

Abstract
Aberrant expression of the epidermal growth factor receptor (EGFR) is a common characteristic of many cancers including non-small cell lung carcinoma (NSCLC), head and neck squamous cell carcinoma, and ovarian cancer. While EGFR is currently a favorite molecular target for treatment of these cancers, inhibition of the receptor with small molecule inhibitors (i.e.- erlotinib) or monoclonal antibodies (i.e.- cetuximab) does not provide long-term therapeutic benefit as standalone treatment. Interestingly, we have found that addition of erlotinib to photodynamic therapy (PDT) can improve treatment response in typically erlotinib-resistant NSCLC tumor xenografts. Ninety-day complete response rates of 63% are achieved when erlotinib is administered in three doses before PDT of H460 human tumor xenografts, compared to 16% after PDT-alone. Similar benefit is found when erlotinib is added to PDT of A549 NCSLC xenografts. Improved response is accompanied by increased vascular shutdown, and erlotinib increases the in vitro cytotoxicity of PDT to endothelial cells. Tumor uptake of the photosensitizer (benzoporphyrin derivative monoacid ring A; BPD) is increased by the in vivo administration of erlotinib; nevertheless, this elevation of BPD levels only partially accounts for the benefit of erlotinib to PDT. Thus, pretreatment with erlotinib augments multiple mechanisms of PDT effect that collectively lead to large improvements in therapeutic efficacy. These data demonstrate that short-duration administration of erlotinib before PDT can greatly improve the responsiveness of even erlotinib-resistant tumors to treatment. Results will inform clinical investigation of EGFR-targeting therapeutics in conjunction with PDT.
Keywords: Erlotinib, photodynamic therapy, non-small cell lung cancer, EGFR, VEGF
Introduction
A leading characteristic of aggressive tumors is aberrant expression of the epidermal growth factor receptor (EGFR), a signaling protein involved in promoting cell proliferation, angiogenesis, and survival. Several EGFR-inhibiting drugs are used to treat malignancies such as non-small cell lung carcinoma (NSCLC), head and neck squamous cell carcinoma (HNSCC), and breast cancer (1, 2). These drugs include EGFR-targeting antibodies (i.e. – cetuximab) and small molecule inhibitors (e.g. – gefitinib, erlotinib). The efficacy of EGFR inhibition varies based on the molecular characteristics of the tumor. For example, tumors harboring EGFR-activating mutations tend to be more sensitive to EGFR-targeted therapy, whereas tumors that exhibit KRAS mutations respond poorly; and while some tumor types are initially sensitive to EGFR inhibition, others will acquire resistance, leaving patients with limited treatment options (1, 3). Thus, while EGFR has gained favor as a molecular target for treating several types of tumors, inhibition of the receptor as standalone treatment does not consistently yield clinically positive results.
Photodynamic therapy (PDT) employs a combination of light, photosensitizer, and oxygen to inflict cell damage. Combinations of PDT with drugs that target EGFR suggest that multimodality therapy can increase cytotoxicity (4, 5). EGFR targeting of photosensitizer reduces cell proliferation after PDT (6), and anti-EGFR antibodies and small molecular inhibitors can increase apoptosis and/or inhibit proliferation in tumors or cell cultures that are treated with PDT (7–10). Therapeutic benefit has been shown in combinations of PDT with cetuximab in tumor models of ovarian, nasopharyngeal, and bladder cancer (7–9). Such results have stimulated clinical interest in combining EGFR inhibition with PDT, but the mechanisms that underlie this added therapeutic benefit are poorly understood.
The goals of the present study are two-fold. First, we establish the therapeutic efficacy of adding erlotinib, a small molecule inhibitor of EGFR, before PDT of NSCLC human tumor xenografts. Erlotinib is an oral drug, so pre-PDT administration is translatable to intraoperative delivery of PDT, where oral drug administration to patients in the intensive care unit postoperatively is not feasible. Second, we have elucidated the mechanisms by which response is improved using this protocol. The effects of erlotinib on PDT-induced tumor cytotoxicity, endothelial cell damage, and vascular function have been determined and studied for their contribution to long-term PDT responsiveness. These findings provide insights that will inform the clinical development of treatment regimens that incorporate targeting of EGFR with PDT.
Materials and Methods
Cell lines
EGFR wild-type, KRAS mutant human NSCLC cell lines, H460 and A549, were obtained from American Type Culture Collection (Manassas, VA). H460-mCherry cells and SVEC (immortalized mouse endothelial cells) were provided by Drs. Jay Dorsey (University of Pennsylvania) and Bin Chen (University of the Sciences, Philadelphia, PA), respectively. Cells were maintained in RPMI-1640 (H460, SVEC) or Dulbecco’s modified Eagle medium (A549) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. All cell lines were authenticated by and confirmed as mycoplasma free using the Cell Check™ Plus authentication service from IDEXX Bioresearch™ (May, 2014).
Tumor models
Tumors were propagated by intradermal injection of 1×106 cells over the right shoulder of female nude mice (Athymic, Ncr-nu/nu; National Cancer Institute, Frederick, MD). Studies were initiated when tumors reached diameters of 6–8 mm. Investigations were approved by the University of Pennsylvania Institutional Animal Care and Use Committee; animal facilities are AAALAC accredited.
PDT
Animals received benzoporphyrin derivative monoacid ring A (BPD, Visudyne®; QLT Ophthalmics, Vancouver, CA), at 1 mg/kg (i.v.), 3h before light delivery (690 ± 5 nm). Light from a diode laser (B&W Tek, Inc.) was delivered through microlens-tipped fibers (Pioneer, Bloomfield, CT) to produce a 1 cm diameter field of uniform intensity. Laser output was measured (LabMaster power meter; Coherent, Auburn, CA) and adjusted to produce a fluence rate of 75 mW/cm2 at the tumor surface. A fluence of 135 J/cm2 was delivered, during which mice were anesthetized by inhalation of isoflurane in medical air (VetEquip anesthesia machine).
Anti-EGFR and anti-VEGF treatment
PDT was combined with erlotinib (Tarceva®, Genentech, San Francisco, CA) or bevacizumab (Avastin®, Genentech). Erlotinib (50 mg/kg) was administered (oral gavage) at either 3h before PDT (single dose) or at 2d, 1d, and 3h before PDT (triple dose). Dosing was chosen from pilot studies in which 50 mg/kg was efficacious in the absence of toxicities, whereas higher doses (up to 100 mg/kg) resulted in dramatic weight loss in the animals. Bevacizumab (10 mg/kg) was administered via intraperitoneal injection at 2d, 1d, and 3h before PDT.
Tumor response assays
After treatment, animals were followed daily to assess long-term outcome. Tumor volume was calculated using the formula volume=diameter × width2 × π/6. Animals were euthanized when tumor volume exceeded its pretreatment measurement or at 90 days after PDT in the absence of tumor recurrence (i.e., complete response).
Phosphorescence lifetime oximetry
Phosphorescence lifetime-based measurements of tumor oxygenation were performed as described previously (11) utilizing intratumoral injection of the probe Oxyphor G4 (20 mM). Erlotinib was administered on a triple-dose schedule. Tumor pO2 was measured before erlotinib (50 mg/kg) administration on each day and then at 3h after the final dose (corresponding to measurements at 0, 24, 48, and 51h relative to the first dose of erlotinib). Phosphorescence lifetime oximetry is based on variations in the probe phosphorescence decay time due to quenching of the probe triplet state by oxygen. The measured quantity (phosphorescence lifetime) is dependent on the concentration of oxygen, but not upon the concentration of the probe, the excitation light intensity or signal collection efficiency. Data represent the average of multiple measurements that were taken at different locations on the tumor.
Flow cytometry
Treated or control H460-mCherry tumors (2–5 pooled/condition) were excised and studied for the presence of treatment-induced apoptosis (labeled by Annexin V) and necrosis (labeled by DAPI). The staining protocol is detailed in Supplemental Materials and Methods. Samples were analyzed on a LSRII Flow Cytometer using Diva software (Becton Dickinson, Mountain View, CA). Data were analyzed in FlowJo (Treestar, San Carlos, CA).
In vivo imaging
To detect BPD, animals were imaged 3h post-BPD injection. For perfusion imaging, IRDye®800 PEG (LICOR, Lincoln, NE) was administered i.v. 15–30 min before imaging at 1h, 4h, and 18h after PDT. Imaging was performed on a LICOR Pearl Impulse using white light, 700 nm, and 800 nm to visualize anatomy, BPD, and IRDye®800, respectively. For analysis, similarly-sized tumor and flank-identifying regions of interest (ROIs) were drawn on the white light images and applied to the near-infrared images. Data are expressed as average fluorescence intensity (FI)/unit area, and for IRDye®800 images, data are normalized to the flank in order to provide for individual standardization of dye circulation. For scab measurements, ROIs were drawn around scab-containing areas in the white light images, and data are expressed as the number of pixels in the scab (or the pre-PDT tumor).
ELISA
Tumor-localized concentrations of human VEGF were assayed by ELISA (R&D Systems). Excised tumors were immediately frozen in a slurry of dry ice and 70% ethanol, then homogenized on dry ice and resuspended in cold 1X PBS. Vortexed samples were freeze-thawed (3 times), centrifuged, and protein concentrations of cleared lysates were detected using BCA reagent (Thermo Scientific, Rockford, IL). Manufacturer’s protocols were subsequently followed. Plates were analyzed using SoftMax Pro software (Molecular Devices, Sunnyvale, CA).
Western blot analysis
EGFR expression in lysed solutions of SVEC, H460, and A549 cells was confirmed by Western blotting. Probing with anti-EGFR antibody (Cell Signaling Technologies) was performed overnight on equivalent amounts of protein electrophoresed on BIs-Tris NuPage gels (Life Technologies) and transferred to nitrocellulose membranes. Anti-β-actin antibody was used as a loading control. Details on the staining protocol are provided in the Supplemental Materials and Methods.
AlamarBlue® assay
Viability/proliferation was assessed in cells that were either left untreated or exposed to erlotinib-alone, light-alone, BPD-alone, DMSO-alone, PDT-alone or erlotinib/PDT. Specific to the condition studied, cells were pretreated with 4 μM erlotinib 2d before PDT. The following day (1d pre-PDT), cells were reseeded into black-walled 96 well plates (4 wells/condition), and incubated overnight. On the day of PDT, BPD (100 ng/ml) and/or erlotinib (4 μM) were added for a 3h incubation, cells were illuminated (5 mW/cm2, 0.5 J/cm2), and replacement medium was added that contained a low dose (1 μM) of Erlotinib (accounting for cell exposure to erlotinib in the short term after PDT in vivo). At 20h after PDT, the medium was replaced with alamarBlue®-containing medium (cells were not reseeded) and the plate was read every hour up to 25h post-PDT. A five hour incubation in alamarBlue® reagent produced a strong but unsaturated signal, thus viability was assessed at the 25h timepoint per the manufacturer’s protocol. Data are expressed as viability relative to untreated controls.
In vitro BPD uptake
Cells were incubated with 4μM erlotinib at 2d, 1d, and 3h before their extraction. BPD (100 ng/ml) was included with the final addition of erlotinib. After 3h, cells were washed 2x in Cell Rinse (0.1 M NaCl, 5 mM KCl, 1 mM NaH2PO4, 6 mM glucose, 20 mM HEPES, pH 7.2), extracted in 1% SDS, passed through a 30 g needle 2–3x to shear the DNA, and stored frozen (−20°C). For analysis, samples were heated (50°C), diluted in PBS, and read on a Fluoromax-4 spectrofluorometer (Horiba Scientific, Edison, NJ) with excitation at 405 nm. Peak emission between 688–693 nm was determined and BPD concentration was calculated by comparison to a standard curve. Protein concentrations were determined using a BCA assay (Pierce) and data are expressed as BPD uptake per unit protein.
Bilirubin tests
Animals received either single- or triple-dose erlotinib treatment. Three hours after the final erlotinib dose was administered, anesthetized animals underwent a terminal cardiac puncture. Blood coagulated at room temperature (~20 min), and plasma was collected by centrifugation and frozen (−20°C). Bilirubin content was analyzed by Antech Diagnostics (Lake Success, NY).
Clonogenic assay
Following the protocol used in the alamarBlue® assay, cells that were untreated, as well as those that were erlotinib and/or PDT-treated, were plated at defined concentrations and incubated to permit colony formation (~7 days). Tests of a single “dose” of erlotinib were performed as a 3h erlotinib exposure before PDT. A slightly higher light dose (0.8 J/cm2) was used for clonogenics due to the more protracted time course of this assay. Surviving fractions were calculated relative to the plating efficacy of cells exposed to neither erlotinib nor PDT.
Statistical analysis
Means and standard errors are reported in figures. Student’s t-tests (JMP, SAS Institute, Inc.; Cary, NC) were used to assess differences in means between groups, with paired tests used where noted. Tumor response data were described using the Kaplan-Meier method, yielding median regrowth times, and 95% confidence intervals (95% CI) noting that in some cases the upper bound was undetermined. We fit a Cox model to the data and determined differences in median survival between groups based on a Wald test. A post-hoc test for trend was used to determine whether median survival increased as a function of erlotinib dose (zero, one or three doses). Hypothesis tests were one-sided for the tumor response data, as there was no scientific interest in determining whether treatment reduced response, and two-sided for all other hypothesis tests. The Type I error rate was 0.05.
Results
Erlotinib improves therapeutic response and enhances shutdown of the tumor vasculature after PDT
We first considered the therapeutic effect of erlotinib administration before PDT. At 2d, 1d, and 3h before light delivery, erlotinib was administered to mice bearing H460 tumors (Figure 1A). All animals treated with erlotinib regrew their tumors in less than one day (i.e., no tumor regression occurred). In mice treated with PDT, median time-to-regrowth was 11 days (95% CI 5,29 days), and impressively, pretreatment with erlotinib increased the median time-to-regrowth to over 90 days after PDT (p<0.001 compared to PDT). This was accompanied by an increase in 90-day complete response rate from 16% (PDT) to 63% (erlotinib/PDT). Erlotinib similarly improved response in mice bearing A549 tumors (Figure 1B). Pretreatment with erlotinib increased median time-to-regrowth to 69 days versus 8 days in animals that received PDT (p=0.033). This was reflected in an increase in 90-day complete response rate from 15% (PDT) to 51% (erlotinib/PDT).
Figure 1.
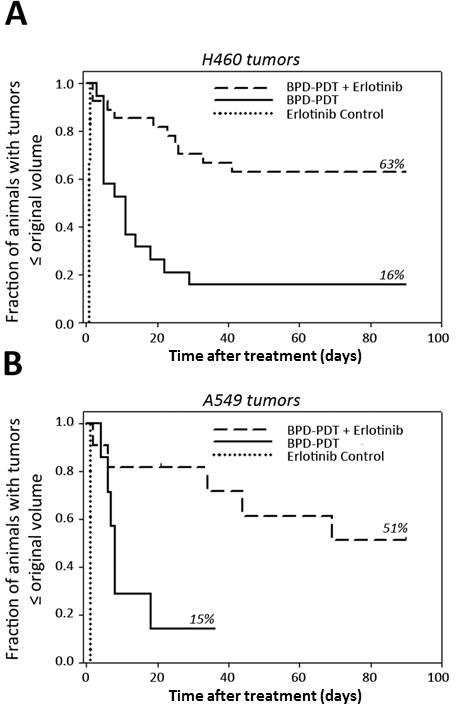
Addition of erlotinib to BPD-PDT improves therapeutic response of mice bearing NSCLC tumor xenografts. (A) Response of H460 tumors to erlotinib (n=6), BPD-PDT (n=19), or erlotinib/BPD-PDT (n=27). (B) Response of A549 tumors (n=7–11) to the same conditions. Three daily doses of erlotinib were administered prior to PDT. Tumor regrowth was monitored up to 90d post-PDT. For (B), a long-term survivor of PDT was euthanized (tumor-free) at 36 days due to unrelated illness.
These findings identify exceptional improvements in therapeutic outcome as a consequence of adding erlotinib to PDT. The results were far superior to the benefit that was expected from our previous in vitro work in which erlotinib augmented the cytotoxic effect of PDT by <1 log of kill (12). As such, we considered the potential contribution of in vivo-specific factors to tumor response with erlotinib/PDT. Others have reported short duration treatment with erlotinib to increase tumor oxygenation (13), which could improve the in vivo effectiveness of PDT relative to in vitro findings (for which oxygen limitation is not an issue). To test for this, tumor oxygenation was measured after the administration of erlotinib. Compared to baseline values in each tumor, no appreciable increases in oxygenation were observed after erlotinib administration (Supplemental Figure 1).
Flow cytometry was performed to study the nature and time course of treatment-induced cytotoxicity (Supplemental Figure 2). In agreement with tumor response results, erlotinib was minimally cytotoxic to H460 tumors. When used as a single agent, erlotinib induced only small, insignificant increases in apoptosis (augmented by the addition of PDT) that did not affect overall viability. In the absence of PDT, erlotinib did not induce necrosis. Moreover, these data revealed erlotinib-mediated increases in PDT cytotoxicity at delayed (18h), but not early (1h) time points after treatment. This time course is consistent with PDT cytotoxicity secondary to in vivo microenvironmental effects, such as vascular damage, and led us to next evaluate the contribution of vascular effects to the erlotinib-enhanced PDT response.
Near-infrared in vivo imaging of tumor perfusion was employed to study the anti-vascular effects of adding erlotinib to the pre-PDT regimen. In vivo imaging incorporated signal that originated mostly from the tumor vasculature, but unavoidably included signal from overlying skin, and, to some extent, underlying tissue. Thus, we compared in vivo to ex vivo imaging of tumors in order to confirm that in vivo imaging was representative of the tumor-specific signal (obtained by ex vivo imaging). The data obtained from ex vivo and in vivo images were extremely well correlated (Supplemental Figure 3), validating use of in vivo imaging to study tumor vascular response.
By in vivo imaging, we could identify localized increases in dye accumulation, indicative of a hyper-permeable state that is characteristic after PDT (14), as well as dye exclusion that demonstrates impairment of tumor blood flow. PDT of H460 tumors produced a hyper-permeable state at 1h that was followed at 4h and 18h by the progressive development of dye-excluding vascular damage (Figure 2). In both H460 and A549 tumors (Figure 2 inset), there was significantly greater dye exclusion at 18h after PDT when animals were pretreated with erlotinib.
Figure 2.
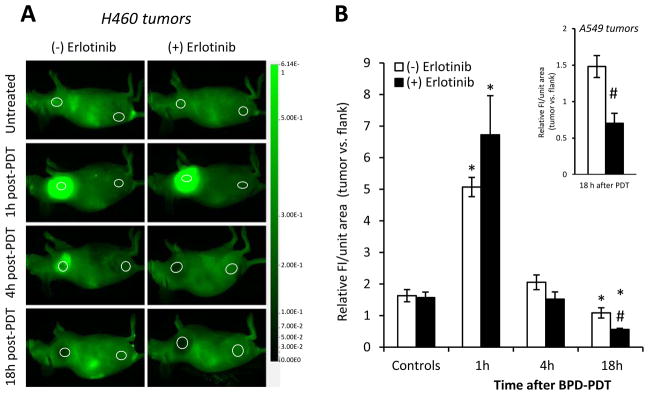
Erlotinib increases PDT-created vascular damage. (A) In H460 tumors, in vivo imaging of IRDye®800-PEG uptake and (B) quantification of dye fluorescence (at 800nm) are shown. Regions of interest (ROIs) that identify the tumor (left) and flank (right) are circled. Data are calculated as the average fluorescence (FI) per unit area of the ROI and then plotted as the ratio of this value in the tumor vs. flank of the same animal (n=9–15). Response of A549 tumors (n=6) is shown in the inset. *p<0.05 compared to untreated-control; #p<0.05 for erlotinib/PDT vs. PDT at the same timepoint.
Erlotinib reduces acute PDT-induced VEGF secretion, but without therapeutic consequence
EGFR inhibition can decrease the expression of vascular endothelial growth factor (VEGF), a protein critical for reestablishment of damaged vasculature (15). Like other cancer therapies, PDT can induce increases in VEGF that contribute to tumor recurrence after treatment (16–18). Therefore, if erlotinib attenuates PDT-induced increases in VEGF, we might expect these decreases to contribute to the stronger vascular response in tumors pretreated with erlotinib. Indeed, PDT produced large increases in tumor concentrations of H460-produced (human) VEGF (Figure 3A). Pretreatment with erlotinib significantly abrogated this effect and VEGF levels were barely elevated after PDT. PDT of A549 tumors led to even larger increases in human VEGF that were reduced nearly two-fold when erlotinib was given before PDT (Supplemental Figure 4).
Figure 3.
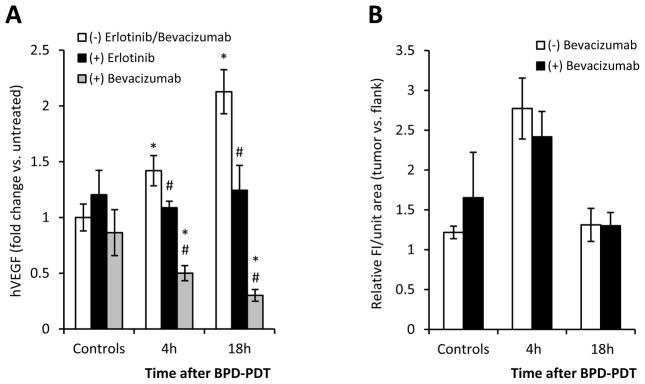
Therapeutic effect of erlotinib/PDT cannot be attributed to abrogation of extracellular VEGF in the early time course after PDT. (A) Relative concentrations of tumor (H460)-localized human VEGF (hVEGF) after treatment with PDT, erlotinib/PDT or bevacizumab/PDT (n=6–8 and 3–5 for erlotinib and bevacizumab conditions, respectively; *p<0.05 compared to untreated-control; #p<0.05 for erlotinib/PDT or bevacizumab/PDT vs. PDT). (B) In vivo imaging of tumor perfusion after PDT or bevacizumab/PDT (n=3–5 for bevacizumab-treated and 2–3 in replication of Figure 2B condition of PDT without molecular-targeting drugs).
To assess if abrogation of VEGF in the early time course after PDT is responsible for augmentation of PDT response by erlotinib, we investigated whether inhibition of extracellular VEGF resulted in similar improvements in PDT response. To accomplish this, we substituted a clinically-approved antibody which specifically targets VEGF, bevacizumab, for erlotinib. These studies followed the rationale that if reduction of extracellular VEGF was responsible for mediating vascular shutdown after treatment with erlotinib/PDT, then bevacizumab/PDT would result in similarly robust vascular shutdown. The H460 tumor model was employed because in this model the administration of erlotinib provided complete abrogation of PDT-induced increases in VEGF. Pre-PDT administration of bevacizumab reduced VEGF levels in H460 tumors after PDT below those observed in untreated controls (Figure 3A). However, despite the dramatic reduction of VEGF in H460 tumors as a result of the pre-PDT addition of bevacizumab, no enhanced vascular damage was observed after PDT (Figure 3B). In contrast to improvements in tumor response after treatment with erlotinib/PDT (see Figure 1A), pretreatment with bevacizumab yielded no improvement and all tumors regrew within 26 days of illumination (median 10 days; data not shown).
Erlotinib increases endothelial cytotoxicity after PDT
To further consider the mechanisms of erlotinib-mediated augmentation of PDT vascular damage, the direct cytotoxic action of erlotinib/PDT on endothelial cells was studied. The viability of mouse endothelial cells (SVEC) was compared after treatment with erlotinib, PDT, or the combination. Prior to initiation of these experiments, EGFR expression by SVEC was confirmed by western blots and was detected at similar levels as were observed in the NSCLC tumor lines studied (Supplemental Figure 5). SVEC viability was significantly reduced by PDT and erlotinib, as well as by the erlotinib/PDT combination (Figure 4A). Moreover, erlotinib pretreatment before PDT significantly increased the cytotoxic effect of PDT.
Figure 4.

Erlotinib/PDT reduces endothelial cell viability and increases BPD uptake. (A) In vitro viability of SVEC endothelial cells after treatment with erlotinib and/or PDT relative to untreated (n=3). Viability assessed at 25h after PDT and analyzed by paired t-test. (B) In vitro BPD uptake assessed by spectrofluorimetry (n=3–4). *p<0.05 compared to untreated, #p<0.05 for erlotinib/PDT vs. PDT.
A mechanism by which erlotinib could increase PDT cytotoxicity is through the capability of receptor tyrosine kinase inhibitors (TKIs) to augment photosensitizer retention. This is mediated via their inhibition of drug transport by ABCG2 (ATP-binding cassette transporter G2). TKIs act through this mechanism to increase tumor levels of several photosensitizers, although BPD is typically less affected (19). Indeed, we previously found no effect of erlotinib on BPD uptake in H460 cells (12). In the present study of endothelial cells, erlotinib did lead to a small but significant increase in BPD levels (Figure 4B) that consisted of a change from 0.76 ± 0.06 nmoles BPD/μg protein in BPD-exposed cells to 0.97 ± 0.07 in erlotinib/BPD-exposed cells. Although small in magnitude, these data did prompt us to question in vivo photosensitizer retention after erlotinib pretreatment.
Increased therapeutic efficacy is due to multiple mechanisms
To elucidate if erlotinib-induced increases in photosensitizer uptake were of significance in vivo, the fluorescence intensity of BPD in H460 tumors was directly measured by in vivo optical imaging at 700 nm. Fluorescence intensity at 700 nm increased by ~50% when animals were pretreated with erlotinib. With erlotinib pretreatment, BPD levels were also elevated in the flanks of the animals, indicating a more systemic impact of erlotinib on photosensitizer uptake (Figure 5A). Moreover, similar elevations were detected in A549 tumors (Supplemental Figure 6).
Figure 5.

Erlotinib increases BPD uptake in vivo as a function of dosing schedule. (A) In vivo images and quantification of BPD uptake in mice receiving three doses of erlotinib prior to BPD administration. Imaging performed 3h post-BPD injection (designated time of light delivery). Regions of interest (ROIs) that identify the tumor (H460; on the left) and flank (right) are circled (n=26–31). (B) Quantification of BPD uptake in mice receiving a single-dose of erlotinib prior to BPD administration. (C) Plasma bilirubin levels in mice receiving single- and triple-dose erlotinib regimens (n=4–7). *p<0.05 for erlotinib-treated versus untreated, #p<0.05 for triple- versus single-dose erlotinib schedules.
The contribution of erlotinib-induced increases in BPD uptake to therapeutic response was tested using an alternative schedule of erlotinib dosing. We found that in contrast to the triple-dosing schedule, a single dose of erlotinib given immediately before BPD administration did not increase BPD uptake (Figure 5B). This finding may reflect the known induction of hepatic side effects by erlotinib (common among TKIs (20, 21)) because patients with mild hepatic dysfunction exhibit increases in the plasma half-life of BPD (22). Indeed, we found that bilirubin, a breakdown product of heme catabolism whose elevation is associated with liver dysfunction, was five-times higher in the plasma of mice given three doses of erlotinib (1.03 mg/dL ± 0.17) than in untreated controls (0.21 mg/dL ± 0.03). In contrast, a single dose of erlotinib administered 3h before PDT (before BPD administration) caused only a small increase in bilirubin concentration (Figure 5C).
Thus, a single dose of erlotinib did not alter tumor BPD levels. Yet, importantly, a single dose of erlotinib does increase PDT-induced cytotoxicity to SVEC, indicating that a single dose is adequate at a molecular level to augment cell death from PDT (Supplemental Figure 7). From these results, we hypothesized that treatment with a single erlotinib dose would produce little improvement in tumor response compared to PDT-alone if erlotinib-induced increases in BPD uptake were the sole mechanism of enhanced tumor response. Conversely, if the causative mechanisms included other factors in addition to enhanced BPD uptake, then the single-dose erlotinib schedule should yield a tumor response that is intermediate to the effects of treatment with PDT and with erlotinib/PDT on a triple-dose schedule. Our data support the second hypothesis. A log-rank test for trend indicated an increasing median regrowth time for triple-dose erlotinib/PDT, followed by single-dose erlotinib/PDT and lastly PDT-alone (p=0.009) (Table 1). Specifically, treatment with PDT after a single dose of erlotinib yielded a median regrowth time of 21 days (95% CI of 10 days and undetermined), a value that was significantly lower than regrowth time after a triple-dose erlotinib schedule (median of >90 days, p=0.042) and higher than regrowth time after PDT-alone (median of 11 days, p=0.053). Since response to erlotinib/PDT on a single-dose schedule is independent of an elevation in BPD levels, these data show that the therapeutic action of erlotinib/PDT includes factors other than photosensitizer uptake, such as erlotinib-induced augmentation of PDT vascular response.
Table 1.
PDT response of H460 tumors after pretreatment with single- vs. triple-dose Erlotinib
| PDT | Erlotinib (single dose) + PDT | Erlotinib (triple dose) + PDT | |
|---|---|---|---|
| Median regrowth time in days (95% CI) | 11 (5,29) | 21 (10, undetermined)* | >90 (undetermined)** |
| 90-day complete response rate | 16% | 48% | 63% |
p=0.053 for comparison to PDT-alone and 0.042 for comparison to triple-dose regimen
p<0.001 for comparison to PDT-alone
Efficacy is not at the expense of selectivity
Since erlotinib-induced enhancement of photosensitizer levels partly contributes to tumor response, and because erlotinib led to general increases in BPD levels, we lastly considered the selectivity of PDT when preceded by erlotinib dosing. Irrespective of whether the animal received erlotinib, scabs that developed 18–30h after PDT encompassed areas that were smaller than their pre-PDT tumor sizes (Figure 6). Thus, the addition of erlotinib to PDT did not promote a nonselective reaction. However, the PDT-induced scabs on animals that received erlotinib/PDT were larger than those on animals that received PDT. Accordingly, they involved a larger area of the treated tumor (without nonselectively extending beyond it), which corroborates the benefit that erlotinib pretreatment provided to tumor response to PDT.
Figure 6.
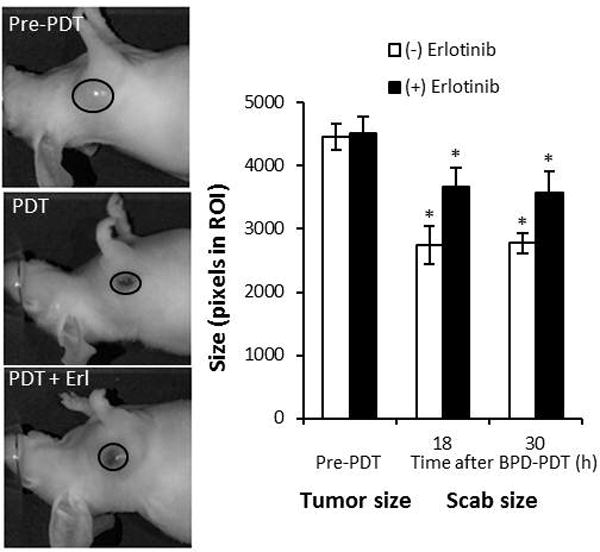
Erlotinib does not reduce PDT selectivity. Tumor size (pre-PDT) and scab size after treatment with PDT or erlotinib/PDT was quantified by in vivo imaging (n=4–11). *p<0.05 for comparison to pre-PDT tumor size of the respective group.
Discussion
Combinations of molecular targeting agents with chemotherapy or radiation are under investigation, especially in treatment of NSCLC (23, 24), following the premise that addition of these agents to standard therapies could provide a multi-faceted treatment approach (25). Similar to other therapies, there exists a benefit to combining PDT with molecular targeting (26, 27). EGFR inhibition using receptor-targeting antibodies and small molecule inhibitors can increase the cytotoxicity of PDT against tumor cells in vitro and reduce tumor burden after PDT in vivo (7–10, 28). Furthermore, molecular targeting can be employed for photosensitizer delivery through the integration of photosensitizers into antibody (e.g. cetuximab)-coated nanoparticles or by coupling of antibodies or molecular targeting agents (e.g. erlotinib) directly to a photosensitizer (29–34). In this way, molecular targeting is exploited to both increase the selectivity of PDT and augment its cytotoxicity. These studies, together with our current work, demonstrate great potential in combining PDT with molecular targeting agents to improve therapeutic outcomes. They emphasize a need to discern the mechanisms that drive improvements in PDT response, toward the goal of informing translation into clinical protocols.
When erlotinib was used as a standalone treatment, it completely failed to impact tumor growth compared to control; indeed, failure rates were identical in the two groups. In contrast, when combined with PDT, erlotinib significantly improved therapeutic outcomes in H460 and A549 tumors, and the effect was larger than PDT alone. The combined effect was, thus, by definition synergistic in nature. Both H460 and A459 cells are relatively resistant to erlotinib (35), although it is reported that a three week dosing schedule can produce tumor growth delay in vivo (36). This schedule is more prolonged than the triple-dose pre-PDT schedule that we employed, which may account for the standalone effect of erlotinib. Effectiveness can increase at higher doses, but 100 mg/kg is the maximum tolerated dose (36) and we found it to be associated with morbidities. Thus, the present studies were conducted at an erlotinib dose of 50 mg/kg, which was well tolerated by the animals.
One mechanism by which erlotinib enhances tumor response to PDT appears to be through the augmentation of PDT vascular effects. Vascular damage contributes to PDT response with many treatment regimens (37, 38). In BPD-PDT, acute vascular shutdown occurs when treatment is performed at short drug-light intervals (e.g., 15 min (38, 39)). Yet, longer drug-light intervals are associated with venodilation (40), followed by reduced perfusion at delayed timepoints (41, 42). For example, a 50% reduction in tumor perfusion is reported at 6h after BPD-PDT with a 3h drug-light interval (41). Based on in vivo imaging of a perfusion marker, we find no decrease in tumor perfusion at 4h after PDT and an ~35% decrease at 18h, compared to controls. Erlotinib pretreatment augmented vascular effects, producing a 65% reduction in perfusion at 18h after PDT. Enhancement of PDT-induced vascular effects by erlotinib may be a result of endothelial cell damage. EGFR inhibition can reduce the proliferation (43) and promote apoptosis (44) of endothelial cells in tumors. We found that erlotinib reduced the viability of endothelial cells after PDT compared to PDT or EGFR inhibition as monotherapies. These results suggest that EGFR inhibition can increase the cytotoxicity of PDT to endothelial cells, thereby contributing to the vascular-damaging effects of BPD-PDT. Thus, our findings uniquely identify the tumor vasculature as an important target when erlotinib is administered in the pre-PDT time course.
Another mechanism for erlotinib’s effect on PDT response stems from observations that TKIs can improve photosensitizer uptake (19). Indeed, we found that erlotinib augmentation of PDT cytotoxicity to endothelial cells was accompanied by small but significant increases in BPD uptake. A triple-dose erlotinib schedule also increased in vivo uptake of BPD, but BPD uptake was minimally affected when the dosing schedule was shorted to a single erlotinib dose. When combined with in vivo PDT, a single erlotinib dose was not as effective as the triple-dose schedule, but still provided for better outcomes than if PDT was performed alone. Single doses of erlotinib also remained effective in augmenting PDT cytotoxicity to endothelial cells. Thus, these data suggest that changes in photosensitizer uptake are one component, but not the sole contributor to erlotinib’s effect in PDT.
Other potential mechanisms were also explored for the therapeutic benefit of erlotinib to PDT. For example, tumor blood flow can improve after TKI therapy (13, 45), which could potentially benefit oxygenation and PDT response. However, erlotinib did not improve oxygenation of H460 tumors in our studies, nor in the models recently published by others (46). EGFR inhibition can also reduce tumor VEGF levels (47), and, in agreement, we found erlotinib pretreatment to reduce PDT-induced acute increases in VEGF. Use of antiangiogenic agents after PDT to decrease VEGF production is associated with better long-term tumor response (16, 26, 48). However, typical combinations of antiangiogenics with PDT include prolonged, post-PDT administration of these drugs, not the exclusive pre-PDT administration of drug that was used in this study. To inform if therapeutic outcome was aided by decreases in extracellular VEGF in the early post-PDT time course, we employed pre-PDT administration of bevacizumab. Results demonstrated that tumor VEGF levels were reduced by bevacizumab in the 18h after PDT, but without benefit to long term PDT response. These results are not in opposition to favorable reports of combining antiangiogenics with PDT due to the pre-PDT and short time course of drug delivery in our studies. However, they do suggest that decreases in extracellular VEGF in the immediate time frame of PDT cannot account for the therapeutic benefit of pretreatment with erlotinib.
Toward the goal of clinical translation, the short pre-PDT time course of erlotinib administration in this study provides a number of benefits. First, a pre-PDT schedule of erlotinib could be adapted to clinical trials of intraoperative PDT, where it is difficult to administer oral agents to post-operative patients who are intubated. Additionally, compared to typical long-term erlotinib dosing, an abbreviated schedule may avoid erlotinib side effects of skin rash and diarrhea, which typically develop in the first month (49). Limited use of erlotinib could further avoid the development of acquired resistance to TKIs that is prevalent with long-term drug use (50). Consequently, an abbreviated dosing schedule might minimize the incidence of adverse events and prevent acquired resistance, thereby improving patient quality of life compared to more typical schedules of erlotinib administration.
In conclusion, these studies uniquely investigate the pre-PDT administration of erlotinib in a clinically-relevant regimen for adding molecular targeting to intraoperative PDT. Given the prevalence of tumor resistance to molecular targeting drugs, studies were conducted in erlotinib-resistant tumor models. Nevertheless, we find that short-term pre-PDT administration of erlotinib produces large improvements in PDT outcome. Multiple mechanisms are responsible for this effect, including erlotinib sensitization of the tumor vasculature to PDT and an erlotinib-induced increase in tumor accumulation of BPD. Given the multiple actions of erlotinib on PDT response and the large magnitude of its effect on this response, these findings suggest a significant potential for PDT with pre-administration of erlotinib in the treatment of NSCLC and other types of malignancies.
Supplementary Material
Acknowledgments
Financial support: National Institutes of Health (CA-129554, CA-087971, CA-085831, and T32-CA-009677).
We acknowledge the assistance of Drs. Julie Czupryna and Jim Delikatny (Small Animal Imaging Facility), Sarah Hagan (spectrofluorometric studies), and Shirron Carter (manuscript preparation).
Footnotes
Conflicts of interest: none
References
- 1.Robinson KW, Sandler AB. EGFR tyrosine kinase inhibitors: difference in efficacy and resistance. Curr Oncol Rep. 2013;15:396–404. doi: 10.1007/s11912-013-0323-7. [DOI] [PubMed] [Google Scholar]
- 2.Baker SJ, Reddy EP. Targeted inhibition of kinases in cancer therapy. Mt Sinai J Med. 2010;77:573–86. doi: 10.1002/msj.20220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Viloria-Petit AM, Kerbel RS. Acquired resistance to EGFR inhibitors: mechanisms and prevention strategies. Int J Radiat Oncol Biol Phys. 2004;58:914–26. doi: 10.1016/j.ijrobp.2003.09.091. [DOI] [PubMed] [Google Scholar]
- 4.Weyergang A, Kaalhus O, Berg K. Photodynamic targeting of EGFR does not predict the treatment outcome in combination with the EGFR tyrosine kinase inhibitor Tyrphostin AG1478. Photochem Photobiol Sci. 2008;7:1032–40. doi: 10.1039/b806209a. [DOI] [PubMed] [Google Scholar]
- 5.Weyergang A, Selbo PK, Berg K. Sustained ERK [corrected] inhibition by EGFR targeting therapies is a predictive factor for synergistic cytotoxicity with PDT as neoadjuvant therapy. Biochim Biophys Acta. 2013;1830:2659–70. doi: 10.1016/j.bbagen.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 6.Gijsens A, Missiaen L, Merlevede W, de Witte P. Epidermal growth factor-mediated targeting of chlorin e6 selectively potentiates its photodynamic activity. Cancer Res. 2000;60:2197–202. [PubMed] [Google Scholar]
- 7.del Carmen MG, Rizvi I, Chang Y, Moor AC, Oliva E, Sherwood M, et al. Synergism of epidermal growth factor receptor-targeted immunotherapy with photodynamic treatment of ovarian cancer in vivo. J Natl Cancer Inst. 2005;97:1516–24. doi: 10.1093/jnci/dji314. [DOI] [PubMed] [Google Scholar]
- 8.Bhuvaneswari R, Gan YY, Soo KC, Olivo M. Targeting EGFR with photodynamic therapy in combination with Erbitux enhances in vivo bladder tumor response. Mol Cancer. 2009;8:94. doi: 10.1186/1476-4598-8-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koon HK, Chan PS, Wong RN, Wu ZG, Lung ML, Chang CK, et al. Targeted inhibition of the EGFR pathways enhances Zn-BC-AM PDT-induced apoptosis in well-differentiated nasopharyngeal carcinoma cells. J Cell Biochem. 2009;108:1356–63. doi: 10.1002/jcb.22366. [DOI] [PubMed] [Google Scholar]
- 10.Postiglione I, Chiaviello A, Aloj SM, Palumbo G. 5-aminolaevulinic acid/photo-dynamic therapy and gefitinib in non-small cell lung cancer cell lines: a potential strategy to improve gefitinib therapeutic efficacy. Cell Prolif. 2013;46:382–95. doi: 10.1111/cpr.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esipova TV, Karagodov A, Miller J, Wilson DF, Busch TM, Vinogradov SA. Two new “protected” oxyphors for biological oximetry: properties and application in tumor imaging. Anal Chem. 2011;83:8756–65. doi: 10.1021/ac2022234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edmonds C, Hagan S, Gallagher-Colombo SM, Busch TM, Cengel KA. Photodynamic therapy activated signaling from epidermal growth factor receptor and STAT3: Targeting survival pathways to increase PDT efficacy in ovarian and lung cancer. Cancer Biol Ther. 2012;13:1463–70. doi: 10.4161/cbt.22256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cerniglia GJ, Pore N, Tsai JH, Schultz S, Mick R, Choe R, et al. Epidermal growth factor receptor inhibition modulates the microenvironment by vascular normalization to improve chemotherapy and radiotherapy efficacy. PLoS One. 2009;4:e6539. doi: 10.1371/journal.pone.0006539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He C, Agharkar P, Chen B. Intravital microscopic analysis of vascular perfusion and macromolecule extravasation after photodynamic vascular targeting therapy. Pharm Res. 2008;25:1873–80. doi: 10.1007/s11095-008-9604-5. [DOI] [PubMed] [Google Scholar]
- 15.Lu YY, Jing DD, Xu M, Wu K, Wang XP. Anti-tumor activity of erlotinib in the BxPC-3 pancreatic cancer cell line. World J Gastroenterol. 2008;14:5403–11. doi: 10.3748/wjg.14.5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gallagher-Colombo SM, Maas AL, Yuan M, Busch TM. Photodynamic therapy-induced angiogenic signaling: consequences and solutions to improve therapeutic response. Israel Journal of Chemistry. 2012;52:681–90. doi: 10.1002/ijch.201200011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhuvaneswari R, Thong PS, Gan YY, Soo KC, Olivo M. Evaluation of hypericin-mediated photodynamic therapy in combination with angiogenesis inhibitor bevacizumab using in vivo fluorescence confocal endomicroscopy. J Biomed Opt. 2010;15:011114. doi: 10.1117/1.3281671. [DOI] [PubMed] [Google Scholar]
- 18.Gomer CJ. Induction of prosurvival molecules during treatment: rethinking therapy options for photodynamic therapy. J Natl Compr Canc Netw. 2012;10 (Suppl 2):S35–9. doi: 10.6004/jnccn.2012.0172. [DOI] [PubMed] [Google Scholar]
- 19.Liu W, Baer MR, Bowman MJ, Pera P, Zheng X, Morgan J, et al. The tyrosine kinase inhibitor imatinib mesylate enhances the efficacy of photodynamic therapy by inhibiting ABCG2. Clin Cancer Res. 2007;13:2463–70. doi: 10.1158/1078-0432.CCR-06-1599. [DOI] [PubMed] [Google Scholar]
- 20.Iacovelli R, Palazzo A, Procopio G, Santoni M, Trenta P, De Benedetto A, et al. Incidence and relative risk of hepatic toxicity in patients treated with anti-angiogenic tyrosine kinase inhibitors for malignancy. Br J Clin Pharmacol. 2014;77:929–38. doi: 10.1111/bcp.12231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–42. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 22.Houle JM, Strong A. Clinical pharmacokinetics of verteporfin. J Clin Pharmacol. 2002;42:547–57. doi: 10.1177/00912700222011607. [DOI] [PubMed] [Google Scholar]
- 23.Mok TS, Lee K, Leung L. Targeting epidermal growth factor receptor in the management of lung cancer. Semin Oncol. 2014;41:101–9. doi: 10.1053/j.seminoncol.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 24.Padda SK, Burt BM, Trakul N, Wakelee HA. Early-stage non-small cell lung cancer: surgery, stereotactic radiosurgery, and individualized adjuvant therapy. Semin Oncol. 2014;41:40–56. doi: 10.1053/j.seminoncol.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 25.Ausborn NL, Le QT, Bradley JD, Choy H, Dicker AP, Saha D, et al. Molecular profiling to optimize treatment in non-small cell lung cancer: a review of potential molecular targets for radiation therapy by the translational research program of the radiation therapy oncology group. Int J Radiat Oncol Biol Phys. 2012;83:e453–64. doi: 10.1016/j.ijrobp.2012.01.056. [DOI] [PubMed] [Google Scholar]
- 26.Gomer CJ, Ferrario A, Luna M, Rucker N, Wong S. Photodynamic therapy: combined modality approaches targeting the tumor microenvironment. Lasers Surg Med. 2006;38:516–21. doi: 10.1002/lsm.20339. [DOI] [PubMed] [Google Scholar]
- 27.Martinez-Carpio PA, Trelles MA. The role of epidermal growth factor receptor in photodynamic therapy: a review of the literature and proposal for future investigation. Lasers Med Sci. 2010;25:767–71. doi: 10.1007/s10103-010-0790-0. [DOI] [PubMed] [Google Scholar]
- 28.Hasan T. Using cellular mechanisms to develop effective combinations of photodynamic therapy and targeted therapies. J Natl Compr Canc Netw. 2012;10 (Suppl 2):S23–6. doi: 10.6004/jnccn.2012.0168. [DOI] [PubMed] [Google Scholar]
- 29.Mir Y, Elrington SA, Hasan T. A new nanoconstruct for epidermal growth factor receptor-targeted photo-immunotherapy of ovarian cancer. Nanomedicine. 2013;9:1114–22. doi: 10.1016/j.nano.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang FL, Huang Q, Zheng K, Li J, Liu JY, Xue JP. A novel strategy for targeting photodynamic therapy. Molecular combo of photodynamic agent zinc(II) phthalocyanine and small molecule target-based anticancer drug erlotinib. Chem Commun (Camb) 2013;49:9570–2. doi: 10.1039/c3cc45487h. [DOI] [PubMed] [Google Scholar]
- 31.Abu-Yousif AO, Moor AC, Zheng X, Savellano MD, Yu W, Selbo PK, et al. Epidermal growth factor receptor-targeted photosensitizer selectively inhibits EGFR signaling and induces targeted phototoxicity in ovarian cancer cells. Cancer Lett. 2012;321:120–7. doi: 10.1016/j.canlet.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rizvi I, Dinh TA, Yu W, Chang Y, Sherwood ME, Hasan T. Photoimmunotherapy and irradiance modulation reduce chemotherapy cycles and toxicity in a murine model for ovarian carcinomatosis: perspective and results. Isr J Chem. 2012;52:776–87. doi: 10.1002/ijch.201200016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spring BQ, Abu-Yousif AO, Palanisami A, Rizvi I, Zheng X, Mai Z, et al. Selective treatment and monitoring of disseminated cancer micrometastases in vivo using dual-function, activatable immunoconjugates. Proc Natl Acad Sci U S A. 2014;111:E933–42. doi: 10.1073/pnas.1319493111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soukos NS, Hamblin MR, Keel S, Fabian RL, Deutsch TF, Hasan T. Epidermal growth factor receptor-targeted immunophotodiagnosis and photoimmunotherapy of oral precancer in vivo. Cancer Res. 2001;61:4490–6. [PMC free article] [PubMed] [Google Scholar]
- 35.Zou Y, Ling YH, Sironi J, Schwartz EL, Perez-Soler R, Piperdi B. The autophagy inhibitor chloroquine overcomes the innate resistance of wild-type EGFR non-small-cell lung cancer cells to erlotinib. J Thorac Oncol. 2013;8:693–702. doi: 10.1097/JTO.0b013e31828c7210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Higgins B, Kolinsky K, Smith M, Beck G, Rashed M, Adames V, et al. Antitumor activity of erlotinib (OSI-774, Tarceva) alone or in combination in human non-small cell lung cancer tumor xenograft models. Anticancer Drugs. 2004;15:503–12. doi: 10.1097/01.cad.0000127664.66472.60. [DOI] [PubMed] [Google Scholar]
- 37.Maas AL, Carter SL, Wileyto EP, Miller J, Yuan M, Yu G, et al. Tumor vascular microenvironment determines responsiveness to photodynamic therapy. Cancer Res. 2012;72:2079–88. doi: 10.1158/0008-5472.CAN-11-3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen B, Pogue BW, Hoopes PJ, Hasan T. Vascular and cellular targeting for photodynamic therapy. Crit Rev Eukaryot Gene Expr. 2006;16:279–305. doi: 10.1615/critreveukargeneexpr.v16.i4.10. [DOI] [PubMed] [Google Scholar]
- 39.Fingar VH, Kik PK, Haydon PS, Cerrito PB, Tseng M, Abang E, et al. Analysis of acute vascular damage after photodynamic therapy using benzoporphyrin derivative (BPD) Br J Cancer. 1999;79:1702–8. doi: 10.1038/sj.bjc.6690271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Major A, Kimel S, Mee S, Milner TE, Smithies DJ, Srinivas SM, et al. Microvascular photodynamic therapy effects determined in vitro using optical doppler tomography. IEEE J Sel Top Quantum Electron. 1999;5:1168–75. [Google Scholar]
- 41.Chen B, Pogue BW, Goodwin IA, O’Hara JA, Wilmot CM, Hutchins JE, et al. Blood flow dynamics after photodynamic therapy with verteporfin in the RIF-1 tumor. Radiat Res. 2003;160:452–9. doi: 10.1667/RR3059. [DOI] [PubMed] [Google Scholar]
- 42.Pogue BW, O’Hara JA, Demidenko E, Wilmot CM, Goodwin IA, Chen B, et al. Photodynamic therapy with verteporfin in the radiation-induced fibrosarcoma-1 tumor causes enhanced radiation sensitivity. Cancer Res. 2003;63:1025–33. [PubMed] [Google Scholar]
- 43.Naumov GN, Nilsson MB, Cascone T, Briggs A, Straume O, Akslen LA, et al. Combined vascular endothelial growth factor receptor and epidermal growth factor receptor (EGFR) blockade inhibits tumor growth in xenograft models of EGFR inhibitor resistance. Clin Cancer Res. 2009;15:3484–94. doi: 10.1158/1078-0432.CCR-08-2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baker CH, Pino MS, Fidler IJ. Phosphorylated epidermal growth factor receptor on tumor-associated endothelial cells in human renal cell carcinoma is a primary target for therapy by tyrosine kinase inhibitors. Neoplasia. 2006;8:470–6. doi: 10.1593/neo.06172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qayum N, Muschel RJ, Im JH, Balathasan L, Koch CJ, Patel S, et al. Tumor vascular changes mediated by inhibition of oncogenic signaling. Cancer Res. 2009;69:6347–54. doi: 10.1158/0008-5472.CAN-09-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weiss A, Bonvin D, Berndsen RH, Scherrer E, Wong TJ, Dyson PJ, et al. Angiostatic treatment prior to chemo- or photodynamic therapy improves anti-tumor efficacy. Sci Rep. 2015;5:8990. doi: 10.1038/srep08990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhuvaneswari R, Yuen GY, Chee SK, Olivo M. Antiangiogenesis agents avastin and erbitux enhance the efficacy of photodynamic therapy in a murine bladder tumor model. Lasers Surg Med. 2011;43:651–62. doi: 10.1002/lsm.21109. [DOI] [PubMed] [Google Scholar]
- 48.Bhuvaneswari R, Yuen GY, Chee SK, Olivo M. Hypericin-mediated photodynamic therapy in combination with Avastin (bevacizumab) improves tumor response by downregulating angiogenic proteins. Photochem Photobiol Sci. 2007;6:1275–83. doi: 10.1039/b705763f. [DOI] [PubMed] [Google Scholar]
- 49.Chen X, Liu Y, Roe OD, Qian Y, Guo R, Zhu L, et al. Gefitinib or erlotinib as maintenance therapy in patients with advanced stage non-small cell lung cancer: a systematic review. PLoS One. 2013;8:e59314. doi: 10.1371/journal.pone.0059314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chong CR, Janne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19:1389–400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.