

SUMMARY
Metabolic adaptation to hypoxia is critical for survival in metazoan species for which reason they have developed cellular mechanisms for mitigating its adverse consequences. Here, we have identified L-2-hydroxyglutarate (L2HG) as a universal adaptive determinant of the hypoxia response. L2HG is a metabolite of previously unknown function produced by the reduction of mitochondrial 2-oxoglutarate by malate dehydrogenase. L2HG accumulates in response to increases in 2-oxoglutarate, which occur as a result of tricarboxylic acid cycle dysfunction and increased mitochondrial reducing potential. These changes are closely coupled to cellular redox homeostasis, as increased cellular L2HG inhibits electron transport and glycolysis to offset the adverse consequences of mitochondrial reductive stress induced by hypoxia. Thus, L2HG couples mitochondrial and cytoplasmic energy metabolism in a new model of cellular redox regulation.
Graphical Abstract
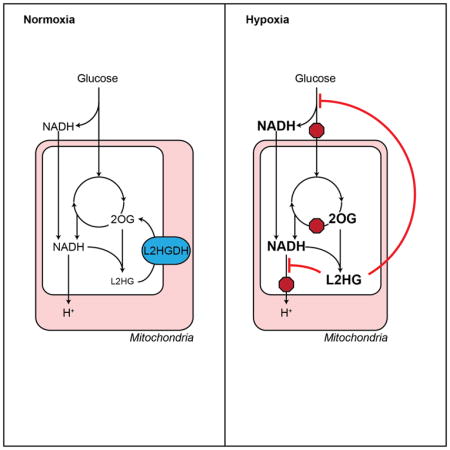
INTRODUCTION
Hypoxia is an important environmental stimulus that causes genetic and metabolic reprogramming in cells to facilitate survival. This programmed response is mediated primarily through stabilization of hypoxia-inducible factor 1α (HIF1α), a transcription factor that coordinates a shift in energy metabolism away from oxidative phosphorylation and toward glycolysis and lactate fermentation through the increased expression of glucose transporters (GLUT1), glycolytic enzymes, lactate dehydrogenase (LDHA), and pyruvate dehydrogenase kinase (Semenza, 2012). These changes limit carbon entry into the tricarboxylic acid cycle (TCA) and mitigate damage from reactive oxygen species (ROS) generated by dysfunctional (uncoupled) electron transport in the mitochondria (Zhang et al., 2008).
While hypoxia limits the proliferation of many cell types, some cancer cells, stem/progenitor cells, and pulmonary vascular cells continue to grow and divide in low oxygen conditions. Metabolic flux analyses suggest that transformed cell lines may utilize reductive carboxylation of glutamine-derived 2-oxoglutarate (2OG) by isocitrate dehydrogenase (IDH) to overcome the limitation of decreased glucose-derived citrate on macromolecular biosynthesis under hypoxic conditions (Gameiro et al., 2013; Scott et al., 2011; Wise et al., 2011). However, the overall contribution of reductive carboxylation to net reductive carbon flux has been questioned (Fan et al., 2013). Indeed, evidence for a glutamine-driven, glucose-independent oxidative TCA cycle under hypoxic conditions has been found in human Burkitt lymphoma cells, which would be consistent with both oxidative and reductive flux through IDH (Le et al., 2012). Notably, these studies have all been performed in transformed cells. Comparatively little is known about the metabolic response to hypoxia in primary cells.
Given that hypoxia is a potent stimulus of pulmonary vascular cell remodeling and proliferation in the pathogenesis of pulmonary hypertension (Cottrill and Chan, 2013), we performed metabolic profiling of human pulmonary arterial endothelial and smooth muscle cells (PAEC and PASMC) exposed to 0.2% oxygen for 24 h. This oxygen tension is below the P50 for mitochondrial respiration (Vemuri et al., 2007), which would enhance any differences between glycolytic and oxidative metabolism. As expected, hypoxia had a significant impact on pathways involved in energy metabolism and precursor biosynthesis.
Interestingly, in contrast to other TCA metabolites, hypoxia caused a 2-fold increase in intracellular 2OG with a concomitant increase in its reduced metabolite, 2-hydroxyglutarate (2HG). Two enantiomers of 2HG, L and D, have been associated with separate, rare inborn errors of metabolism resulting in increased urinary excretion of 2HG linked to neurological deficits in children (Chalmers et al., 1980; Duran et al., 1980; Kranendijk et al., 2012). While the roles of L2HG and D2HG in normal human metabolism are unknown, L2HG has recently been shown to accumulate in renal cell carcinoma (Struys, 2013) and in children and cell lines with electron transport chain defects (Mullen et al., 2014; Reinecke et al., 2012; Worth et al., 2015). By contrast, D2HG has been identified as the product of cancer-associated mutant IDH enzymes (Cairns and Mak, 2013; Dang et al., 2009; Ward et al., 2010). Both enantiomers inhibit 2OG-dependent dioxygenases, such as prolyl hydroxylases (PHD), TET family 5-methylcytosine hydroxylases, and JmjC domain-containing histone demethylases (Figueroa et al., 2010; Lu et al., 2012; Xu et al., 2011). While a previous study reported hypoxia-mediated increases in 2HG in the SF188 glioblastoma cell line, the authors suggested this was a consequence of incomplete reductive carboxylation of 2OG by wild type IDH to produce D2HG (Wise et al., 2011). Here, we (1) demonstrate that hypoxia specifically increases L2HG in a variety of cell types, (2) provide insight into the molecular mechanisms of L2HG accumulation, (3) describe an important role for L2HG metabolism in maintaining cellular and mitochondrial redox homeostasis, and (4) offer a mechanism for the coordinated regulation of energy metabolism by L2HG through inhibition of glycolysis and oxygen consumption. These observations provide important insights into the critical functional role of L2HG metabolism in normal cell physiology.
RESULTS
Global Metabolic Reprogramming of Pulmonary Vascular Cells in Response to Hypoxia
Hypoxia is a potent stimulus for pulmonary vascular remodeling in patients with pulmonary hypertension, characterized by smooth muscle and endothelial cell proliferation and metabolic dysregulation (Cottrill and Chan, 2013). For this reason, we initially measured levels of extracellular and intracellular metabolites by liquid chromatography-mass spectrometry (LC-MS) in PAEC and PASMC after 24 h exposure to 0.2% oxygen and compared those results with 21% oxygen control samples. As expected, an increase in extracellular lactate and a decrease in extracellular glucose were observed in hypoxia-treated cells, consistent with increased glycolytic flux (Figure S1). Similarly, hypoxia affected a broad range of metabolic pathways (Figure 1A and Supplementary Tables 1–3), including precursor biosynthesis; energy production (glycolysis, TCA cycle, electron transport chain); and redox homeostasis (malate-aspartate shuttle, glycerol phosphate shuttle, nicotinamide metabolism, glutathione metabolism), as identified by metabolite set enrichment analysis (Xia et al., 2012; Xia et al., 2009). Similar changes were observed in PAEC and PASMC.
Figure 1. Hypoxia Increases 2OG and 2HG.

(A) Volcano plot of metabolite changes in PASMC treated with 0.2% oxygen for 24 h compared to 21% oxygen controls. AA, amino acids; NA, nucleic acid pathway intermediates; PPP, pentose phosphate pathway intermediates; TCA, tricarboxylic acid cycle intermediates. (B and C) Targeted LCMS reveals that hypoxia-mediated increases in 2OG (B) and 2HG (C) occur in a variety of cell types. PAEC, human pulmonary artery endothelial cells; PASMC, human pulmonary artery smooth muscle cells; LF, human lung fibroblasts; HEK, human embryonic kidney 293 cells; HepG2, human liver hepatocellular carcinoma cells; MSC, human mesenchymal stem cells. (D) Intracellular 2OG and 2HG vary inversely with oxygen tension in PASMC (p < 0.0001 for trend for each). Data are mean ± SEM. See also Figure S1.
A pathway topological analysis identified the TCA cycle as among the most strongly modulated by hypoxia based on the number of differentially regulated metabolites and the location of these metabolites within the pathway (Xia et al., 2012; Xia et al., 2009) (Figure 1A and Supplementary Tables 1–3). Consistent with decreased TCA cycle flux, the levels of most TCA metabolites were decreased by hypoxia, with the notable exception of 2OG (Figure 1A). Mitochondrial 2OG is produced in the TCA cycle by oxidative decarboxylation of isocitrate by IDH2 and IDH3. 2-Oxoglutarate then undergoes oxidative decarboxylation to succinate by the 2OG dehydrogenase complex (OGDHC). Both upstream TCA metabolites (aconitate, citrate, and isocitrate) and downstream metabolites (succinate, fumarate, and malate) are decreased or unchanged in hypoxia-treated cells, suggesting that 2OG may be a reservoir of TCA carbon. Interestingly, hypoxia increased levels of the reduced 2OG metabolite, 2HG, in both cell types. Given the unknown role of D2HG and L2HG in normal cell physiology, their demonstrated effects on 2OG-dependent dioxygenases, and their connection to mitochondrial redox shuttling, we sought to understand the role of 2HG metabolism in the cellular response to hypoxia.
Hypoxia Increases L-2-hydroxyglutarate
A targeted LC-MS assay confirmed the hypoxia-mediated increases in 2OG and 2HG in cell lysates of PAEC and PASMC (Figures 1B and 1C), and several other cell types, including lung fibroblasts (LF), immortalized cell lines (HEK293 and HepG2), and mesenchymal stromal cells. This diversity of cell types suggests that these metabolic changes are a common fundamental feature of the hypoxia response. Basal intracellular concentrations of 2OG and 2HG were approximately 0.4–2 mM and 0.05–0.24 mM, respectively, similar to previous estimates (Losman and Kaelin, 2013; MacKenzie et al., 2007; Matsunaga et al., 2012). Hypoxia induced between a 1.5–2.5-fold increase in 2OG and a 1.7–8.8-fold increase in 2HG depending on the cell type. These increases vary inversely with oxygen tension (Figure 1D).
Cell extracts were derivatized with diacetyl-L-tartaric anhydride (DATAN) to enable separation of 2HG enantiomers for quantification by LC-MS (Struys et al., 2004a) (Figures 2A and 2B). Normoxic levels of D2HG and L2HG were similar; however, L2HG preferentially increases with hypoxia in all cell types examined except PASMC (Figure 2B). In PASMC, D2HG and L2HG increased to a similar degree (1.5 ± 0.2 and 1.7 ± 0.2-fold, respectively, p = 0.45). Interestingly, the magnitude of the L2HG increase was markedly greater in HEK cells compared to other cell types.
Figure 2. Hypoxia Preferentially Increases the L Entantiomer of 2HG.

(A) Extracted ion chromatograms (XIC) of underivatized 2HG (purple) and derivatized enantiomerically pure standards of D2HG (green), L2HG (blue), or a racemic mixture (red). Chromatograms are offset for clarity. The XIC of a derivatized cell extract is also shown (black) with intensity values on the right y-axis. (B) Fold change after hypoxia treatment of D2HG and L2HG in derivatized cell extracts. (C) Immunoblot of D2HGDH and L2HGDH levels in LF treated with targeting siRNA compared to a non-targeting control siRNA (siCTL). (D) Total 2HG in cell extracts treated with D2HGDH or L2HGDH siRNA. (E) Immunoblot of LF cell extracts overexpressing D2HGDH or L2HGDH. (F) Total 2HG in LF overexpressing D2HGDH or L2HGDH compared to Vector control. Data are mean ± SEM. See also Figure S2.
As L2HG appears to be a common feature of the metabolic response to hypoxia, LF were selected as the model cell line for additional exploration of L2HG metabolism as they are primary cells that demonstrate robust increases in 2HG in response to hypoxia and are likely more representative of characteristic hypoxia responses than either PAEC or PASMC. PAEC, and endothelial cells generally, have a glycolytic phenotype, which likely accounts for the low abundance of total 2HG in these cells. PASMC proliferate under hypoxic conditions, demonstrate smaller increases in L2HG, and were the only primary cells studied with hypoxia-mediated increases in D2HG. Thus, LF appeared to be the most ideal model system for further analysis; however, many of the subsequent results could also be extended to other cell lines (as illustrated throughout this report).
L2HG has a specific dehydrogenase, L2HGDH, the only enzyme known to metabolize L2HG. L2HGDH is a mitochondrial protein that catalyzes the oxidation of L2HG to 2OG coupled to the reduction of FAD (Rzem et al., 2006; Rzem et al., 2004; Topcu et al., 2004) (Figure 3A). Similarly, D2HG has a specific mitochondrial dehydrogenase, D2HGDH, that catalyzes its oxidation to 2OG and transfers electrons to FADH2 (Achouri et al., 2004; Struys et al., 2005). In confirmation of the biochemical derivatization, LF treated with siRNA targeting L2HGDH, but not D2HGDH, demonstrated a potentiation of the hypoxia-mediated increase in total 2HG (Figures 2C and 2D). This was also observed in PAEC and in PASMC despite the results of the biochemical derivatization suggesting an increase in both enantiomers in the PASMC (Figure S2G and S2H). L2HGDH silencing also increased 2HG levels in aerobic conditions. In the complementary experiment, overexpression of L2HGDH, but not D2HGDH, blocked the hypoxia-mediated increase in 2HG (Figures 2E and 2F). 2HGDH knockdown and overexpression had little effect on 2OG levels (Figures S2A and S2B). Derivatization of LF cell extracts confirmed that changes in total 2HG specifically reflect changes in L2HG in these experimental conditions (Figures S2C–S2F). Notably, siD2HGDH treatment did not result in significant changes in D2HG, contrary to a previous study of MCF7 breast cancer cells where siD2HGDH treatment induced a 4-fold increase in total 2HG (enantiomeric quantification was not performed) (Matsunaga et al., 2012). Together, these data demonstrate a hypoxia-mediated increase in L2HG and no change in the levels of (cancer-associated) D2HG, in the metabolic response of primary cells to hypoxia.
Figure 3. Potential Mechanisms of L2HG Accumulation in Hypoxia.

(A) Enzymatic and cofactor requirements for L2HG metabolism. (B and C) Intracellular 2OG (B) and 2HG (C) in normoxia and hypoxia measured after treatment with a cell-permeable 2OG analogue, TFMB-2OG. (D and E) 2OG (D) and 2HG (E) in cells treated with the OGDHC inhibitor KMV (20 mM) ± 0.2% oxygen. (F) Correlation between 2OG and 2HG from experiments illustrated in B-E. (G) Immunoblot of MDH1 and MDH2 in siRNA-treated LF. (H–J) 2OG (H), 2HG (I), and 2OG/2HG (J) in MDH knockdown cells. (K) Relative changes in mRNA levels of 2HG metabolic enzymes in response to hypoxia. (L) Immunoblot of LF cell lysates of proteins involved in L2HG metabolism. (M) Intracellular malate determined by targeted LC-MS. (M) Cellular NADH/NAD+ determined by enzymatic cycling assay. Data are mean ± SEM. See also Figure S3.
Mechanisms of L2HG Accumulation in Hypoxia
Given that 2OG is the only known substrate for L2HG synthesis, we next examined whether increased intracellular 2OG could increase L2HG in normoxia or potentiate the increase in hypoxia. In LF treated for 24 h with a cell-permeable analogue of 2OG, TFMB-2OG (MacKenzie et al., 2007), no increase in total 2HG was observed in normoxia or hypoxia despite increased levels of intracellular 2OG (Figures 3B and 3C). Mass isotopomer studies of lymphoblasts derived from patients with D2HG or L2HG aciduria suggest that the mitochondrial pool of 2OG is reduced in 2HG synthesis (Struys et al., 2007; Struys et al., 2004b). We, therefore, treated cells with an inhibitor of OGDHC, α-keto-β-methylvaleric acid (KMV) (Patel, 1974). KMV increased 2OG and 2HG in normoxia while potentiating the hypoxia-mediated increases in both (Figures 3D and 3E). Indeed, the increases in 2HG were highly correlated with increased 2OG in KMV-treated cells, while no correlation was observed in cells treated with TFMB-2OG (Figure 3F). These data suggest that compartmental metabolism plays an important role in 2HG synthesis. OGDHC activity is decreased by chronic hypoxia or oxidative stress, suggesting a potential mechanism by which to account for the increases in 2OG and 2HG in hypoxia (Lai et al., 2003; Starkov, 2013; Tretter and Adam-Vizi, 1999).
Malate dehydrogenases (MDH) 1 and 2 are the only enzymes known to produce L2HG from the reduction of 2OG, which is coupled to the oxidation of NADH (Rzem et al., 2007) (Figure 3A). Both MDH enzymes catalyze the reversible oxidation of malate to oxaloacetic acid (OAA) coupled to the reduction of NAD+. MDH1 is localized to the cytoplasm where its kinetics favor production of malate by the reduction of OAA. MDH2 is localized to the mitochondria where production of OAA by the oxidation of malate is favored. Together, MDH1 and MDH2 form critical components of the malate-aspartate shuttle by which cells transport electrons from the cytoplasmic NADH pool to the mitochondrial NADH pool. MDH2 also catalyzes a key step in the TCA cycle. 2-Oxoglutarate reductase activity co-fractionated with MDH activity in rat liver cytosolic and mitochondrial extracts and purified MDH enzymes demonstrated NADH-dependent reduction of 2OG to L2HG in vitro (Rzem et al., 2007). To determine the role of MDH1 and MDH2 in hypoxia-mediated 2HG accumulation, LF were treated with siRNA targeting these enzymes separately and in combination (Figures 3G–J). Knocking down either MDH enzyme potentiated the hypoxia-mediated increase in 2OG and impaired the hypoxia-mediated increase in 2HG, resulting in an increase in the substrate/product (i.e., 2OG/2HG) ratio. These findings were confirmed in HEK293 cells (Figure 3S) and were similar to results from mouse embryonic fibroblasts isolated from l2hgdh−/− mice (Rzem et al., 2015). This supports the role of both MDH enzymes in the synthesis of L2HG from 2OG. No evidence of increased MDH enzyme expression or protein was observed (Figure 3K and 3L). L2HG production may be favored in hypoxia by decreased competition by malate for MDH2 owing to a reduction of the concentration of this metabolite (Figure 3M) and an increase in cellular NADH (Figure 3N).
In addition to increased synthesis, hypoxia caused a 50% reduction in L2HGDH expression (Figure 3K) associated with a small reduction in L2HGDH protein (Figure 3L), suggesting a role for decreased oxidation of L2HG to 2OG in hypoxia. Comparatively smaller hypoxia-mediated decreases in D2HGDH expression and protein were observed (Figure 3K and 3L).
HIF1α Is Sufficient But Not Necessary for 2HG Accumulation
HIF1α and HIF2α are constitutively expressed and degraded under normoxic conditions through the combined actions of PHD and VHL, an E3 ubiquitin ligase that recognizes hydroxylated HIF and targets it for proteasomal degradation (Kaelin and Ratcliffe, 2008). To test the role of HIF in 2HG metabolism, LF were treated with siRNA targeting VHL to prevent the degradation of HIF1α and HIF2α in normoxic conditions (Figures 4A–C). Consistent with normoxic HIF activity, siVHL-treated cells had increased expression of the HIF1α target GLUT1 and HIF2α target SERPINE1 (Figure 4B). These cells also demonstrated an increase in 2OG and 2HG in normoxia, suggesting that HIF stabilization is sufficient for these metabolic changes (Figure 4C). Notably, VHL silencing had no impact on the expression of L2HGDH (Figure 4B).
Figure 4. Role of HIF in 2HG Metabolism.

(A) Immunoblot of LF treated with siRNA targeting VHL mRNA compared to siCTL demonstrating stabilization of HIF1α and HIF2α in normoxia. (B) Normoxic stabilization of HIF1α and HIF2α increases GLUT1 and SERPINE1 mRNA, but has no effect on L2HGDH mRNA. (C) HIF stabilization through VHL knockdown is sufficient to increase 2HG and 2OG in normoxia. (D) Immunoblot demonstrating effective knockdown of HIF1α and HIF2α in normoxia and hypoxia. (E) Silencing HIF1α blunts the hypoxia-mediated increase in GLUT1 expression. (F) Silencing HIF2α blunts the hypoxia-mediated increase in SERPINE1 expression. (G and H) 2HG and 2OG in cells treated with HIF siRNA in normoxia and hypoxia. (I) Levels of L2HGDH mRNA in cells treated with siRNA targeting hypoxia-regulated transcription factors. Data are mean ± SEM. See also Figure S4.
To test whether HIF is necessary for these metabolic changes in hypoxia, cells were treated with siRNA targeting HIF1α and HIF2α alone or in combination (Figures 4D–I). Surprisingly, siHIF1 potentiated the hypoxia-mediated increases in 2OG and 2HG despite blunting the hypoxia-mediated increase in GLUT1 mRNA (Figures 4E, 4G, 4H). Treatment with siHIF2 had no effect on 2OG or 2HG levels despite blunting the hypoxia-mediated increase in SERPINE1 expression (Figures 4F, 4G, 4H). Derivatization with DATAN confirmed that L2HG is the enantiomer increased by hypoxia in the HIF1α knockdown (Figures S4A and S4B). Elevations in succinate and malate were observed in siHIF1-treated cells in hypoxia, suggesting that persistent TCA carbon flux in the absence of HIF1α activation accounts for the potentiation of the hypoxia-mediated increase in L2HG (Figures S4C and S4D). Silencing of HIF had little impact on the hypoxia-mediated decrease in L2HGDH mRNA (Figure 4I). Thus, while HIF stabilization and activation of a hypoxia-like gene expression program are associated with increases in 2OG and L2HG in normoxia, these data suggest an alternative mechanism, not dependent on HIF, that causes these metabolic changes in hypoxia. The discrepancies between L2HGDH expression and 2HG concentration support a stronger role for an alternative mechanism to decreased L2HGDH activity (e.g., OGDHC inhibition, increased mitochondrial NADH) causing L2HG accumulation in hypoxia.
In contrast to LF, VHL knockdown in HEK cells did not result in normoxic increases in 2HG or 2OG (Figure S4E and S4F). Additionally, siHIF2 blunted the hypoxia-mediated increases in HEK, while siHIF1 had little effect (Figures S4G–I).
2HGDH Knockdown Activates HIF1α Independent of 2HG Levels
Both L2HG and D2HG have been shown to inhibit the function of 2OG-dependent dioxygenases, like PHD (Xu et al., 2011), although D2HG has also been shown to activate PHD (Koivunen et al., 2012; Losman et al., 2013). Thus, we hypothesized that hypoxia-mediated increases in L2HG may potentiate HIF1α stabilization through PHD inhibition. To test this, LF were treated with siRNA targeting D2HGDH and L2HGDH. The expression of HIF1α target genes GLUT1 and LDHA were measured by RT-qPCR (Figure 5A and 5B) and HIF1α protein was measured by immunoblot (Figure 5C and 5D) Under normoxic conditions, both siD2HGDH and siL2HGDH treatment increased HIF1α protein and target gene expression, and these changes were potentiated by hypoxia. These findings were similar in PAEC and PASMC treated with siRNA targeted to the 2HGDH enzymes (Figures S5A–E), although PASMC did not demonstrate changes in LDHA expression in response to siRNA (Figure S5E).
Figure 5. D2HGDH and L2HGDH Knockdown Activate HIF Target Gene Expression.

(A–B, E–F) Expression of HIF target genes, GLUT1 (A and E) and LDHA (B and F) in LF treated with siD2HGDH and siL2HGDH (A and B) or D2HGDH and L2HGDH (E and F). (C) Representative immunoblot demonstrating normoxic HIF1α stabilization by siD2HGDH and siL2HGDH. (D) Quantification of HIF1α densitometry. (G) Representative micrographs of MitoSOX-stained cells. Images were inverted and leveled for presentation. Scale bar is 20 μm. (H) Quantification of MitoSOX staining. Data are mean ± SEM. See also Figure S5.
By contrast, 2HGDH overexpression had no effect on GLUT1 or LDHA mRNA (Figures 5E and 5F). This finding, together with the observation that D2HGDH knockdown had no impact on cellular 2HG (Figure 2D and S2C), suggested an alternative mechanism of HIF1α stabilization independent of direct PHD inhibition by 2HG. Superoxide through its metabolism to H2O2 is a known activator of HIF (Gorlach and Kietzmann, 2007). As D2HGDH and L2HGDH are localized to the inner mitochondrial membrane, we measured whether 2HGDH suppression increased superoxide production as a potential mechanism of HIF1α activation. Both siD2HGDH and siL2HGDH increased mitochondrial superoxide measured by MitoSOX (Figures 5G and 5H) in a pattern similar to the changes in HIF1α target gene expression. While treatment with N-acetylcysteine (NAC) had no impact on MitoSOX fluorescence or HIF1α target gene expression under normoxia, it did decrease the signal under hypoxia (Figures S5F–H). These data support an alternative mechanism for HIF stabilization beyond direct PHD inhibition by 2HG. While the correlation with superoxide production is compelling, these data suggest ROS generation is not the only mechanism linking 2HGDH silencing and HIF activation.
L2HG Metabolism Contributes to Cellular Redox Homeostasis
The metabolites 2OG and L2HG form a redox couple linked to NADH/NAD+ and FADH2/FAD. Moreover, both 2HG enantiomers appear to be biochemical cul-de-sacs that do not participate in other reactions beyond oxidation back to 2OG. These features raise the possibility that 2HG may provide cells with a reservoir of reducing equivalents, and that 2HG-2OG interconversions may buffer cellular redox potential.
To examine this, whole cell NADH/NAD+ ratios were measured by an enzymatic cycling assay in 2HGDH knockdown or overexpressing cells (Figure 6A and B). We found that increased cellular L2HG increases NADH/NAD+. To test if exogenous L2HG could also increase cellular NADH/NAD+, LF expressing a genetically encoded fluorescence sensor of NADH/NAD+ in situ, SoNar (Zhao et al., 2015), were treated with permeable trifluoromethylbenzyl ester (TFMB) derivatives of 2OG, D2HG, and L2HG (Losman et al., 2013; MacKenzie et al., 2007) (Figures 6C and S6). Both D2HG and L2HG derivatives increased the fluorescence ratio, indicating an increase in NADH/NAD+, while the permeable 2OG compound had the opposite effect. These increases in NADH/NAD+ were dose-dependent in HEK cells (Figure S6). This evidence lends support to the hypothesis that 2HG is a source of cellular reducing equivalents.
Figure 6. 2HG Metabolism Is Coupled to Cellular Redox State.
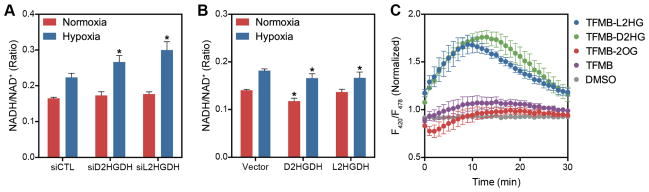
(A and B) NADH/NAD+ ratio determined by enzymatic cycling assay in LF treated with siRNA targeting D2HGDH or L2HGDH expression (A) or overexpressing D2HGDH or L2HGDH (B). (C) F420/F485 fluorescence ratio, corresponding to cytoplasmic NADH/NAD+, in LF treated with permeable analogues of 2OG, D2HG, and L2HG (500 μM) at time 0 normalized to the fluorescence ratio of untreated cells. Data are mean ± SEM. See also Figure S6.
L2HGDH Regulates Bioenergetic Metabolic Pathways
The TCA cycle and glycolysis are the primary pathways producing NADH in the mitochondrion and cytoplasm, respectively. Similar to succinate dehydrogenase (Complex II), both D2HGDH and L2HGDH are mitochondrial flavoproteins that couple electron transfer from 2HG to FAD and subsequently to coenzyme Q. One mechanism leading to NADH accumulation in siRNA-treated cells may be impaired electron transport. To test this hypothesis, siRNA-treated LF were subjected to a mitochondrial stress assay, where oxygen consumption rate (OCR) is measured as cells are treated sequentially with oligomycin to inhibit ATP synthesis, FCCP to collapse the inner membrane potential (ΔΨm), and antimycin A plus rotenone to inhibit all flux through the respiratory chain (Figures 7A and 7B). Compared to siCTL, siL2HGDH decreased basal OCR and both siD2HGDH and siL2HGDH decreased maximal OCR assessed after FCCP treatment. No difference in mitochondrial coupling efficiency or non-mitochondrial OCR was observed. To explore further the role of 2HG metabolism under hypoxic conditions, siRNA-treated cells were labeled with tetramethylrhodamine methyl ester (TMRM), a fluorescent indicator of ΔΨm. While no differences were observed in normoxia, siD2HGDH and siL2HGDH reversed the hypoxia-mediated increase in ΔΨm (Figures 7C and 7D). These data are consistent with the OCR measurements, and, together, indicate that both 2HGDH enzymes contribute to the generation of the mitochondrial membrane potential under stressed (e.g., FCCP or hypoxia) conditions.
Figure 7. D2HGDH and L2HGDH Silencing Affects Oxidative Phosphorylation and Glycolysis.

(A) Oxygen consumption rate (OCR) of LF treated with siRNA targeting D2HGDH or L2HGDH after treatment with the indicated compounds (Oligo, oligomycin; AMA, antimycin A). (B) Summarized data from (A). (C) Representative micrographs of TMRM-stained LF. Images were inverted and leveled for presentation. Scale bar is 20 μm. Scale bar is 20 μm. (D) Quantification of TMRM staining in normoxia and hypoxia in siRNA-treated LF. (E) Extracellular acidification rate (ECAR) of LF treated with siRNA targeting D2HGDH or L2HGDH before and after treatment with oligomycin to measure glycolytic capacity (2DG, 2-deoxyglucose). (F) Summarized data from (E). (G) Cell-permeable analogues of D2HG and L2HG inhibit oligomycin-stimulated ECAR. Data are mean ± SEM. See also Figure S7.
As glycolysis is a major determinant of cytoplasmic reducing potential under hypoxic conditions, we next measured the effect of 2HGDH knockdown on glycolytic flux by measuring the extracellular acidification rate (ECAR) in response to glucose and oligomycin (Wu et al., 2007). Cells treated with siL2HGDH had a modest decrease in basal ECAR and a marked decrease in oligomycin-stimulated ECAR compared to siCTL-treated cells (Figures 7E and 7F). In contrast, siD2HGDH increased glycolytic capacity while having no effect on basal glycolytic flux. To test if exogenous L2HG could inhibit oligomycin-stimulated ECAR, LF were treated with the cell permeable 2HG analogues. Both TFMB-D2HG and TFMB-L2HG reduced glycolytic capacity by 12 and 19%, respectively, compared to vehicle (Figure 7G). These findings were confirmed with alternative siRNA sequences targeting the 2HGDH enzymes (Figure S7A). PAEC treated with siRNA targeting the 2HGDH enzymes recapitulated the findings in LF, while PASMC demonstrated only inhibition of ECAR by siL2HGDH (Figures S7B and S7C). The latter finding is consistent with the inability of siD2HGDH to increase LDHA expression in PASMC (Figure S5E). NAC had little impact on oligomycin-stimulated ECAR in siD2HGDH-treated cells while further reducing glycolytic flux in siL2HGDH-treated cells (Figure S7D).
DISCUSSION
Hypoxia is an important stimulus that requires metabolic adaptation to sustain energy production, mitigate ROS production, maintain cellular redox homeostasis, and, in some cases, facilitate cell proliferation. Much of our understanding of the cellular response to hypoxia is derived from studies of the HIF1α transcriptional program or of metabolic flux in malignant cell lines. The goal of this study was to identify novel features of the hypoxia response in primary cultured cells. Here, we demonstrate that hypoxia specifically and dose-dependently induces the accumulation of L2HG in a variety of cell types and that L2HG metabolism is closely coupled to cellular redox state and energy metabolism through inhibition of glycolytic flux and oxygen consumption.
These studies confirm that the MDH enzymes are likely the primary enzymes responsible for L2HG generation in hypoxia. The most important mechanism for increased L2HG synthesis is likely related to increased substrate (2OG and NADH) availability. Hypoxia lowers L2HGDH, although siVHL treatment increased 2OG and 2HG in normoxia without affecting L2HGDH mRNA. The contrasting effects of VHL and HIF knockdown in LF and HEK cells specifically suggest different biochemical mechanisms leading to L2HG accumulation. MDH knockdown inhibited L2HG production in both cell types. In LF, VHL knockdown could partially recapitulate the hypoxia-mediated changes under normoxia, while HEK cells were insensitive siVHL despite demonstrable HIF1α stabilization. Indeed, HEK cells were also unaffected by siHIF1 in hypoxia, while this treatment markedly enhanced L2HG production in LF under hypoxia. Previous studies have highlighted the importance of glutaminolysis in malignant cells under hypoxic conditions (Gameiro et al., 2013; Scott et al., 2011; Wise et al., 2011). We speculate that the primary carbon source for L2HG production in LF is glycolysis while in HEK the primary carbon source may be glutaminolysis, thereby accounting for the differential consequences of HIF knockdown.
In addition to hypoxia, mutations in cytb in osteosarcoma cells (Complex III deficiency) and fumarate hydratase deficiency in renal carcinoma cells and Complex I inhibition with rotenone increase L2HG (Mullen et al., 2014; Worth et al., 2015). A metabolomics study of urinary organic acids in children with electron transport chain disorders also identified increases in total 2HG, presumed by the authors to be L2HG (Reinecke et al., 2012). These observations indicate that L2HG increases in response to mitochondrial reductive stress caused by respiratory chain or TCA dysfunction. All of these perturbations increase mitochondrial NADH, and likely 2OG, thereby providing the substrates for L2HG production. Given that the only known metabolic fate of L2HG is oxidation to 2OG, we considered that L2HG might serve as a sink for mitochondrial reducing equivalents under conditions of reductive stress. To test this, we measured whole cell NADH/NAD+ and found that this ratio varied directly with L2HG (and inversely with L2HGDH activity) in hypoxia. Results from the intracellular fluorescent NADH/NAD+ sensor indicate that exogenously administered L2HG can transfer electrons to NAD+, consistent with this potential role for L2HG as an electron sink (and carrier). Together, these data raise the question as to the mechanism of L2HG oxidation, particularly in the context of L2HGDH knockdown, as this is the only known enzyme with L2HG oxidase activity.
To examine the impact of L2HG redox shuttling on mitochondrial reducing potential, we measured OCR and TMRM fluorescence. These studies suggest that L2HGDH contributes to the generation of ΔΨm as shown by reduced FCCP-stimulated OCR in normoxia and decreased TMRM fluorescence in hypoxia. The data also suggest that L2HGDH downregulation in hypoxia may mitigate mitochondrial membrane hyperpolarization and attendant ROS generation. Indeed, siL2HGDH blunted the hypoxia-mediated increase in MitoSOX fluorescence accounting for elevated superoxide production in normoxia. Previous work has shown no direct effect of L2HG on electron flux through the respiratory chain in rat cortical homogenates and striated muscle cells (da Silva et al., 2002; Latini et al., 2005); however, L2HG was able to decrease the respiratory control ratio (RCR), suggesting uncoupling of electron transport from ATP production (Latini et al., 2005). Our data from whole cells may also be consistent with a decrease in RCR based on the reduction in FCCP-stimulated OCR (Brand and Nicholls, 2011); however, additional studies are necessary to clarify if this is a consequence of too much L2HG (and uncoupling) or too little electron transport via L2HGDH (and decreased ΔΨm).
Perhaps the most striking finding in the present work is the marked impact of L2HGDH knockdown on glycolytic flux and oligomycin-stimulated glycolytic capacity (25% and 35% reductions, respectively). This observation supports the compelling hypothesis that L2HG serves to couple the two main components of cellular NADH production, glycolysis and the TCA cycle, through a redox sensitive mechanism. As L2HG accumulates in response to reductive stress, it initiates a negative feedback loop to limit additional NADH production by glycolysis. Whether this occurs as a consequence of direct inhibition of glycolytic enzymes by L2HG or through accumulation of cytoplasmic NADH remains to be determined. The observation that cell permeable analogues of both D2HG and L2HG had similar effects on oligomycin-stimulated ECAR may support the latter hypothesis. To our knowledge, this is the first evidence of a mitochondrial metabolite regulating cytoplasmic energy production and represents a novel mechanism of cellular redox control.
Recently, investigators have described an l2hgdh−/− mouse characterized by neurological deficits (Rzem et al., 2015). A limited metabolomics analysis identified markedly increased concentrations of lysine and arginine associated with decreases in saccharopine, glutamine, and glutamate in the brains of adult l2hgdh−/− mice. L2HG inhibits lysine/2OG reductases, which converts lysine to saccharopine, in one pathway of lysine degradation. A role for L2HG inhibition of lysine/2OG reductases in hypoxia is not clear from our data as lysine levels decreased in both PAEC and PASMC. Interestingly, the lysine degradation pathway was identified in our (network-based) analyses, and lysine metabolism is a source of NADH production through the functions of sacchariopine dehydrogenase and α-aminoadipate semialdehyde dehydrogenase.
In contrast to L2HGDH silencing, D2HGDH silencing increased glycolytic capacity, presumably as a consequence of HIF1α activation and upregulation of glucose transporters and glycolytic genes. While the correlation between superoxide production and HIF stabilization suggested one potential mechanism linking these observations, NAC was unable to abrogate the changes in HIF target gene expression or glycolytic flux in the 2HGDH knockdown cells. While studies have shown that both D2HG and L2HG can inhibit PHD (Figueroa et al., 2010; Lu et al., 2012; Xu et al., 2011), the observations that HIF is stabilized in siD2HGDH-treated cells despite no change in intracellular D2HG levels and that blocking L2HG accumulation in hypoxia through L2HGDH overexpression has no effect on HIF target gene expression argue against this mechanism. The regulation of HIF is complex and a variety of mechanisms have been implicated in its stabilization, including micro RNA, other post-translational modifications, and intracellular signaling cascades (e.g., ERK, AKT), in addition to ROS and PHD inhibition (Movafagh et al., 2015; Yee Koh et al., 2008). Indeed, the incompletely understood etiology of HIF activation under aerobic conditions is of critical importance to our understanding of the pathogenesis of cancer and pulmonary vascular disease.
Manipulations of D2HG metabolism resulted in similar effects on NADH/NAD+, mitochondrial superoxide production, and OCR as manipulations of L2HG metabolism, which may be a consequence of similarities in the enzymology of D2HGDH and L2HGDH and the structural similarities of D2HG and L2HG. Importantly, these data clearly indicate that hypoxia affects L2HG metabolism exclusively, indicating that exquisite enantiomeric specificity is provided by the biology of the hypoxia response. PASMC were the only cell type in which hypoxia-mediated increases in D2HG were observed by biochemical derivatization; however, siD2HGDH was unable to potentiate the hypoxia-mediated increases in total 2HG in these cells. Given that PASMC proliferate in response to hypoxia, one interesting hypothesis is that the D2HG represents incomplete reductive carboxylation by IDH in these cells as has been observed in a variety of cancer cell lines (Gameiro et al., 2013; Scott et al., 2011; Wise et al., 2011).
While D2HG is a so-called “oncometabolite” produced by mutations in IDH associated with an increasing number of human malignancies (Cairns and Mak, 2013), these data reinforce the notion that D2HG and L2HG are separate and distinct metabolites with unique effects on metabolic regulation (Struys, 2013). Given the prominence of regional hypoxia in tumor biology, this work also highlights the importance of enantiomeric characterization of 2HG elevations, particularly as L2HG has recently been identified in association with renal cell carcinoma tissues, many of which contained VHL mutations (Shim et al., 2014).
L2HGDH is found in bacteria, plants, and animals (Engqvist et al., 2014). Despite its evolutionary conservation, the only ascribed role for this enzyme is the recovery of 2OG mistakenly reduced by non-specific enzymatic activity (Kalliri et al., 2008; Rzem et al., 2007). Indeed, L2HG aciduria, caused by mutations in L2HGDH, has been described as a “disorder of metabolite repair” (Rzem et al., 2007). In this work, we propose that L2HG serves an important role in the regulation of cellular redox homeostasis and cellular energy metabolism through its regulation of glycolysis and oxidative phosphorylation. Future studies will focus on the relationships between L2HG, glycolytic enzymes and intermediates, and the regulation of compartmental NADH/NAD+ to test this hypothesis. Importantly, our data show that L2HG is much more sensitive to metabolic perturbations and hypoxia than D2HG in the cells studied here and may play a critical role in the adaptive response to hypoxia.
EXPERIMENTAL PROCEDURES
Cell lines and cell culture
Primary human pulmonary artery endothelial cells (PAEC), pulmonary artery smooth muscle cells (PASMC), lung fibroblasts (LF), and mesenchymal stem cells (MSC) were cultured in their respective growth media without antibiotics (Lonza). HEK293 and HepG2 cells were cultured in High Glucose DMEM (Life Technologies) supplemented with 10% FBS (Mediatech).
Hypoxia treatment
Cells were placed in a modular incubator chamber (Billups-Rothenberg) flushed with 100 L of a gas mixture containing 0.2% or 2% O2, 5% CO2, and balanced with N2 (Airgas) then incubated at 37 °C for 24 h.
Targeted LC-MS
Metabolites were extracted in cold 80% methanol. Prior to scraping, [13C4]-2OG (Cambridge Isotope Laboratories) was added as an internal standard (ISTD). Metabolites were separated using a ZIC-HILIC stationary phase (150 mm×2.1 mm×3.5 μm; Merck). The MS parameters were optimized using a 2HG standard solution. Monitored mass transitions were 147 → 129 (2HG, Rt 10.3 min), 145 → 01 (2OG, Rt 10.1 min), 133 → 115 (malate, Rt 10.6 min), 115 → 73 + 99 (succinate, Rt 4.7 min), and 149 → 105 (ISTD, Rt 10.1 min). Retention time windows were confirmed by the analysis of neat and matrix-spiked standards. Peak areas were quantified by Xcalibur Software (Thermo) and manually reviewed. Concentrations were determined by interpolating the peak area ratio of analyte to ISTD from a standard curve (0.1–10 μM neat standards). Concentrations were normalized to cell count from plates of cells treated in parallel.
Quantification of 2HG enantiomers
Metabolite extracts were derivatized with diacetyl-L-tartaric anhydride (DATAN) using an adaptation of a previously described method (Struys et al., 2004a). In-source fragmentation of the derivatized species was observed, and the mass transition for 2HG, 147 → 129, was monitored. The derivatization reaction proceeded to completion, and 2HG and DATAN-2HG were well resolved chromatographically with an Rt shift from 10.5 min (D2HG) to 11.4 (L2HG) (Figure 2A). Standard solutions of enantiomerically pure D2HG and L2HG were derivatized with 100 μM lactate as a catalyst in parallel to generate a standard curve (0–2 μM). Concentrations of D2HG and L2HG were normalized to cell count.
NADH/NAD+ enzymatic assay
Cellular NAD+ and NADH were measured using an adaptation of a previously published enzymatic fluorimetric cycling assay (Umemura and Kimura, 2005; Zhu and Rand, 2012).
Reactive oxygen species and ΔΨm measurements
Cells were stained with 5 μM MitoSOX (Life Technologies) or TMRM (2 nM) for 30 min at 37 °C. T en random images per well were recorded using a Nikon TE2000 epifluorescent microscope. Mean cell fluorescence intensity for each image was determined using CellProfiler (Kamentsky et al., 2011). For presentation, the micrographs were inverted and leveled using the same range for all images.
Extracellular flux analyses
Cells were treated with siRNA 48 h prior to the experiment and seeded at a density of 25,000 cells per well 24 h prior to the assay. NAC (10 mM) was added at the time of seeding in the assay plate. OCR and ECAR were measured on an XF24 or XFe24 Analyzer (Seahorse Biosciences). Mitochondrial and glycolytic stress assays were performed according to the manufacturer’s protocols. Permeable compounds were injected following oligomycin in a standard glycolysis stress assay and ECAR was measured after 4 h and prior to injection of 2-deoxyglucose. OCR and ECAR were normalized to cell count measured after assay completion.
Statistical analyses
Calculations were performed with GraphPad Prism 6. Data are the mean ± SEM of a minimum of three biological replicates. Data were analyzed by paired Student’s t test, one-way ANOVA, or two-way ANOVA as appropriate. The significance threshold used was p < 0.05. Same treatment normoxia-hypoxia differences are indicated by † and same oxygen treatment-control differences are indicated by *.
Supplementary Material
HIGHLIGHTS.
Hypoxia increases cellular L-2-hydroxyglutarate (L2HG)
L2HG accumulates in response to mitochondrial dysfunction
This accumulation is independent of hypoxia-inducible factor activation
L2HG inhibits electron transport and glycolysis to mitigate reductive stress
Acknowledgments
This work was supported by the NIH/NHLBI (HL007633 to WMO; HL048743, HL061795, and HL108630 to JL), the Natural Science Foundation of China (31225008 and 91313301 to YY), and the Brigham and Women’s Hospital Department of Medicine.
Footnotes
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Achouri Y, Noel G, Vertommen D, Rider MH, Veiga-Da-Cunha M, Van Schaftingen E. Identification of a dehydrogenase acting on D-2-hydroxyglutarate. Biochem J. 2004;381:35–42. doi: 10.1042/BJ20031933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov. 2013;3:730–741. doi: 10.1158/2159-8290.CD-13-0083. [DOI] [PubMed] [Google Scholar]
- Chalmers RA, Lawson AM, Watts RW, Tavill AS, Kamerling JP, Hey E, Ogilvie D. D-2-hydroxyglutaric aciduria: case report and biochemical studies. J Inherit Metab Dis. 1980;3:11–15. doi: 10.1007/BF02312516. [DOI] [PubMed] [Google Scholar]
- Cottrill KA, Chan SY. Metabolic dysfunction in pulmonary hypertension: the expanding relevance of the Warburg effect. Eur J Clin Invest. 2013;43:855–865. doi: 10.1111/eci.12104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva CG, Ribeiro CA, Leipnitz G, Dutra-Filho CS, Wyse AA, Wannmacher CM, Sarkis JJ, Jakobs C, Wajner M. Inhibition of cytochrome c oxidase activity in rat cerebral cortex and human skeletal muscle by D-2-hydroxyglutaric acid in vitro. Biochim Biophys Acta. 2002;1586:81–91. doi: 10.1016/s0925-4439(01)00088-6. [DOI] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran M, Kamerling JP, Bakker HD, van Gennip AH, Wadman SK. L-2-Hydroxyglutaric aciduria: an inborn error of metabolism? J Inherit Metab Dis. 1980;3:109–112. doi: 10.1007/BF02312543. [DOI] [PubMed] [Google Scholar]
- Engqvist MK, Esser C, Maier A, Lercher MJ, Maurino VG. Mitochondrial 2-hydroxyglutarate metabolism. Mitochondrion. 2014 doi: 10.1016/j.mito.2014.02.009. [DOI] [PubMed] [Google Scholar]
- Fan J, Kamphorst JJ, Rabinowitz JD, Shlomi T. Fatty acid labeling from glutamine in hypoxia can be explained by isotope exchange without net reductive IDH flux. J Biol Chem. 2013 doi: 10.1074/jbc.M113.502740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gameiro PA, Yang J, Metelo AM, Perez-Carro R, Baker R, Wang Z, Arreola A, Rathmell WK, Olumi A, Lopez-Larrubia P, et al. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 2013;17:372–385. doi: 10.1016/j.cmet.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlach A, Kietzmann T. Superoxide and derived reactive oxygen species in the regulation of hypoxia-inducible factors. Methods Enzymol. 2007;435:421–446. doi: 10.1016/S0076-6879(07)35022-2. [DOI] [PubMed] [Google Scholar]
- Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Kalliri E, Mulrooney SB, Hausinger RP. Identification of Escherichia coli YgaF as an L-2-hydroxyglutarate oxidase. J Bacteriol. 2008;190:3793–3798. doi: 10.1128/JB.01977-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamentsky L, Jones TR, Fraser A, Bray MA, Logan DJ, Madden KL, Ljosa V, Rueden C, Eliceiri KW, Carpenter AE. Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics. 2011;27:1179–1180. doi: 10.1093/bioinformatics/btr095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranendijk M, Struys EA, Salomons GS, Van der Knaap MS, Jakobs C. Progress in understanding 2-hydroxyglutaric acidurias. J Inherit Metab Dis. 2012;35:571–587. doi: 10.1007/s10545-012-9462-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai JC, White BK, Buerstatte CR, Haddad GG, Novotny EJ, Jr, Behar KL. Chronic hypoxia in development selectively alters the activities of key enzymes of glucose oxidative metabolism in brain regions. Neurochem Res. 2003;28:933–940. doi: 10.1023/a:1023235712524. [DOI] [PubMed] [Google Scholar]
- Latini A, da Silva CG, Ferreira GC, Schuck PF, Scussiato K, Sarkis JJ, Dutra Filho CS, Wyse AT, Wannmacher CM, Wajner M. Mitochondrial energy metabolism is markedly impaired by D-2-hydroxyglutaric acid in rat tissues. Mol Genet Metab. 2005;86:188–199. doi: 10.1016/j.ymgme.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15:110–121. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman JA, Kaelin WG., Jr What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27:836–852. doi: 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, Kaelin WG., Jr (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie ED, Selak MA, Tennant DA, Payne LJ, Crosby S, Frederiksen CM, Watson DG, Gottlieb E. Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol Cell Biol. 2007;27:3282–3289. doi: 10.1128/MCB.01927-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga H, Futakuchi-Tsuchida A, Takahashi M, Ishikawa T, Tsuji M, Ando O. IDH1 and IDH2 have critical roles in 2-hydroxyglutarate production in D-2-hydroxyglutarate dehydrogenase depleted cells. Biochem Biophys Res Commun. 2012;423:553–556. doi: 10.1016/j.bbrc.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Movafagh S, Crook S, Vo K. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: new developments in an old debate. J Cell Biochem. 2015;116:696–703. doi: 10.1002/jcb.25074. [DOI] [PubMed] [Google Scholar]
- Mullen AR, Hu Z, Shi X, Jiang L, Boroughs LK, Kovacs Z, Boriack R, Rakheja D, Sullivan LB, Linehan WM, et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 2014;7:1679–1690. doi: 10.1016/j.celrep.2014.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MS. Inhibition by the branched-chain 2-oxo acids of the 2-oxoglutarate dehydrogenase complex in developing rat and human brain. Biochem J. 1974;144:91–97. doi: 10.1042/bj1440091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinecke C, Koekemoer G, van der Westhuizen F, Louw R, Lindeque J, Mienie L, Smuts I. Metabolomics of urinary organic acids in respiratory chain deficiencies in children. Metabolomics. 2012;8:264–283. [Google Scholar]
- Rzem R, Achouri Y, Marbaix E, Schakman O, Wiame E, Marie S, Gailly P, Vincent MF, Veiga-da-Cunha M, Van Schaftingen E. A mouse model of L-2-hydroxyglutaric aciduria, a disorder of metabolite repair. PLoS One. 2015;10:e0119540. doi: 10.1371/journal.pone.0119540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzem R, Van Schaftingen E, Veiga-da-Cunha M. The gene mutated in l-2-hydroxyglutaric aciduria encodes l-2-hydroxyglutarate dehydrogenase. Biochimie. 2006;88:113–116. doi: 10.1016/j.biochi.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Rzem R, Veiga-da-Cunha M, Noel G, Goffette S, Nassogne MC, Tabarki B, Scholler C, Marquardt T, Vikkula M, Van Schaftingen E. A gene encoding a putative FAD-dependent L-2-hydroxyglutarate dehydrogenase is mutated in L-2-hydroxyglutaric aciduria. Proc Natl Acad Sci U S A. 2004;101:16849–16854. doi: 10.1073/pnas.0404840101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzem R, Vincent MF, Van Schaftingen E, Veiga-da-Cunha M. L-2-hydroxyglutaric aciduria, a defect of metabolite repair. J Inherit Metab Dis. 2007;30:681–689. doi: 10.1007/s10545-007-0487-0. [DOI] [PubMed] [Google Scholar]
- Scott DA, Richardson AD, Filipp FV, Knutzen CA, Chiang GG, Ronai ZA, Osterman AL, Smith JW. Comparative metabolic flux profiling of melanoma cell lines: beyond the Warburg effect. J Biol Chem. 2011;286:42626–42634. doi: 10.1074/jbc.M111.282046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim EH, Livi CB, Rakheja D, Tan J, Benson D, Parekh V, Kho EY, Ghosh AP, Kirkman R, Velu S, et al. L-2-Hydroxyglutarate: An Epigenetic Modifier and Putative Oncometabolite in Renal Cancer. Cancer Discov. 2014 doi: 10.1158/2159-8290.CD-13-0696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkov AA. An update on the role of mitochondrial alpha-ketoglutarate dehydrogenase in oxidative stress. Mol Cell Neurosci. 2013;55:13–16. doi: 10.1016/j.mcn.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struys EA. 2-Hydroxyglutarate is not a metabolite; D-2-hydroxyglutarate and L-2-hydroxyglutarate are! Proc Natl Acad Sci U S A. 2013;110:E4939. doi: 10.1073/pnas.1318777110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struys EA, Gibson KM, Jakobs C. Novel insights into L-2-hydroxyglutaric aciduria: mass isotopomer studies reveal 2-oxoglutaric acid as the metabolic precursor of L-2-hydroxyglutaric acid. J Inherit Metab Dis. 2007;30:690–693. doi: 10.1007/s10545-007-0697-5. [DOI] [PubMed] [Google Scholar]
- Struys EA, Jansen EE, Verhoeven NM, Jakobs C. Measurement of urinary D- and L-2-hydroxyglutarate enantiomers by stable-isotope-dilution liquid chromatography-tandem mass spectrometry after derivatization with diacetyl-L-tartaric anhydride. Clin Chem. 2004a;50:1391–1395. doi: 10.1373/clinchem.2004.033399. [DOI] [PubMed] [Google Scholar]
- Struys EA, Salomons GS, Achouri Y, Van Schaftingen E, Grosso S, Craigen WJ, Verhoeven NM, Jakobs C. Mutations in the D-2-hydroxyglutarate dehydrogenase gene cause D-2-hydroxyglutaric aciduria. Am J Hum Genet. 2005;76:358–360. doi: 10.1086/427890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struys EA, Verhoeven NM, Brunengraber H, Jakobs C. Investigations by mass isotopomer analysis of the formation of D-2-hydroxyglutarate by cultured lymphoblasts from two patients with D-2-hydroxyglutaric aciduria. FEBS Lett. 2004b;557:115–120. doi: 10.1016/s0014-5793(03)01459-5. [DOI] [PubMed] [Google Scholar]
- Topcu M, Jobard F, Halliez S, Coskun T, Yalcinkayal C, Gerceker FO, Wanders RJ, Prud’homme JF, Lathrop M, Ozguc M, et al. L-2-Hydroxyglutaric aciduria: identification of a mutant gene C14orf160, localized on chromosome 14q22.1. Hum Mol Genet. 2004;13:2803–2811. doi: 10.1093/hmg/ddh300. [DOI] [PubMed] [Google Scholar]
- Tretter L, Adam-Vizi V. Inhibition of alpha-ketoglutarate dehydrogenase due to H2O2-induced oxidative stress in nerve terminals. Ann N Y Acad Sci. 1999;893:412–416. doi: 10.1111/j.1749-6632.1999.tb07867.x. [DOI] [PubMed] [Google Scholar]
- Umemura K, Kimura H. Determination of oxidized and reduced nicotinamide adenine dinucleotide in cell monolayers using a single extraction procedure and a spectrophotometric assay. Anal Biochem. 2005;338:131–135. doi: 10.1016/j.ab.2004.10.020. [DOI] [PubMed] [Google Scholar]
- Vemuri GN, Eiteman MA, McEwen JE, Olsson L, Nielsen J. Increasing NADH oxidation reduces overflow metabolism in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2007;104:2402–2407. doi: 10.1073/pnas.0607469104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A. 2011;108:19611–19616. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worth AJ, Gillespie KP, Mesaros C, Guo L, Basu SS, Snyder NW, Blair IA. Rotenone stereospecifically increases (S)-2-hydroxyglutarate in SH-SY5Y neuronal cells. Chem Res Toxicol. 2015 doi: 10.1021/tx500535c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292:C125–136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- Xia J, Mandal R, Sinelnikov IV, Broadhurst D, Wishart DS. MetaboAnalyst 2.0--a comprehensive server for metabolomic data analysis. Nucleic Acids Res. 2012;40:W127–133. doi: 10.1093/nar/gks374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Psychogios N, Young N, Wishart DS. MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009;37:W652–660. doi: 10.1093/nar/gkp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee Koh M, Spivak-Kroizman TR, Powis G. HIF-1 regulation: not so easy come, easy go. Trends Biochem Sci. 2008;33:526–534. doi: 10.1016/j.tibs.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhao Y, Hu Q, Cheng F, Su N, Wang A, Zou Y, Hu H, Chen X, Zhou HM, Huang X, et al. SoNar, a Highly Responsive NAD(+)/NADH Sensor, Allows High-Throughput Metabolic Screening of Anti-tumor Agents. Cell Metab. 2015;21:777–789. doi: 10.1016/j.cmet.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu CT, Rand DM. A hydrazine coupled cycling assay validates the decrease in redox ratio under starvation in Drosophila. PLoS One. 2012;7:e47584. doi: 10.1371/journal.pone.0047584. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.