (Figure S3C), the mRNA expression of both BDNF and NT-3 was significantly higher in WT brain than Ppara null brain. We have shown that both simvastatin and pravastatin are able to cross the blood-brain barrier (BBB) with variable efficiencies (Ghosh et al., 2009). Oral administration of both pravastatin (Figures S3D and S3E) and simvastatin (Figures S3F and S3G) significantly stimulated the expression of BDNF and NT-3 in vivo in the midbrain of WT, but not Ppara null, mice. However, in accordance with their BBB permeabilities, simvastatin was more effective than pravastatin in stimulating the expression of neurotrophins in vivo in the brain.
Statins Interact with the Ligand-Binding Domain of PPARα
Next, we were prompted to study how statins employ PPARα in the expression of neurotrophic factors. It is not known whether statins are ligands of PPARα. In order to determine whether simvastatin directly interacts with the ligand-binding domain (LBD) of PPARα, simvastatin-treated astrocytic nuclear extracts were pulled down by the PPARα-LBD followed by electrospray ionization tandem mass spectrometry (ESI-MS) analysis. Interestingly, we detected the peak for simvastatin itself in the PPARα-LBD pull-down fraction of simvastatin-treated (Figures 3B and 3D), but not control (Figures 3A and 3C), astrocytes. Time-resolved fluorescence resonance energy transfer (TR-FRET) is a widely accepted technology for monitoring the physical interaction between protein and ligand spaced at a distance as low as 5 μm. We adopted TR-FRET for analyzing the interaction between PPARα and different statins (Figure 3E). Our fluorescence dose-response curve clearly indicated that simvastatin (Figure 3F), mevastatin (Figure 3G), and pravastatin (Figure 3H) dose-dependently induced the TR-FRET signal from PPARα-PGC1α complex, suggesting that statins serve as ligand of PPARα. For comparison, we also used atorvastatin and rosuvastatin and found that the signal strength of emitted fluorescence was the highest for simvastatin among simvastatin, mevastatin, pravastatin, atorvastatin, and rosuvastatin (Figure 3I), suggesting that simvastatin is the strongest ligand of PPARα among different statins. Our observation was further corroborated once we compared EC50 value of different statins after 30 min of interaction with PPARα. The EC50 value of simvastatin (4.26) was lower than pravastatin, mevastatin, atorvastatin, and rosuvastatin further suggesting that simvastatin is the strongest ligand of PPARα among different statins. GW7642, a well-known PPARα agonist, displayed similar dose-response curve (Figure 3F) under similar condition with much lower EC50 value (1.11), indicating that the binding affinity of statins to PPARα is still lower than its prototypic ligands.
Figure 3. A High Throughput Analysis to Study the Interaction between PPARα and Statin.
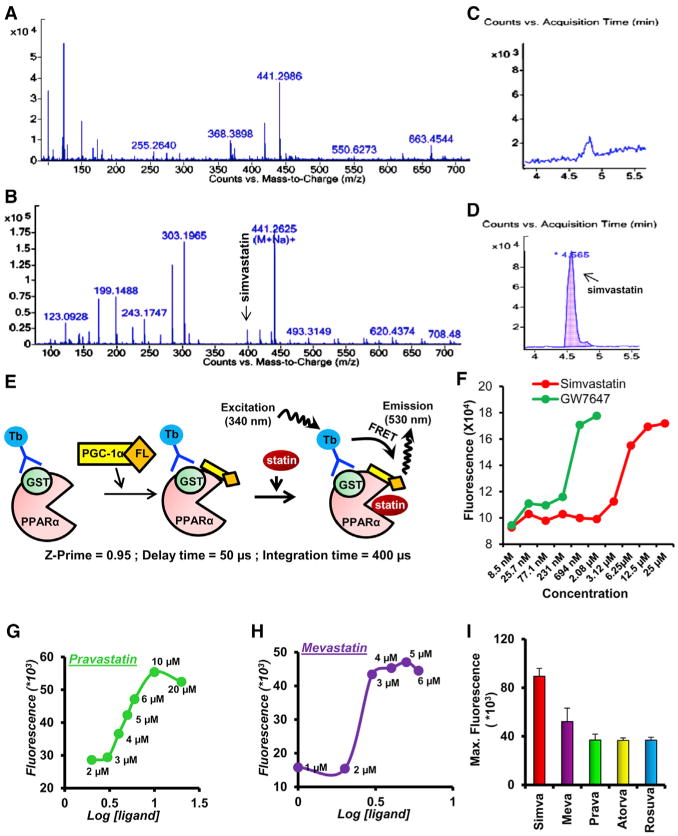
(A–D) PPARα-LBD affinity-purified nuclear fraction extracted from control (A) and simvastatin-treated (B) primary astrocytes was analyzed by electrospray ionization tandem mass spectrometry. Counts versus retention time shows the peak of simvastatin at 4.56 min in simvastatin-treated, but not control, sample. The magnified view of counts versus mass-to-charge ratio clearly shows the peak of simvastatin in the simvastatin-treated (D), but not control (C), sample.
(E) To identify stain group of drugs as ligand of PPARα, time-resolved fluorescence energy transfer (TR-FRET) technology was adopted. Successful binding of statin with PPARα would transfer fluorescence energy from Terbium (Tb)-tagged anti-GST antibody to Fluorescein (FL)-tagged PGC-1α co-activator. The optimum level of emitted fluorescence was measured at 400 μ-second integration time and 40 μ-second delay time set in Molecular Device Analyst instrument.
(F–H) Fluorescein emission was recorded with increasing doses of (F) simvastatin, (G) pravastatin, and (H) mevastatin.
(I) TR-FRET was also performed for atorvastatin and rosuvastatin and maximum emitted fluorescence was compared among different statins. Results represent three independent analysis.
To characterize the interaction between simvastatin and PPARα in a molecular level, we carried out in silico docking studies of simvastatin with PPARα-LBD. These studies generated a reasonable docked pose of the simvastatin molecule in the ligand-binding pocket (Figure 4A). The docked pose of simvastatin showed two potential hydrogen bonds between the statin ligand and two active-site residues, Tyr334 (Y334) and Leu331 (L331) (Figure 4B), of the PPARα-LBD. After obtaining a suitable docked pose of the simvastatin, we attempted to compute the apparent binding energy of the ligand along with the strain energy. Using the MM-GBSA approach, we found the binding energy of simvastatin to be –36.0 Kcal/mol with minimal strain energy of 0.71 Kcal/mol, suggesting a strong interaction between simvastatin and PPARα. Imposing the most stringent docking protocols, a reasonable docked pose of simvastatin was obtained for PPARα, with a total score of 8.27, a polar score of 1.54, and a crash score of –0.71. Interestingly, by applying similar docking protocols, we failed to obtain any dock pose of simvastatin for both PPARβ and PPARγ, suggesting that the interaction of simvastatin with PPARα-LBD is specific and not possible with other PPAR isoforms. However, in silico modeling of protein-ligand interaction is hypothetical and requires rigorous experimental analysis for further validation.
Figure 4. Proteomic Analysis of Interaction between PPARα and Simvastatin.
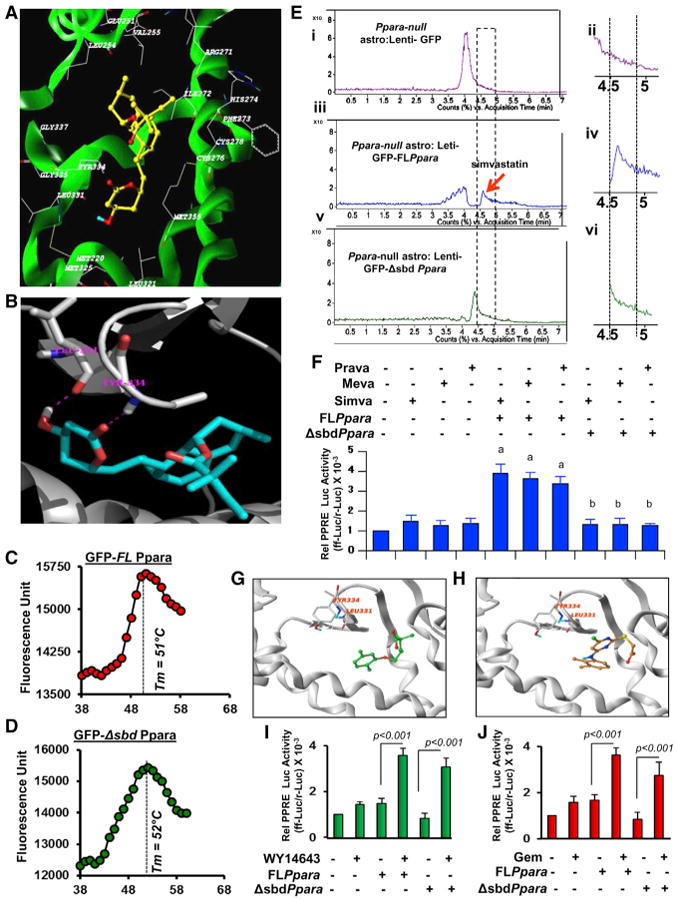
(A) Ribbon representations of the docked complex of PPARα (3VI8.pdb, X-ray, green) and simvastatin show the distribution of amino acids within a 5.00-Å distance of simvastatin.
(B) Magnified view of the docking site shows the interaction between simvastatin and Leu331 and Tyr334.
(C and D) Thermal shift assay of (C) full-length (GFP-FLPpara) and (D) statin-binding domain mutated (GFP-ΔsbdPpara) PPARα was performed.
(E) De novo binding assay of simvastatin where mouse astrocytes transduced with FLPpara and ΔsbdPpara constructs were treated with simvastatin for 2 hr, nuclear extracts were isolated, passed through GFP column for affinity-purification, and then analyzed by ESI-MS. Representative counts versus acquisition time ratio of simvastatin analyzed in the nuclear extracts of GFP-transduced (i and ii), GFP-FLPpara-transduced (iii and iv), and GFP-ΔsbdPpara-transduced (v and vi) astroglial cells.
(F) PPRE-driven luciferase activity in FLPpara and ΔsbdPpara-transduced Ppara null astrocytes after treatment with simvastatin (top), pravastatin (middle), and mevastatin (bottom). Results are mean ± SD of three independent experiments. ap <0.001 versus control; bp < 0.001 versus FLPpara.(G and H) Docked poses of gemfibrozil (G) and WY14673 (H) in PPARα ligand binding core.
(I and J) PPRE-driven luciferase activity in FLPpara and ΔsbdPpara-transduced Ppara null astrocytes after treatment with WY14643 (I) and gemfibrozil (J). Results are mean ± SD of three independent experiments.
Therefore, we performed lentivirus-mediated de novo expression studies, where we overexpressed wild-type (GFP-FLPpara) and statin-binding domain (SBD) mutated PPARα (GFP-ΔsbdPpara) gene in Ppara null astrocytes followed by the binding analyses with simvastatin. Briefly, we performed site-directed mutagenesis in mouse PPARα gene where we replaced Leu331 residue with methionine (L331M) and Tyr334 residue with aspartate (Y334D). After that, we cloned the entire mouse GFP-Ppara gene and L331M/Y334D Ppara (GFP-ΔsbdPpara) gene in the pLenti6∕V5-TOPO lentiviral expression vector, packaged in lentivirus particle with the help of HEK293FT cells, transduced mouse astrocytes with lentiviral particles for 48 hr, purified full-length (GFP-FLPPARα) and mutated PPARα (GFP-ΔsbdPPARα) proteins in a GFP-affinity column, and finally performed thermal shift assay in order to analyze their conformational stability. Both full-length (Figure 4C) and mutated (Figure 4D) proteins displayed similar pattern of thermal shift with equivalent melting temperature (Tm), suggesting that mutations in L331 and Y334 residues did not alter the conformational stability of PPARα. In another experiment, Ppara null astrocytes transduced with different lentiviral PPARα constructs were incubated with simvastatin for 2 hr and then performed ESI-MS analyses in the GFP-affinity purified nuclear fraction of astrocytes. Interestingly, we observed a very specific peak of simvastatin in the nuclear extract of GFP-PPARα (Figures 4Ei and 4Eii), but neither in empty GFP vector-transduced (Figures 4Eiii and 4Eiv) nor GFP-ΔsbdPpara-transduced (Figures 4Ev and 4Evi) nuclear extracts of astrocytes, suggesting that Leu331 and Tyr334 residues are indeed essential for binding with statin. Next, we examined if these two amino acids were required for statin-mediated activation of PPARα. We measured PPRE-driven luciferase activity in both FLPpara and Δsbd Ppara-transduced Ppara null astrocytes after treatment of simvastatin, mevastatin, and pravastatin. Interestingly, we observed that different statins stimulated PPRE-luciferase activity in FLPpara-transduced, but not in ΔsbdPpara-transduced, astrocytes (Figure 4F), suggesting that statins require these two amino acids for the activation of PPARα.
Next, we investigated if WY14643 and gemfibrozil, classical ligands of PPARα, also required Leu331 and Tyr334 residues for the activation PPARα. Gemfibrozil (Figure 4G) and WY14643 (Figure 4H) were allowed to dock in the LBD of PPARα using the similar docking protocols used to dock simvastatin. We found that both the compounds docked very nicely with a good docked scores and with very little penalty scores. However, the binding poses of gemfibrozil (Figure 4G) and WY14673 (Figure 4H) were very different from that of simvastatin. Simvastatin showed potential hydrogen bonds with L331 and Y334 but gemfibrozil and WY14673 were ∼6 Å and ∼5 Å away from these residues, suggesting that these two known ligands of PPARα do not interact with L331 and Y334 residues. Consistently, gemfibrozil (Figure 4I) and WY14643 (Figure 4J) stimulated PPRE-luciferase activity in ΔsbdPpara-transduced Ppara null astrocytes almost similar to that found FLPpara-transduced cells, confirming that these two ligands do not require L331 and Y334 residues for the activation of PPARα.
Statins Increase CREB via PPARα-Mediated Transcriptional Regulation
Different statins (Figure 5A for mevastatin, pravastatin, and simvastatin; Figure 5B for atorvastatin and rosuvastatin) induced PPARα response element (PPRE)-driven reporter activity, revealing that different statins are capable of activating PPARα in astrocytes. However, detailed analysis of BDNF and NT-3 promoters showed no PPRE, ruling out the direct involvement of PPARα in the upregulation of neurotrophic factors. On the other hand, a number of CRE sequences were found in BDNF and NT-3 promoters. Accordingly, antisense knockdown of CREB suppressed the expression of CREB and inhibited the simvastatin-induced expression of BDNF and NT-3 in mouse astrocytes (Figure S4A). Similarly, we observed that CREB siRNA, but not control siRNA, inhibited simvastatin-induced expression of BDNF in astrocytes (Figure S4B). These results indicate the involvement of CREB in simvastatin-mediated upregulation of neurotrophic factors.
Figure 5. The Role of PPARα, PPARβ, and PPARγ in Transcriptional Regulation of CREB.
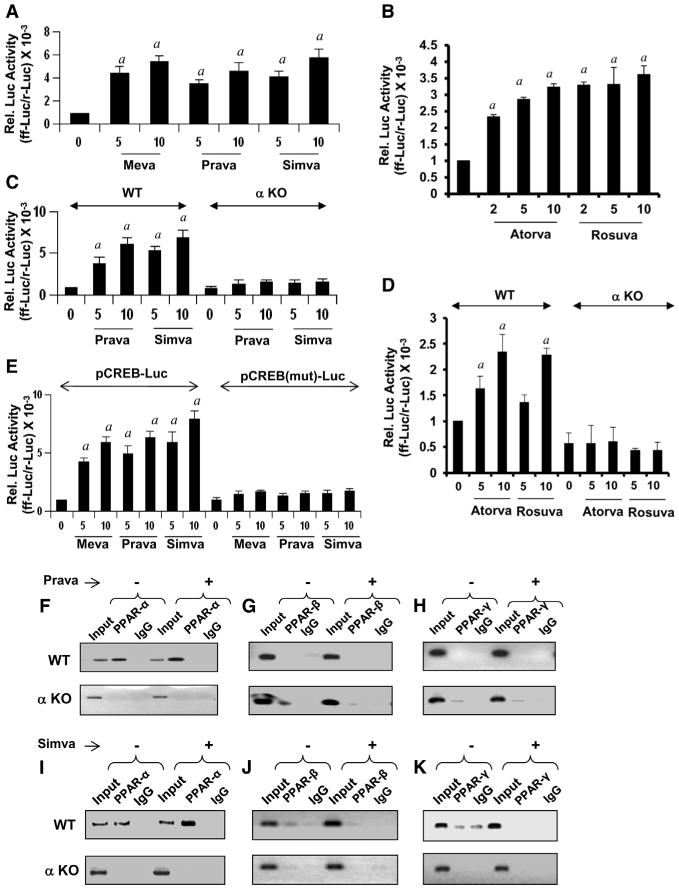
(A–E) Mouse astrocytes were transfected with PPRE luciferase and after a 24-hr transfection, cells were treated with mevastatin, pravastatin, simvastatin (A), atorvastatin, and rosuvastatin (B) for 4 hr, followed by the luciferase assay. Activation of WT CREB promoter by pravastatin, simvastatin (C), atorvastatin, and rosuvastatin (D) in astrocytes isolated from WT and Ppara null (αKO) mice. Activation of WT and mutated CREB promoter by mevastatin, pravastatin, and simvastatin in mouse astrocytes (E). Results are means ± SD of three independent experiments. ap < 0.001 versus control.
(F–H) The ChIP assay for PPARα (F), PPARβ (G) and PPARγ (H) in pravastatin-treated WT and αKO astrocytes.
(I–K) The ChIP assay for PPARα (I), PPARβ (J), and PPARγ (K) in simvastatin-treated WT and αKO astrocytes. Results represent three separate analyses.
See also Figures S3 and S5.
Although we did not find any PPRE in BDNF and NT-3 promoters, analysis of the CREB promoter revealed the presence of a conserved PPRE (–1,152 to –1,164 bp upstream of the start sequence) prompting us to study the involvement of PPARα in the transcriptional regulation of CREB. Different statins stimulated the expression of CREB mRNA in mouse astrocytes and microglia (Figure S4C). However, statins upregulated the expression of CREB mRNA in astrocytes and microglia isolated from WT, but not Ppara null, mice (Figure S4D). Immunofluorescence analysis also showed that simvastatin upregulated CREB protein in astrocytes isolated from WT, but not Ppara null, mice (Figure S4E). Similarly, simvastatin also upregulated the expression of CREB mRNA (Figure S4F) and protein (Figure S4G) in neurons isolated from WT, but not Ppara null, mice. A lower abundance of CREB in different parts of the brain of Ppara null animals than of WT and Pparb null animals further shows that PPARα, but not PPARβ, controls the expression of CREB in vivo in the brain (Figures S3H and S3I). Oral administration of simvastatin was also observed to stimulate the expression of CREB in vivo in the cortex of WT, but not Ppara null, mice (Figure S3J), indicating the role of PPARα in the statin-stimulated expression of CREB in the brain.
Next, we investigated whether reinstatement of PPARα helps statins to induce the expression of CREB in Ppara null cells. PPARα overexpression increased the basal level of CREB in Ppara null astrocytes (Figure S4H). Although simvastatin was unable to increase the mRNA expression of CREB in Ppara null neurons, we observed stimulation of CREB mRNA expression by simvastatin in Ppara null neurons after PPARα overexpression (Figure S4H). Immunocytochemical analyses also revealed that ectopic overexpression of PPARα in primary human neurons stimulated basal expression of CREB (Figure S4K) compared with untransfected (Figure S4I) or pcDNA-transfected (Figure S4J) cells and that simvastatin stimulation further increased the level of CREB in PPARα-overexpressed cells (Figure S4K). On the other hand, a dominant-negative mutant of human PPARα (ΔPPARα) abrogated the expression of CREB, and simvastatin also failed to stimulate the expression of CREB in ΔPPARα-expressed human neurons (Figure S4L).
To further analyze the role of PPARα in the transcription of Creb, we cloned the mouse Creb promoter and then performed site-directed mutagenesis to mutate the PPRE as described (Roy et al., 2013). Different statins markedly induced Creb promoter-driven luciferase activity in astrocytes isolated from WT, but not Ppara null, mice (Figure 5C for pravastatin and simvastatin; Figure 5D for atorvastatin and rosuvastatin). However, in WT astrocytes, statins remained unable to induce luciferase activity driven by a Creb promoter in which PPRE was mutated (Figure 5E). Next, we performed chromatin immunoprecipitation (ChIP) analysis in which the recruitment of PPARα to the Creb promoter was monitored in untreated and statin-treated astrocytes. After immunoprecipitation of chromatin fragments by antibodies against PPARα, we were able to amplify a 144-bp fragment encompassing the PPRE of the Creb promoter in astrocytes isolated from wild-type, but not Ppara null, mice (Figures 5F and 5I). On the other hand, no amplification product of the Creb promoter was observed in any of the immunoprecipitates obtained with PPARβ, PPARγ, or control IgG (Figures 5G, 5H, 5J, and 5K), suggesting that PPARα, but neither PPARβ nor PPARγ, is recruited to the PPRE of the Creb promoter in astrocytes. Both pravastatin (Figure 5F) and simvastatin (Figure 5I) increased the recruitment of PPARα, but neither PPARβ nor PPARγ (Figures 5G, 5H, 5J, and 5K), to the Creb promoter in astrocytes.
Evaluating the Role of Leu331 and Tyr334 of PPARα in the Statin-Induced Expression of BDNF in Cultured Astrocytes and In Vivo in Mouse Brain
Next, we examined if Leu331 and Tyr 334 residues of PPARα were required for statin-mediated upregulation of CREB and BDNF in the cultured astrocytes as well as in vivo in the brain. Mouse primary astrocytes isolated from Ppara null mice were transduced with EGFP-constructed lentiviral particles containing either FLPpara or ΔsbdPpara. Transduction efficiency was more than 90% for both the constructs (Figure 6A). Interestingly, simvastatin significantly upregulated the expression of CREB and BDNF (Figures 6B and 6C) in FLPpara-transduced astrocytes, but neither in ΔsbdPpara- nor empty vector-transduced astrocytes, which we further confirmed by BDNF ELISA from the astroglial supernatants (Figure 6D). Since BDNF is the major neurotrophic factor that controls the function of hippocampus, we targeted the hippocampus for lentiviral manipulation of the Ppara gene. GFP-FLPpara and GFP-ΔsbdPpara lentiviral particles were infused bilaterally in the pyramidal layer of the CA2 region and in the subgranular layer of the dentate gyrus (DG) of 6- to 8-week-old Ppara null mice as described earlier (Roy et al., 2013). Three weeks after the infusion, we observed a marked distribution of EGFP constructed PPARα in the entire pyramidal (CA1, CA2, and CA3) and DG region (Figure 6E), indicating the distribution efficiency of FLPpara and ΔsbdPpara in our proposed injection coordinates. Simvastatin feeding was started after 6 weeks of lentiviral injection and continued for 2 weeks. Consistent with cell culture results, our immunofluorescence and immunoblot analyses revealed that the bilateral infusion of lentiviral GFP-FLPpara, but neither empty vector nor GFP-ΔsbdPpara, strongly upregulated the expression of CREB (Figures 6G and 6H) and BDNF (Figures 6F–6I) in the hippocampus of simvastatin-fed mice, suggesting that both L331 and Y334 residues of PPARα are critical for the statin-mediated up-regulation of CREB and BDNF.
Figure 6. Lentiviral Manipulation of FLPpara and ΔsbdPpara in the Adult Brain Hippocampus and Its Role in Statin-Stimulated Expression of BDNF.
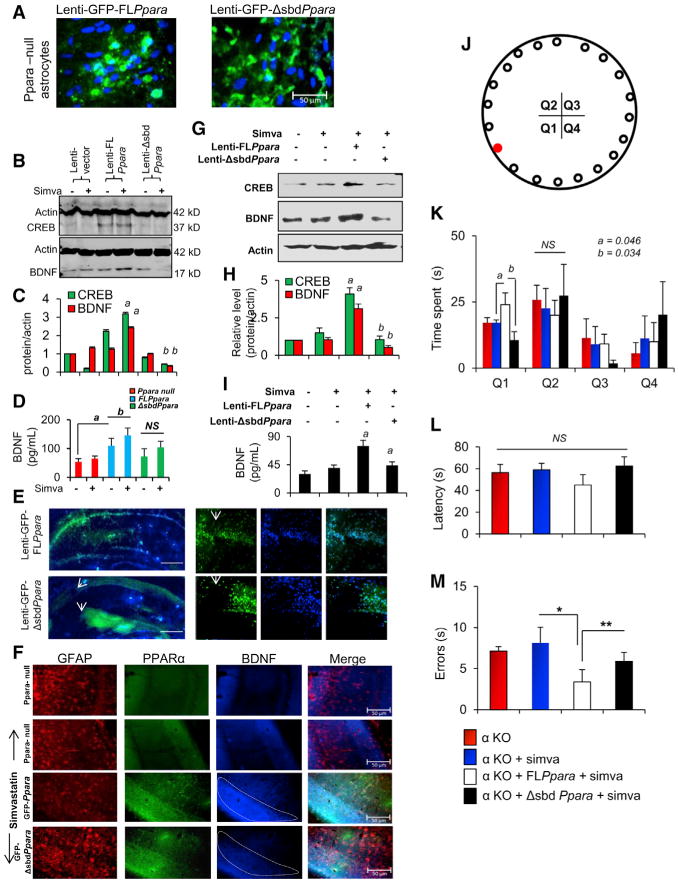
(A) Astrocytes isolated from Ppara null (αKO) mice were transduced with GFP-FLPpara and GFP-ΔsbdPpara to analyze the transduction efficiency of our plasmids.
(B) Expression of CREB and BDNF were checked by immunoblot analysis.
(C) Bands were scanned and presented as relative to control. Results are mean ± SD of three independent experiments. ap < 0.001 versus lenti-FLPpara control-CREB; bp < 0.001 versus lenti-FLPpara control-BDNF.
(D) BDNF expression was confirmed by ELISA from the respective supernatants. ap < 0.001 versus lenti-vector control; bp < 0.05 versus lenti-FLPpara control.
(E) Both of these constructs were injected bilaterally in the hippocampus of 6- to 8-week-old male C57BL/6 mice with coordinates of 2.54 mm AP axis, 1.30 mm ML (CA2 layer) and 1.80 mm ML (SGlayer of DG) axis, and 2.4 mm DV axis. Each animal received four injections with two injections perhemisphere. After 3 weeks, the distribution of lentivirus was detected by microscopic analysis of GFP. Injection sites were magnified and shown inside panels.
(F) After 2 weeks of simvastatin feeding, the expression of BDNF in GFAP-immunoreactive astrocytes were analyzed (green, GFP-PPARα; red, GFAP; blue, BDNF).
(G) CREB and BDNF expression were analyzed by immunoblot in the hippocampal extracts of GFP, GFP-FLPpara, and GFP-ΔsbdPpara animals.
(H) Bands were scanned and presented as relative to control. ap < 0.05 versus simvastatin; bp < 0.01 versus lenti-FLPpara-simvastatin.
(I) Expression of BDNF was confirmed by ELISA. ap < 0.01 versus simvastatin; bp < 0.01 versus lenti-FLPpara-simvastatin.
(J) Performances of statin-fed FLPpara and ΔsbdPpara animals (n = 5 per group) in Barnes maze were compared with statin-fed αKO mice. Target hole placed in first quadrant (Q1) is shown as red.
(K–M) Total time spent in all four quadrants (Q1–Q4), (L) latency and (M) errors were calculated. ap < 0.05 (= 0.041) versus statin-fed Ppara null and bp < 0.05 (= 0.037) versus statin-fed FLPpara. *p < 0.01 versus statin-fed αKO and **p < 0.05 (= 0.045) versus statin-fed ΔsbdPpara (0.0015) animals.
Next, we wanted to determine whether the simvastatin-mediated upregulation of BDNF production could improve hippocampus-dependent spatial learning and memory in these animals. Therefore, we performed Barnes maze analyses after oral administration of simvastatin (1 mg/kg body weight/day) to FLPpara and ΔsbdPpara animals for 2 weeks. Since these animals were all on the Ppara null background, they spent similar time before entering into the hole (Figure 6L). However, FLPpara mice spent more time in the target quadrant (Q1) after statin feeding when compared to statin-fed Ppara null mice (F3,28 = 2.87; p ≤ 0.05) or statin-fed ΔsbdPpara mice (F3,28 = 3.57; p ≤ 0.05) (Figures 6J and 6K). Moreover, the number of errors is significantly less in statin-fed FLPpara mice (Figure 6M) as compared to statin-fed ΔsbdPpara (F3,28 = 3.14; p ≤ 0.05) and statin-fed Ppara null mice (F3,28 = 13.07; p ≤ 0.01), suggesting that both L331 and Y334 residues of PPARα are crucial for the statin-induced hippocampus-dependent learning and memory.
Validating the PPARα-Mediated Neurotrophic Effect of Statin in FAD5X Mouse Model of AD
In order to analyze the therapeutic importance of PPARα in statin-mediated upregulation of BDNF, we used FAD5X (B6SJL-Tg(APPSwFlLon,PSEN1*M146L*L286V)6799Vas/J) transgenic mouse model of AD. We generated FAD5X mice in the homozygous Ppara null background (Figure 7A). Five-month-old non-Tg, FAD5X, Ppara null, and FAD5X/Ppara null mice were fed simvastatin (1 mg/kg body weight/day) for 2 weeks. Interestingly, we observed that simvastatin increased the astroglial expression of BDNF in cortex (Figures 7B–7E) of WT (Figure 7B) and FAD5X animals (Figure 7D), but not in Ppara null (Figure 7C) and FAD5X/Ppara null mice (Figure 7E), suggesting that PPARα regulates the expression of BDNF during AD-like pathology. Similarly, our immunoblot (Figures 7F and 7G) and ELISA (Figure 7H) analyses revealed that both WT and FAD5X, but neither Ppara null nor FAD5X/Ppara null, mice showed upregulated expression of BDNF after simvastatin treatment, suggesting the therapeutic prospect of PPARα in AD. Next, we validated the effect of statin in the process of learning and memory in these animals by analyzing their performance in Barnes maze. Upon simvastatin treatment, both wild-type and FAD5X mice exhibited significant improvement in memory performance on Barnes maze test as shown by errors (Figure 7I) (F1,14 = 1.4 [>Fc = 0.34] between unfed and simvastatin-fed WT; F1,14 = 1 [>Fc = 0.26] between unfed and simvastatin-fed FAD5X) and latency (Figure 7J) (F1,14 = 3.59 [>Fc = 1.72] between unfed and simvastatin-fed WT; F1,14 = 3.78 [>Fc = 2.87] between unfed and simvastatin-fed FAD5X). However, Ppara null and FAD5X/Ppara null mice showed no improvement after simvastatin feeding (Figures 7I and 7J). Simvastatin treatment did not significantly alter body weight (Figure 7K), stereotypy (Figure 7L), total distance (Figure 7M), and horizontal activity (Figure 7N) in all groups of mice, suggesting that either simvastatin treatment or genetic alteration do not modulate gross metabolic and motor activities.
Figure 7. Simvastatin Increases BDNF in the Brain of FAD5X Mice via PPARα.
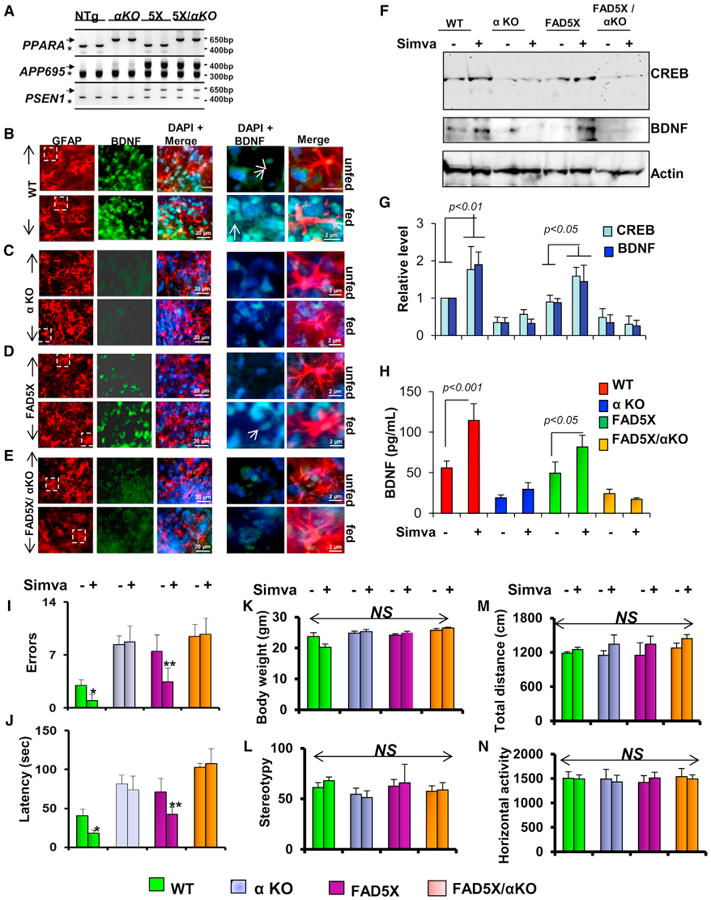
(A–E) Genotyping of FAD5X/Ppara null (5X/αKO) mice. Five-month-old mice were fed simvastatin for 2 weeks followed by immunofluorescence analysis of astroglial BDNF (GFAP, red; BDNF, green) in the cortices of (B) wild-type (WT), (C) αKO, (D) FAD5X, and (E) FAD5X/αKO animals.
(F and G) Immunoblot (F) and densitometric (G) analyses of CREB and BDNF were performed in the hippocampal extracts.
(H) ELISA of BDNF in the hippocampal extracts.
(I and J) Number of errors (I) and latency (J) in Barnes maze for statin unfed and fed WT, αKO, FAD5X, and FAD5X/αKO.
(K–N) All groups of mice were also monitored for body weight (K), stereotypy (L), total distance (M), and horizontal activity (N). Eight mice (n = 8) were used in each group. *p < 0.05 versus statin-unfed wild-type control and **p < 0.05 versus statin-unfed FAD5X mice.
Discussion
Statins Induce Neurotrophins in Brain Cells
Both BDNF and NT-3 are important for preserving the complex neuronal architecture of hippocampal and cortical circuits. Therefore, increasing the levels of neurotrophins in the CNS is considered to be an important step in halting the progression of several neurodegenerative and neurocognitive diseases. Because these neurotrophic molecules do not cross the BBB, gene therapy and stereotaxic injection directly into the brain are now the only available options. However, from the therapeutic perspective, it seems that the best option would be to stimulate/induce the production of BDNF and NT-3 in vivo in the CNS of patients with neurodegenerative disorders. Here, we describe the finding that statins are capable of upregulating neurotrophic factors in brain cells and in vivo in the brain. Although previous studies have reported the neurotrophic role of statins in stroke (Chen et al., 2005) and depression (Tsai, 2007), nothing is known about the role of statins in controlling the expression of neurotrophic factors. According to Tsai (2007), statins may be involved in the upregulation of plasmin (t-PA) and cleavage of pro-BDNF to mature BDNF. However, this study does not provide any data on the transcriptional upregulation of BDNF by statins.
A Tale of Two Independent Pathways Regulated by Statins
Statins inhibit HMG-CoA reductase, the regulatory enzyme in the cholesterol biosynthesis pathway and thereby lower the level of cholesterol in patients with hypercholesterolemia. However, statins are also anti-inflammatory (Cordle and Landreth, 2005; Pahan et al., 1997). Although this anti-inflammatory activity of statins is independent of cholesterol, intermediates of the mevalonate pathway abrogate this anti-inflammatory effect (Pahan, 2006; Roy and Pahan, 2011), and statins employ the isoprenylation-p21ras/p21rac pathway to upregulate proinflammatory molecules (Cordle et al., 2005; Roy and Pahan, 2011). Currently, in addition to cholesterol-lowering, statins are known to inhibit small G protein activation, suppress proinflammatory molecules, modulate the adaptive immune response, stimulate endothelial NOS, attenuate migration and proliferation of smooth muscle cells, lower the production of reactive oxygen species, destabilize amyloid-β fibrils, attenuate α-syn aggregation, and mitigate dyskinesia (Pahan, 2006; Roy and Pahan, 2011; Weber et al., 2005). In fact, all these functions are dependent on their ability to suppress either intermediates or the end product of the mevalonate pathway. By contrast, here, we have found that metabolites of the cholesterol biosynthetic pathway, including FPP and mevalonate, inhibitors of isoprenoid transferring enzymes (FPTi and GGTi), and dominant-negative mutants of p21ras and p21rac, are unable to modulate statin-mediated upregulation of neurotrophic factors, suggesting that statin-mediated inhibition of the cholesterol biosynthetic pathway and/or the isoprenoid-p21ras/p21rac pathway is/are not involved in the upregulation of neurotrophic factors.
On the other hand, as expected, CREB, a classical transcription factor regulating the synthesis of many neurotrophic factors, was found to be involved in statin-mediated upregulation of neurotrophic factors. Next, we explored the effect of statins on the transcriptional regulation of CREB. Interestingly, promoter analysis of CREB, but not BDNF or NT-3, revealed the presence of a conserved PPRE sequence in its promoter, and we found the involvement of PPARα, but not PPARβ or PPARγ, in statin-mediated upregulation of CREB and neurotrophic factors. ChIP analysis also showed that simvastatin induced the recruitment of PPARα, but not PPARβ or PPARγ, to the CREB promoter. Together, our results suggest that statins upregulate neurotrophic factors via a novel PPARα-CREB pathway that is independent of their inhibition of the mevalonate pathway. Therefore, statins employ two distinct pathways to execute their biological functions: suppressing the mevalonate pathway for their previously known biological functions and stimulating the PPARα-CREB pathway for their neurotrophic function (Graphical Abstract).
Apart from neurotrophins, CREB-regulated genes are also involved in the modulation of our basic metabolic processes, including glucose metabolism, fatty acid oxidation, and hepatic lipid mobilization. CREB activates hepatic gluconeogenesis and fatty acid β-oxidation by inducing the expression of PGC-1 and Hes-1 (Herzig et al., 2003) genes. CREB-mediated upregulation of the hepatocyte nuclear factor-4 (HNF-4) gene stimulates the lipid mobilization process in liver (Dell and Hadzopoulou-Cladaras, 1999). Therefore, downregulation of CREB and many other CREB-regulated genes may contribute to different metabolic disorders, including diabetes, arthritis, and obesity. Thus, the importance of our finding is that statins, one of the most commonly used groups of drugs, may be used to modulate multiple metabolic states via PPARα-mediated upregulation of CREB.
Statins Serve as Ligands of PPARα
For the last 30 years, statins have been widely considered to be competitive inhibitors of HMG-CoA reductase. However, until now there has been no receptor protein identified for statins. Here, we report that PPARα, but not PPARβ or PPARγ, serves as a receptor for statins. Detailed energy calculations for simvastatin:PPAR complexes shows greater thermodynamic stability of simvastatin:PPARα than either simvastatin:PPARβ or simvastatin:PPARγ. Our TR-FRET analysis with increasing doses of different statins in PPARα-PGC-1α complex, in silico interaction studies, de novo expression studies of full-length and mutated constructs PPARα followed by ESI-MS analyses, and ESI-MS analysis of PPARα-LBD pull-down from the nuclear fraction confirm the feasibility and stability of the simvastatin: PPARα complex. Among simvastatin, mevastatin, pravastatin, atorvastatin, and rosuvastatin, simvastatin was found to be the strongest ligand of PPARα followed by mevastatin. On the other hand, ligand-binding efficacies of pravastatin, atorvastatin, and rosuvastatin were almost same. According to our detailed structural and molecular techniques, we confirmed that Leu331 and Tyr334 residues of PPARα LBD are important for statin-binding as L331M/Y334D PPARα did not show any interaction with statins. Interestingly, known ligands of PPARα such as WY-14643 and gemfibrozil did not show interaction with Leu331 and Tyr334 residues. While WY-14643 and gemfibrozil docked nicely at the ligand-binding pocket, these ligands were ∼6 Å and ∼5 Å away from Leu331 and Tyr334 residues and mutation of these residues also did not abrogate the ability of these ligands to activate PPARα. These results identify statins as a different group of PPARα ligands that differ from classical ligands in terms of binding to the PPARα LBD. Since PPARα plays an important role in fatty acid oxidation, lipoprotein metabolism, and peroxisome proliferation (Pahan, 2006), our present finding suggests that statins may also regulate these biological processes via PPARα.
Simvastatin Increases BDNF and Improves Memory in an Animal Model of AD via PPARα
Simvastatin treatment increased BDNF in hippocampus and cortex and improved memory and learning in the FAD5X mouse model of AD. In contrast, simvastatin remained unable to stimulate BDNF in CNS tissues and increase memory and learning in FAD5X/Ppara null mice, suggesting a critical role of PPARα in the upregulation of neurotrophins in the CNS and protection of memory during AD pathology. Since upregulation of BDNF in the hippocampus and the cortex has therapeutic potential for AD, simvastatin may be beneficial for AD. However, a number of evidences from randomized clinical trials using simvastatin consistently report no benefit in cognition or dementia prevention (Sano et al., 2011; Stadelmann et al., 2002). In some cases, studies failed to provide clear evidence for its therapeutic efficacy in AD (Burgos et al., 2012), calling into question the relevance of preclinical and epidemiologic findings. Again, decreased neuronal cholesterol synthesis has been implicated as a contributor to neurodegeneration (Karasinska and Hayden, 2011; Liu et al., 2010; Valenza et al., 2010) and impaired CNS function in the context of diabetes/insulin resistance (Suzuki et al., 2010). Here, our findings disclose a new site of action (PPARα) for statin that could be predictive of therapeutic benefit in AD patients. However, a recent study indicates that women with high level of LDL cholesterol show increased loss of visual memory when prescribed with fenofibrate, a known ligand of PPARα, over 7 years (Ancelin et al., 2012). This may be due to the facts that higher LDL cholesterol is associated with better memory functioning among the elderly without ApoE4 allele (West et al., 2008) and that long-term use of fenofibrate affects visual memory via reducing LDL cholesterol. Therefore, the relevance of the statin-PPARα-CREB connection in humans remains to be established.
Experimental Procedures
Isolation of Mouse Primary Astroglia and Microglia
Microglia and astroglia were isolated from mixed glial cultures of 7-day-old mouse pups according to the procedure of Giulian and Baker (1986) as described earlier (Dasgupta et al., 2003; Roy et al., 2006).
Isolation of Human Fetal Neurons
Primary human neurons were isolated from human fetal brain tissues as described by us earlier (Jana and Pahan, 2004a, 2004b). All of the experimental protocols were reviewed and approved by the Institutional Review Board of the Rush University Medical Center.
Transfection of Neurons
Primary neurons were transfected with Lipofectamine PLUS (Invitrogen) and Nupherin-neuron (Biomol) as described by us earlier (Saha and Pahan, 2007). Briefly, each well of 6-well plate was transfected with 0.25 μg of DNA complexed with Neupherin peptide and Lipofectamine PLUS.
ChIP
ChIP was performed as described earlier (Roy et al., 2013).
RT-PCR Analysis
Total RNA was digested with DNase and RT-PCR was carried out as described earlier (Ghosh et al., 2009; Roy et al., 2013).
Real-Time PCR Analysis
Quantitative real-time PCR was performed in the ABI-7500 standard PCRs (Applied Biosystems) as described earlier (Ghosh et al., 2009; Roy et al., 2013) using TaqMan Universal Master mix and FAM-labeled probes and primers (Applied Biosystems).
Cloning of the Creb Promoter and Site-Directed Mutagenesis
CREB promoter was cloned and subjected to site-directed mutagenesis as described earlier (Roy et al., 2013).
Lentiviral Cloning of FL PPARα and L331M/Y334D PPARα
Please see the Supplemental Experimental Procedures.
Lentiviral Administration of FLPPARα and ΔPPARα in the Adult Mouse Hippocampus
Lentiviral administration was performed as described earlier (Roy et al., 2013).
Breeding and Development of FAD5X/Ppara Null Animals
Please see the Supplemental Experimental Procedures.
ESI-MS Analysis of PPARα-Simvastatin Interaction
Please see the Supplemental Experimental Procedures.
In Silico Structural Analyses of PPARα, PPARβ, and PPARγ Complexed with Simvastatin
Please see the Supplemental Experimental Procedures.
TR-FRET Analysis
TR-FRET was performed using Lanthascreen TR-FRET PPAR-alpha coactivator assay kit. In this assay, statins were incubated with GST-tagged recombinant PPARα LBD, Terbium (Tb)-tagged anti GST antibody, and fluorescein (FL)-tagged PGC-1α as directed in the manufacturer's protocol. The entire reaction was set up in corning 384-well plate by an automated robotic injector. Plate was centrifuged, incubated in dark for 30 min, and then analyzed in molecular devices analyst equipped with dichroic mirror. The excitation and emission were set at 340 nm and 540 nm, respectively.
Thermal Shift Assay
Thermal shift assay was performed in Applied Biosystems 7500 standard realtime thermal cycler with thermal shift dye kit (Life Technologies). For each reaction, purified protein (0.5 μg to 1 μg) was added to 18 μl of thermal shift buffer and 1–2 μl of dye. Reaction was set 96-well PCR plate in dark and then placed in the thermal cycler using the following two-stage program ([25°C for 2 min] 1 cycle; [27°C for 15 s, 26°C for 1 min] 70 cycles; auto increment 1 °C for both stages). The filter was set at ROX with no quencher filter and no passive filter.
Statistical Analyses
All values are expressed as the mean ± SD. Differences among means were analyzed using one- or two-way ANOVA with time or genotype as the independent factors. Differences in behavioral measures were examined by independent one-way or repeated-measures ANOVAs using SPSS. Homogeneity of variance between test groups was examined using Levene's test. Post hoc analyses of between-subjects effects were conducted using Scheffe's, Tukey's or Games-Howell tests, where appropriate. p < 0.05 was considered statistically significant.
Supplementary Material
Highlights.
Upregulation of neurotrophins by statins independent of the mevalonate pathway
Statins bind with Leu331 and Tyr334 residues of the ligand-binding domain of PPARα
Statins produce neurotrophins via the PPARα-CREB pathway
Simvastatin increases CREB and improves memory in an animal model of AD via PPARα
Acknowledgments
This study was supported by grants from NIH (AT6681 and NS83054) and Alzheimer's Association (IIRG-12-241179). The authors would like to thank ChemCore at the Center for Molecular Innovation and Drug Discovery, Northwestern University, funded by the Chicago Biomedical Consortium.
Footnotes
Supplemental Information: Supplemental Information includes Supplemental Experimental Procedures and four figures and can be found with this article online at http://dx.doi.org/10.1016/j.cmet.2015.05.022.
References
- Abe K. Therapeutic potential of neurotrophic factors and neural stem cells against ischemic brain injury. J Cereb Blood Flow Metab. 2000;20:1393–1408. doi: 10.1097/00004647-200010000-00001. [DOI] [PubMed] [Google Scholar]
- Ancelin ML, Carrière I, Barberger-Gateau P, Auriacombe S, Rouaud O, Fourlanos S, Berr C, Dupuy AM, Ritchie K. Lipid lowering agents, cognitive decline, and dementia: the three-city study. J Alzheimers Dis. 2012;30:629–637. doi: 10.3233/JAD-2012-120064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos JS, Benavides J, Douillet P, Velasco J, Valdivieso F. How statins could be evaluated successfully in clinical trials for Alzheimer's disease? Am J Alzheimers Dis Other Demen. 2012;27:151–153. doi: 10.1177/1533317512442998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhang C, Jiang H, Li Y, Zhang L, Robin A, Katakowski M, Lu M, Chopp M. Atorvastatin induction of VEGF and BDNF promotes brain plasticity after stroke in mice. J Cereb Blood Flow Metab. 2005;25:281–290. doi: 10.1038/sj.jcbfm.9600034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B, Mattson MP. NT-3 and BDNF protect CNS neurons against metabolic/excitotoxic insults. Brain Res. 1994;640:56–67. doi: 10.1016/0006-8993(94)91857-0. [DOI] [PubMed] [Google Scholar]
- Cordle A, Landreth G. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors attenuate beta-amyloid-induced microglial inflammatory responses. J Neurosci. 2005;25:299–307. doi: 10.1523/JNEUROSCI.2544-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordle A, Koenigsknecht-Talboo J, Wilkinson B, Limpert A, Landreth G. Mechanisms of statin-mediated inhibition of small G-protein function. J Biol Chem. 2005;280:34202–34209. doi: 10.1074/jbc.M505268200. [DOI] [PubMed] [Google Scholar]
- Dasgupta S, Jana M, Liu X, Pahan K. Role of very-late antigen-4 (VLA-4) in myelin basic protein-primed T cell contact-induced expression of proinflammatory cytokines in microglial cells. J Biol Chem. 2003;278:22424–22431. doi: 10.1074/jbc.M301789200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell H, Hadzopoulou-Cladaras M. CREB-binding protein is a transcriptional coactivator for hepatocyte nuclear factor-4 and enhances apolipoprotein gene expression. J Biol Chem. 1999;274:9013–9021. doi: 10.1074/jbc.274.13.9013. [DOI] [PubMed] [Google Scholar]
- Endo A, Kuroda M, Tsujita Y. ML-236A, ML-236B, and ML-236C, new inhibitors of cholesterogenesis produced by Penicillium citrinium. J Antibiot. 1976;29:1346–1348. doi: 10.7164/antibiotics.29.1346. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Roy A, Matras J, Brahmachari S, Gendelman HE, Pahan K. Simvastatin inhibits the activation of p21ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson's disease. J Neurosci. 2009;29:13543–13556. doi: 10.1523/JNEUROSCI.4144-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Hedrick S, Morantte I, Koo SH, Galimi F, Montminy M. CREB controls hepatic lipid metabolism through nuclear hormone receptor PPAR-gamma. Nature. 2003;426:190–193. doi: 10.1038/nature02110. [DOI] [PubMed] [Google Scholar]
- Jana A, Pahan K. Fibrillar amyloid-beta peptides kill human primary neurons via NADPH oxidase-mediated activation of neutral sphingomyelinase. Implications for Alzheimer's disease. J Biol Chem. 2004a;279:51451–51459. doi: 10.1074/jbc.M404635200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana A, Pahan K. Human immunodeficiency virus type 1 gp120 induces apoptosis in human primary neurons through redox-regulated activation of neutral sphingomyelinase. J Neurosci. 2004b;24:9531–9540. doi: 10.1523/JNEUROSCI.3085-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Albertz J, Guo Z, Peng Q, Rudow G, Troncoso JC, Ross CA, Duan W. Neuroprotective effects of PPAR-γ agonist rosiglitazone in N171-82Q mouse model of Huntington's disease. J Neurochem. 2013;125:410–419. doi: 10.1111/jnc.12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasinska JM, Hayden MR. Cholesterol metabolism in Huntington disease. Nat Rev Neurol. 2011;7:561–572. doi: 10.1038/nrneurol.2011.132. [DOI] [PubMed] [Google Scholar]
- Kariharan T, Nanayakkara G, Parameshwaran K, Bagasrawala I, Ahuja M, Abdel-Rahman E, Amin AT, Dhanasekaran M, Suppiramaniam V, Amin RH. Central activation of PPAR-gamma ameliorates diabetes induced cognitive dysfunction and improves BDNF expression. Neurobiol Aging. 2015;36:1451–1461. doi: 10.1016/j.neurobiolaging.2014.09.028. [DOI] [PubMed] [Google Scholar]
- Kells AP, Fong DM, Dragunow M, During MJ, Young D, Connor B. AAV-mediated gene delivery of BDNF or GDNF is neuroprotective in a model of Huntington disease. Mol Ther. 2004;9:682–688. doi: 10.1016/j.ymthe.2004.02.016. [DOI] [PubMed] [Google Scholar]
- Kerschensteiner M, Gallmeier E, Behrens L, Leal VV, Misgeld T, Klinkert WE, Kolbeck R, Hoppe E, Oropeza-Wekerle RL, Bartke I, et al. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J Exp Med. 1999;189:865–870. doi: 10.1084/jem.189.5.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JP, Tang Y, Zhou S, Toh BH, McLean C, Li H. Cholesterol involvement in the pathogenesis of neurodegenerative diseases. Mol Cell Neurosci. 2010;43:33–42. doi: 10.1016/j.mcn.2009.07.013. [DOI] [PubMed] [Google Scholar]
- Maisonpierre PC, Belluscio L, Squinto S, Ip NY, Furth ME, Lindsay RM, Yancopoulos GD. Neurotrophin-3: a neurotrophic factor related to NGF and BDNF. Science. 1990;247:1446–1451. doi: 10.1126/science.247.4949.1446. [DOI] [PubMed] [Google Scholar]
- Morse JK, Wiegand SJ, Anderson K, You Y, Cai N, Carnahan J, Miller J, DiStefano PS, Altar CA, Lindsay RM, et al. Brain-derived neurotrophic factor (BDNF) prevents the degeneration of medial septal cholinergic neurons following fimbria transection. J Neurosci. 1993;13:4146–4156. doi: 10.1523/JNEUROSCI.13-10-04146.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med. 2009;15:331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahan K. Lipid-lowering drugs. Cell Mol Life Sci. 2006;63:1165–1178. doi: 10.1007/s00018-005-5406-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahan K, Sheikh FG, Namboodiri AM, Singh I. Lovastatin and phenylacetate inhibit the induction of nitric oxide synthase and cytokines in rat primary astrocytes, microglia, and macrophages. J Clin Invest. 1997;100:2671–2679. doi: 10.1172/JCI119812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Pahan K. Prospects of statins in Parkinson disease. Neuroscientist. 2011;17:244–255. doi: 10.1177/1073858410385006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Fung YK, Liu X, Pahan K. Up-regulation of microglial CD11b expression by nitric oxide. J Biol Chem. 2006;281:14971–14980. doi: 10.1074/jbc.M600236200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Liu X, Pahan K. Myelin basic protein-primed T cells induce neurotrophins in glial cells via alphavbeta3 [corrected] integrin. J Biol Chem. 2007;282:32222–32232. doi: 10.1074/jbc.M702899200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Jana M, Corbett GT, Ramaswamy S, Kordower JH, Gonzalez FJ, Pahan K. Regulation of cyclic AMP response element binding and hippocampal plasticity-related genes by peroxisome proliferator-activated receptor α. Cell Rep. 2013;4:724–737. doi: 10.1016/j.celrep.2013.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha RN, Pahan K. Differential regulation of Mn-superoxide dismutase in neurons and astroglia by HIV-1 gp120: Implications for HIV-associated dementia. Free Radic Biol Med. 2007;42:1866–1878. doi: 10.1016/j.freeradbiomed.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano M, Bell KL, Galasko D, Galvin JE, Thomas RG, van Dyck CH, Aisen PS. A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology. 2011;77:556–563. doi: 10.1212/WNL.0b013e318228bf11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadelmann C, Kerschensteiner M, Misgeld T, Brück W, Hohlfeld R, Lassmann H. BDNF and gp145trkB in multiple sclerosis brain lesions: neuroprotective interactions between immune and neuronal cells? Brain. 2002;125:75–85. doi: 10.1093/brain/awf015. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Lee K, Jing E, Biddinger SB, McDonald JG, Montine TJ, Craft S, Kahn CR. Diabetes and insulin in regulation of brain cholesterol metabolism. Cell Metab. 2010;12:567–579. doi: 10.1016/j.cmet.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SJ. Statins may enhance the proteolytic cleavage of proBDNF: implications for the treatment of depression. Med Hypotheses. 2007;68:1296–1299. doi: 10.1016/j.mehy.2006.09.043. [DOI] [PubMed] [Google Scholar]
- Valenza M, Leoni V, Karasinska JM, Petricca L, Fan J, Carroll J, Pouladi MA, Fossale E, Nguyen HP, Riess O, et al. Cholesterol defect is marked across multiple rodent models of Huntington's disease and is manifest in astrocytes. J Neurosci. 2010;30:10844–10850. doi: 10.1523/JNEUROSCI.0917-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber MS, Prod'homme T, Steinman L, Zamvil SS. Drug Insight: using statins to treat neuroinflammatory disease. Nat Clin Pract Neurol. 2005;1:106–112. doi: 10.1038/ncpneuro0047. [DOI] [PubMed] [Google Scholar]
- West R, Beeri MS, Schmeidler J, Hannigan CM, Angelo G, Grossman HT, Rosendorff C, Silverman JM. Better memory functioning associated with higher total and low-density lipoprotein cholesterol levels in very elderly subjects without the apolipoprotein e4 allele. Am J Geriatr Psychiatry. 2008;16:781–785. doi: 10.1097/JGP.0b013e3181812790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Lu D, Jiang H, Xiong Y, Qu C, Li B, Mahmood A, Zhou D, Chopp M. Simvastatin-mediated upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway, and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J Neurotrauma. 2008;25:130–139. doi: 10.1089/neu.2007.0369. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.