

Abstract
In vivo rat kidney tissue metabolites of an anticancer drug, cisplatin (cis-diamminedichloroplatinum [II]) (CP) which is used for the treatment of testicular, ovarian, bladder, cervical, esophageal, small cell lung, head and neck cancers, have been identified and characterized by using liquid chromatography positive ion electrospray ionization tandem mass spectrometry (LC/ESI-MS/MS) in combination with on line hydrogen/deuterium exchange (HDX) experiments. To identify in vivo metabolites, kidney tissues were collected after intravenous administration of CP to adult male Sprague-Dawley rats (n = 3 per group). The tissue samples were homogenized and extracted using newly optimized metabolite extraction procedure which involves liquid extraction with phosphate buffer containing ethyl acetate and protein precipitation with mixed solvents of methanol-water-chloroform followed by solid-phase clean-up procedure on Oasis HLB 3cc cartridges and then subjected to LC/ESI-HRMS analysis. A total of thirty one unknown in vivo metabolites have been identified and the structures of metabolites were elucidated using LC-MS/MS experiments combined with accurate mass measurements. Online HDX experiments have been used to further support the structural characterization of metabolites. The results showed that CP undergoes a series of ligand exchange biotransformation reactions with water and other nucleophiles like thio groups of methionine, cysteine, acetylcysteine, glutathione and thioether. This is the first research approach focused on the structure elucidation of biotransformation products of CP in rats, and the identification of metabolites provides essential information for further pharmacological and clinical studies of CP, and may also be useful to develop various effective new anticancer agents.
Introduction
Identification of metabolites is crucial in the drug discovery and development process to optimize lead compounds for further development. The information generated in the early discovery phase of metabolic identification can be used to identify lead compounds and undesirable metabolic products followed by optimization of pharmacokinetic and safety profiles. Identification of reactive or toxic metabolites is essential to avoid the toxicity and helps to modify the structure by means of chemical transformations. These studies are usually carried out by employing in vitro and in vivo systems followed by using modern analytical instruments. Liquid chromatography tandem mass spectrometry (LC-MS/MS, MSn) combined with online H/D exchange (HDX) experiments and accurate mass measurements is the most popular analytical technique for metabolite identification [1–13]. Accurate and reproducible identification of metabolites in tissue homogenate samples is of high importance to characterize animal models and to identify metabolic changes that occur in different tissue types in specific diseases. However, the extraction of metabolites from tissue homogenates is one of the most labor intensive steps for metabolomic studies to yield reproducible results between repeated samples.
Cisplatin (cis-diamminedichloroplatinum(II)) (CP) is the most potent chemotherapeutic antitumor drug used in the treatment of testicular, ovarian, bladder, cervical, esophageal, small cell lung, head and neck cancers [14,15]. The mechanistic pathway of CP involves the formation of adducts with DNA nucleobases by covalent interactions which result in cross links between adjacent nucleobases that block DNA replication, transcription and ultimately cell division [16]. The hydrolysis of CP yields the monohydrated CP complex (cis-diammineaquachloroplatinum (II); MHC) which is most important for anticancer activities as well as side effects of CP [17, 18]. Numerous side effects are related to CP therapy like ototoxicity, gastrointestinal toxicity, tubular necrosis, oxidative stress, inflammation, apoptosis, myelosuppression, neurotoxicity, nephrotoxicity (kidney injury) and nausea [19–22]. The clinical use of CP is often more complicated by dose-limiting major side effect nephrotoxicity [23]; and the mechanism of nephrotoxicity remains obscure. Even though the mechanism of nephrotoxicity remains unclear, kidney CP levels have been shown to be several fold higher than plasma levels which were found in other organs following administration of CP to animals [24, 25]. It was reported earlier that kidney CP concentrations are higher in patients with evidence of nephrotoxicity than patients without kidney damage [26]. After intravenous administration of CP, it reacts extensively with proteins, nucleotides and low molecular mass compounds to form fixed and mobile metabolites, respectively. Furthermore, some unchanged CP also present in the initial distribution phase after intravenous administration of CP [27]. Moreover, unchanged CP and several low molecular mass metabolites (mobile metabolites) are actively secreted and/or reabsorbed by the kidney during early phase [27–30]. It has been shown that protein bound CP (fixed metabolites) does not cause the nephrotoxicity in rats [31]. Kidneys represent the main route of CP and its metabolites excretion with proximal tubule cells and tissues which are the primary sites of CP accumulation [32]. Currently, there is a great deal of interest in the analysis of CP and its metabolites, since it increases the cure rates dramatically for various types of human cancers in the chemotherapy [33]. However, the dose of CP is limited because of its major toxic side effects such as nephrotoxicity and severe nausea. The nephrotoxicity of CP may be directly or indirectly linked to specific metabolites [34]. To understand the relationship between toxic effects and CP disposition, it is essential to demonstrate the distribution and biotransformation of CP in the organs showing side effects. Several studies have been reported on HPLC analysis [35–42] and pharmacokinetic studies of CP in animals and humans [21, 43–45]; but most of them are in plasma and/or urine. EI-Khateeb et al., [46] have investigated a study on the optimized conditions for HPLC separation of CP and its two hydrolyzed products. Ehrsson et al., have demonstrated the separation and identification of CP, transplatin and their hydrated complexes using porous graphitic carbon and electrospray ionization mass spectrometry (ESI-MS) [47]. Recently, Du et al., have investigated a study on the hydrolysed products of CP in various buffer conditions and the effect of the buffer on hydrolysis of CP by using Fourier transform ion cyclotron resonance mass spectrometry (FTICR-MS) [48]. Some researchers have demonstrated that the production of metabolites is a necessary prerequisite for the nephrotoxicity in the chemotherapy of CP [49–51]. Several studies have been under taken to understand the cause of nephrotoxicity and to correct the problems [52–54]. However, none of these studies provided essential information on the intact CP and its metabolites. It was reported earlier that chlorine atoms from CP can be readily replaced by water and other nucleophiles resulting the formation of platinum complexes [55–57]. Until recent investigations into the mechanism and extent of metabolism of CP were limited by the lack of a suitable LC-MS method and metabolite identification. Thus, it is necessary to develop the LC-MS/MS method for the identification and characterization of metabolites of CP which will become very important for the elucidation of its mode of action, toxicity and optimization of the chemotherapy.
In the present study, we have developed and optimized the LC-MS/MS method for the identification and characterization of unknown in vivo metabolites of CP after intravenous administration of drug to adult male Sprague-Dawley rats by employing following steps. (i) Removal of whole kidney from rats (ii) Homogenization of kidney tissues (iii) Extraction of CP and its metabolites from kidney tissue homogenate samples using newly optimized metabolite extraction procedure (iv) LC/ESI-HR-MS analysis of extracted tissue homogenate sample solutions containing drug and its metabolites and (v) Structural characterization of CP and its metabolites using LC-MS/MS experiments combined accurate mass measurements in conjunction with online HDX experiments.
Experimental
Chemicals and reagents
High pure CP was a gift sample from JW Pharmaceuticals, Seoul, Republic of Korea. HPLC grade methanol, chloroform, water and analytical reagent grade formic acid, ethyl acetate, triethylamine, phosphate buffer, deuterium oxide (D2O, 100% atom ‘D’), deuterated methanol and D-formic acid (98% atom ‘D’) used in the present study were purchased from Sigma Aldrich, Seoul, Republic of Korea and used without further purification.
Animal experiments
Animals were supplied by SAMTACO BIOKOREA, Osan, Republic of Korea. All animal experiments were carried out according to the guidelines by Institutional Animal Care and Use Committee (IACUC), Pusan National University, Busan, Republic of Korea. The study was approved by the IACUC (Approval No: 2014–062), Pusan National University, Busan, Republic of Korea. The animal house is maintained at temperature 22 ± 2°C with relative humidity of 50 ± 15% and 12 h dark/light cycle. Animals were kept in an environmentally controlled breeding room with standard laboratory food and water for three days prior to the experiments. The animals were fasted for 12 h with free access to water prior to the drug administration. Three sets (n = 3 per group for biological replicates) of adult male Sprague-Dawley rats (290–310g) with similar weight and age were utilized to carry out the present study for unique results and to avoid the variation in the total number of metabolites. Another set of rats (n = 3) with same weight and age was used as control specimens. Each rat from three sets were intravenously administered with 3mg/kg dose of CP and all the rats were sacrificed by decapitation method at different time points, 2h, 6h, 12h, 24h and 36h after dosing. All the animals were monitored carefully for every hour after the administration of drug. Whole kidney was excised, washed with normal saline, blotted dry with filter paper and then weighed accurately. The homogenization and extraction procedure of tissues has been explained below in the context of the sample preparation section.
Instrumentation
LC-MS/MS equipment and conditions
The HPLC analysis was performed on an Agilent 1290 infinity series HPLC instrument (Agilent Technologies, USA) equipped with a binary pump (G4220A, USA), a variable wavelength detector (VWD) (G1314F, USA), an auto sampler (G4226A, USA), a thermostat (G1330B, USA), and a column compartment (G1316C, USA). Effective chromatographic separation was achieved on a Agilent ZORBAX SB C-18 column (2.1 X 50 mm, 1.8 μm) using the mobile phase consisting of 0.1% formic acid in water (Solvent A) and methanol (Solvent B) in a gradient elution mode. The optimized gradient program was set as follows: (Tmin / % solution of B): 0/15, 6/30, 12/65, 20/65, 30/90, 35/15. The flow rate of the mobile phase was 0.4 mL/min, the column temperature was at 30°C and the injection volume was 5μL.
LC-MS analysis was performed on a quadrupole time-of-flight (Q-TOF) mass spectrometer (Q-TOF LC/MS 6550 series classic G6550A, Agilent Technologies, USA) equipped with an ESI source. The data acquisition was under the control of Mass Hunter workstation software. The typical operating source conditions for MS scan in positive ion ESI mode were optimized as follows; the fragmentor voltage was set at, 175 V; the capillary at, 3000–3500 V; sheath gas temp at, 350°C; sheath gas flow at, 11 L/min; nitrogen was used as the drying (250°C; 13 L/min) and nebulizing (45 psi) gas. For full scan MS mode, the mass range was set at m/z 100–3000. For collision-induced dissociation (CID) experiments, keeping MS1 static, the precursor ion of interest was selected using the quadrupole analyzer and the product ions were analyzed using a time-of-flight (TOF) analyzer. Ultra high pure nitrogen was used as collision gas. All the spectra were recorded under identical experimental conditions and are average of 20–25 scans. The elemental compositions from the accurate mass measurements of m/z values and data processing of total ion chromatograms (TICs) were also carried out by using Mass Hunter workstation software. The trace level metabolites were verified by extracting the masses of protonated metabolites using extracted ion chromatograms (EICs) after the post run analysis. The metabolites were obtained from the metabolic pathways in Agilent Metabolite ID (Agilent Technologies). The elemental compositions of protonated metabolites and proposed MS/MS fragment ions were confirmed by accurate mass measurements.
For online HDX LC-MS experiments, deuterated water was used to prepare the mobile phase i.e. deuterium oxide (D2O, 100% atom ‘D’) as Solvent A with 0.1% D-formic acid (98% atom ‘D’) and deuterated methanol as solvent B.
Sample Preparation
In this study, liquid extraction with phosphate buffer containing ethyl acetate and protein precipitation (PPT) with mixed solvents of methanol-water-chloroform followed by solid-phase clean-up procedure on Oasis HLB 3cc cartridges was used for the extraction of CP and its metabolites from kidney tissue homogenates. The following steps are involved in the process of extraction of metabolites from tissue homogenates. The frozen tissues of kidney were taken out from -80°C storage, washed with normal saline, blotted dry with filter paper, finely diced with scissors and weighed accurately after fat and connective tissue had been trimmed away. A 100mg of dissected tissue was placed into precooled (dry ice) 2ml homogenization tubes containing ceramic beads (1.4 mm diameter). The pre-cooled extraction solvents were used for the extraction of metabolites from homogenized tissues. A total of 1mL solvent volume was used for 100 mg of tissue. The homogenized tissue sample was transferred into a cleaned poly-propylene centrifuge tube and added 600 μL of phosphate buffer (10mM) followed by 400μL of ethyl acetate. The mixture was vortexed for 2 min, sonicated for 5 min (the tube was refrigerated in an ice-bath) and centrifuged at 13,000 rpm for 10 min at 4°C. Both aqueous and organic layers were carefully collected into separate polypropylene tubes. For more precipitation of proteins from the collected supernatant (200 μL), 400 μL of methanol, 100 μL of water followed by 300 uL of chloroform were added and vortexed the mixture for 1 min, and then centrifuged for 10 min at 4000 rpm at 4°C. A 200 μL of supernatant was collected into another cleaned poly-propylene centrifuge tube and added 400μL each of chloroform and water. A biphasic mixture was formed after vortexed for 1 min and centrifuged for 10 min at 4000 rpm. The upper polar (water) and lower nonpolar (chloroform) layers were carefully collected. The lower nonpolar layer was frozen and upper polar layer was collected into another tube and then it is subjected to solid phase extraction (SPE) using Oasis HLB 3cc cartridges. Simultaneously, the initial aqueous layer which was collected in the first stage (collected from ethyl acetate/phosphate buffer) was also subjected to SPE. The SPE column was conditioned by washing with 1 ml each of methanol-triethylamine (93:7, v/v), water and phosphate buffer (5mM; pH 7.4). After that, the sample was loaded to the column, washed with 1 mL each of water and methanol-water (50:50, v/v), and the analytes were eluted with methanol-triethylamine (93:7, v/v). As the main aim of our study is focused on the identification of total in vivo metabolites of CP, the aliquots from both SPE’s were pooled together and they were finally pooled with the acetonitrile layer. Further, the samples were filtered and evaporated to dryness under a gentle stream of nitrogen at 45°C. After that, the residue was reconstituted in 100 μL methanol-water (50:50, v/v) and only 5 μL aliquots were injected into LC-MS system. The extracted tissue homogenates were stored at -20°C before and after analysis.
Optimization of the Extraction Method
The extraction of analytes from solid biological matrices like tissues require more vigorous approaches than those used for biological fluids such as urine and plasma. Thus, the homogenization is a critical step in the method development for separation and quantitative determination of drug in tissues, because it greatly affects the extraction recovery and optimization of analytical method. The majority of the previous researchers have used the PPT method for the extraction of CP from the biological matrices. For PPT, they used different solvents like hot water [39], methanol [42], acetonitrile [45] and the mixture of acetonitrile/dichloromethane [45] for the extraction of CP from the biological matrices. Some researchers have used sucrose containing phosphate buffer for the extraction of CP from tissues [27].
In our study, for the extraction of CP and its metabolites from kidney tissues, initially we tried PPT and liquid-liquid extraction (LLE) methods with various solvents like methanol, chloroform, methanol-water, chloroform-water, methanol-water-chloroform, phosphate buffer (10mM), methanol/phosphate buffer, ethanol/phosphate buffer, water/ethyl acetate to check the potential differences and to remove the biological interferences, and for better extraction of analytes from tissues. We also tried individual SPE method using Oasis HLB 3cc cartridges for the extraction of CP with different solvents. From these methods, we observed that no single solvent and single method worked best for the extraction of all the metabolites from kidney tissues without showing any significant matrix interference. Furthermore, not these tissues behaved similarly with regard to different extraction solvents in different extraction methods (PPT, LLE and SPE) that were tried. After several trials, liquid extraction with phosphate buffer containing ethyl acetate and protein precipitation with mixed solvents of methanol-water-chloroform followed by solid-phase clean-up procedure on Oasis HLB 3cc cartridges was found to be suitable for the extraction of CP and its metabolites from kidney tissue homogenates since this extraction method was able to detect the more number of metabolites with cleaner chromatogram without showing any significant biological interferences. Moreover, the protocol that we developed here could detect even very low level metabolites with good recovery which was not possible with the individual PPT, LLE and SPE methods that were used for the extraction of metabolites of CP.
The reproducibility of both homogenization and extraction protocol was tested in three independent experiments using kidney tissue samples of the same rat as well as different rats from each group. Each tissue homogenate was analyzed three times to obtain the reproducible results and to avoid the variation in the total number of metabolites. It can be noted that the results obtained from independent experiments are very similar to each other.
Results and Discussion
CP undergoes metabolic activation in the kidney to more potent toxic materials. This process initiates with the formation of glutathione conjugates in the circulation which may presumably mediated by glutathione-S-transferase [58, 59]. As the glutathione-conjugates pass through the kidney, they cleaved to cysteinyl-glycine-conjugates by gamma glutamyl transpeptidase (GGT) expressed on the surface of the proximal tubule cells [60, 61]. The cysteinyl-glycine-conjugates are further metabolized to cysteine-conjugates by aminodipeptidases, also expressed on the surface of the proximal tubule cells [60]. The cysteine-conjugates are transported into the proximal tubule cells, where they are further metabolized by cysteine-S-conjugate beta-lyase to highly reactive thiols [60–62]. The dissociation of one of the chlorine atoms from CP results in a positive charge on the platinum that will attract the negatively charged sulfur present on the active thio group containing molecules like methionine, cysteine, acetylcysteine, glutathione and thioether to form various toxic metabolites.
Fig 1 shows the LC/ESI-MS-TIC of metabolites of CP formed in kidney tissue homogenate samples. All the metabolites (M1-M31) were eluted within 35 min on C-18 column and they were well resolved from parent drug as well as from each other. The positive ion ESI-MS spectrum of CP gives moderately abundant [M+H]+, [M+NH4]+ ions and abundant [M+Na]+ ion, and low abundance [2M+H]+, [2M+NH4]+ [2M+Na]+ ions, whereas the positive ion ESI-MS spectra of all metabolites give abundant [M+H]+ ions and moderately abundant [M+Na]+ ions. To elucidate the structures of metabolites, we examined the fragmentation of the [M+H]+ ions of CP and its metabolites by using online LC/ESI-MS/MS experiments in combination with accurate mass measurements. Further, online HDX LC-MS/MS experiments were also carried out to support the structural identification of metabolites. The product ions of all the metabolites were compared with the product ions of CP, to assign most probable structures for the observed metabolites (Fig 2). The elemental compositions of thirty one metabolites and their majority of product ions have been confirmed by accurate mass measurements data.
Fig 1. LC/ESI-MS-TIC of metabolites of CP formed in kidney tissue homogenate sample.
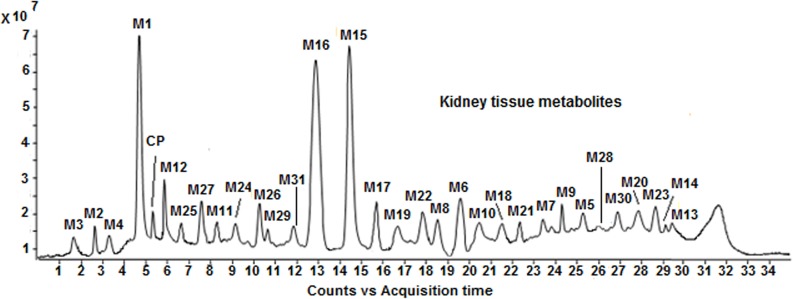
Fig 2. Proposed in vivo ligand exchange biotransformation pathway of CP.

To perform online HDX LC-MS experiments, deuterated solvents (Deuterated water and deuterated methanol) are used as a mobile phase instead of normal mobile phase (deionized water and methanol). The HDX experiments are performed by using D2O as sheath liquid for the LC-MS analysis of drug metabolites. They are useful for determining the numbers and positions of exchangeable hydrogens present in the metabolites to characterize them [13]. The exchangeable hydrogen atoms are usually bound to N-, O- and S- atoms in different functional groups such as-NH-,-NH2,-OH,-COOH and-SH [11]. The examination of the mass shift of the deuteriated molecule from that of the protonated molecule allow the total number of exchangeable protons to be determined. Interpretation of the product ion spectra helps to determine the location of the exchanged protons and assignment of the site(s) of modification for each metabolite. The structure elucidation of metabolites can be effectively achieved by using deuterated precursor ions compared to protonated precursor ions where the protonated precursor ions do not give accurate information in characterizing the metabolites. Thus, by using HDX, some of the unambiguous metabolite identification, which can be very useful in the early stages of drug discovery, can be achieved without the use of large scale preparation for NMR or other analytical characterizations. We have carried out these HDX experiments to further support the structural characterization of in vivo drug metabolites formed in rat kidney tissues. In this study, the HDX experiments allowed us to distinguish the hydroxylated CP metabolites from other metabolites as well as methionine, cysteine and acetylcysteine straight chain containing CP metabolites from their respective chelated ring containing CP metabolites with the help of numbers and positions of exchangeable hydrogens. Furthermore, these HDX studies also allowed us to identify some metabolites that have the same nominal mass and identical elemental compositions which is not possible to characterize by normal LC-MS/MS spectra.
Characterization of CP and its metabolites using LC/ESI-MS/MS experiments
MS/MS of protonated CP (m/z 300.9620)
The ESI-MS/MS spectrum of [M+H]+ ion (m/z 300.9620) of CP displays abundant product ions at m/z 264.4732 (loss of HCl), m/z 247.5031 (loss of NH3 from m/z 264.4732), m/z 228.4812 (loss of HCl from m/z 264.4732) and low abundance product ions at m/z 211.4922 (loss of HCl from m/z 247.5031) and m/z 195.1754 (platinum cation) (Fig 3A) (Fig 4). It can be noted that the product ions observed in the MS/MS spectrum are consistent with the presence of two chlorine atoms and two ammine groups attached to platinum. All the above fragmentation pathways have been confirmed by MSn experiments and most probable structures are proposed for the product ions based on elemental compositions derived from accurate mass measurements.
Fig 3. LC/ESI-MS/MS spectra of (a) Protonated CP (b) M1 and (c) M2 at 30 eV.

Fig 4. Proposed fragmentation mechanism for CP and its metabolites M1-M4.

MS/MS of metabolites of CP
M1 (m/z 282.4860): The most abundant metabolite M1 at m/z 282.4860 with an elemental composition of PtN2H8OCl (Error: 1.92 ppm), was eluted at 4.8 min. The elemental composition data showed that M1 lacks one chlorine atom and inclusion of one aqua molecule as compared to CP, suggesting that it is a mono aqua CP metabolite. To elucidate the structure of metabolite M1, the LC/ESI-MS/MS of M1 was examined (Fig 3B). The CID spectrum of M1 (Fig 3B) gives the base peak at m/z 264.4732 by the loss of H2O as evidenced by its HRMS data (PtN2H6Cl; 2.41 ppm), clearly confirms the presence of aqua molecule in M1 (Fig 4). This is also supported by the fact that protonated CP loses HCl to form the base peak at m/z 264.4732 (Fig 3A) (Fig 4). In addition, the MS/MS spectrum shows other product ions at m/z 247.5031, m/z 228.4812, m/z 211.4922 and m/z 195.1754 (Fig 3B and Fig 4) which were also observed in the MS/MS of protonated CP (Fig 3A) (Fig 4), supports the proposed structure for M1. It was reported earlier that CP forms major metabolite, mono aqua CP [17, 18], based on which, the structure of M1 can be assigned as a mono aqua CP metabolite.
M2 (m/z 264.4728): The metabolite M2 at m/z 264.4728 having the elemental composition of PtN2H9O2 (3.21 ppm), was detected at 2.7 min. Its online HDX LC-MS spectrum shows the deuterated molecular ion at m/z 265.4944 with an elemental composition of PtN2H8O2D, suggesting the presence of one exchangeable hydrogen (Pt-OH group) in its structure. These elemental compositions data revealed that M2 lacks two chlorine atoms when compared to CP, indicating that it is a des-chlorinated CP metabolite. The LC-MS/MS spectrum (Fig 3C) of M2 shows abundant product ions at m/z 246.5177 (PtN2H7O; 1.88 ppm) and m/z 228.4812 (PtN2H5; 3.08 ppm) formed by the loss of one and two water molecules (Fig 4), respectively, from M2, unlike the LC-MS/MS of protonated CP and M1 [which show the characteristic fragment ions at m/z 264.4732 and m/z 247.5031 (Fig 4) diagnostic for the presence of chlorine atom], clearly indicating the presence of hydroxy group (Pt-OH) and aqua molecule (Pt-OH2) in the structure of M2. Based on these data, the structure of M2 was characterized to be a mono hydroxylated mono aqua CP metabolite.
M3 ([M+H] + ; m/z 264.4728): The low level metabolite M3 at m/z 264.4728 ([M+H]+; PtN2H9O2; 3.21 ppm) got eluted at 1.7 min. Its deuterated molecular ion at m/z 267.4932 (PtN2O2H6D3) indicates the presence of three exchangeable hydrogens (two hydroxy groups and mobile proton) in its structure. These HRMS data clearly indicate the absence of two chlorine atoms and presence of two hydroxy groups in M3 as compared to CP. Based on these data, M3 could be a dihydoxylated metabolite of CP. This can be seen from the LC-MS/MS spectrum of protonated M3 which shows abundant product ions at m/z 246.5177 and m/z 228.4812 formed by the loss of one and two water molecules (Fig 4), respectively.
M4 ([M+H] + ; m/z 282.4860): The metabolite M4 at m/z 282.4860 ([M+H]+; PtN2H8OCl; 1.92 ppm) was eluted at 3.3 min. Its deuterated molecular ion at m/z 284.5132 with an elemental composition of PtN2H6OClD2 indicates the presence of two exchangeable hydrogens (Pt-OH group and mobile proton) in its structure. The elemental composition data showed that M4 lacks one chlorine atom and inclusion of one hydroxy group as compared to CP, suggesting that it is a mono hydroxylated CP metabolite. The LC-MS/MS spectrum of protonated M4 gives the base peak at m/z 264.4732 (PtN2H6Cl; 2.41 ppm) by the loss of H2O as evidenced by its HRMS data, confirms the presence of hydroxy group (Pt-OH) in M4 (Fig 4). This is also supported by the fact that protonated CP and M1 lose HCl and H2O, respectively, to form the base peak at m/z 264.4732 (Fig 4). Further support comes from the appearance of other structure indicative product ions at m/z 247.5031, m/z 228.4812, m/z 211.4922 and m/z 195.1754 which were already discussed in protonated CP and M1.
It is worthy to note that the online HDX experiments used in this study allowed us to distinguish the metabolites, M1-M4 from each other. The metabolites, M1 & M4 (m/z 282.4860) and M2 & M3 (m/z 264.4728) that have the same nominal mass and identical elemental compositions were unambiguously distinguished and characterized by HDX LC-MS/MS experiments. These metabolites were distinguished from each other mainly based on the presence of numbers of exchangeable hydrogens in their structures that were different for each metabolite. In HDX experiments, M4 showed its deuterated molecular ion at m/z 284.5132 (two exchangeable hydrogens; hydroxy group & mobile proton), whereas M1 detected at m/z 282.4860 (No exchangeable hydrogens). Similarly, the online HDX LC-MS spectrum of M3 showed its deuterated molecular ion at m/z 267.4932 with three exchangeable hydrogens (two hydroxy groups & mobile proton), whereas M2 showed it’s deuterated molecular in at m/z 265.4944 with one exchangeable hydrogen (mobile proton). It is important to note that the structures of these metabolites (M1-M4) were proposed mainly based on HDX LC-MS experiments. The comparison of the product ion spectra obtained using the precursor ions formed from ‘with’ and ‘without’ HDX experiments clearly indicate that the structure elucidation of hydroxylated metabolites can be effectively achieved by using deuterated precursor ions where the protonated precursor ions have not been given accurate information to characterize them. Besides, these HDX experiments were also helped to further support the structural elucidation of some other metabolites (identified in this work) that has been discussed in appropriate places in the succeeding text.
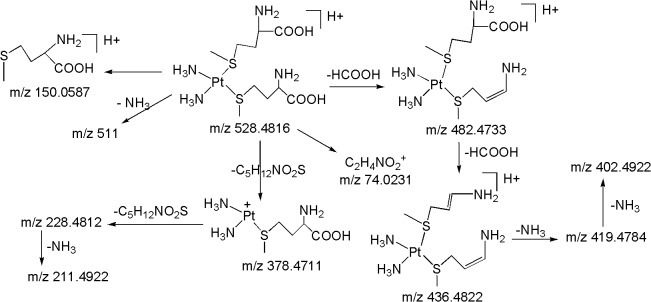
M5 ([M+H] + ;m/z 414.1479): The metabolite M5 at m/z 414.1479 ([M+H]+; C5H18ClN3O2PtS; 3.22 ppm) got eluted at 25.2 min. The appearance of diagnostic product ion at m/z 264.4732 (base peak) (Fig 5A) corresponding to the loss of C5H12NO2S (Fig 6) in the LC-MS/MS spectrum of protonated M5, which is also a characteristic product ion observed in the LC-MS/MS of protonated CP, M1 and M4 (Fig 4), clearly indicating the presence of Pt+(NH3)2(Cl) moiety [two ammine groups, one chlorine atom and platinum atom] in its structure. The LC-MS/MS spectrum of protonated M5 shows diagnostic product ion at m/z 150.0587 corresponding to protonated methionine, confirms the presence of methionine moiety in its structure. This was further substantiated by MS/MS experiments on deuterated derivative M5 in which the m/z 150.0587 ion got shifted to m/z 154.0816 due to incorporation of four deuterium atoms (-NH2,-COOH and mobile proton). It can be noted that the neutral loss fragments of C5H12NO2S (m/z 264.4732) and HCl (m/z 378.1751) (Fig 6) from the protonated molecular ion, also substantiates the presence of methionine moiety and chlorine atom, respectively in M5. Besides, the LC-MS/MS spectrum of protonated M5 shows other fragment ions at m/z 368.1872 (loss of HCOOH), m/z 351.2663 (loss of NH3 from m/z 368.1872) and m/z 74.0231 (C2H4NO2 +), supports the presence of methionine moiety in its structure. Based on these data, M5 was identified as a methionine metabolite of CP.
Fig 5. LC/ESI-MS/MS spectra of (a) Protonated M5 (b) Protonated M6 and (c) Protonated M8 at 33 eV.

Fig 6. Proposed fragmentation mechanism for metabolites M5 and M6.

M6 ([M+H] + ; m/z 396.1561): The metabolite M6 at m/z 396.1561 ([M+H]+; C5H19N3O3PtS; -1.88 ppm) was eluted at 19.6 min. The HRMS data revealed that M6 lacks a chlorine atom and inclusion of hydroxy group as compared to M5, suggesting that it is a hydroxylated methionine CP metabolite. The appearance of significant product ion at m/z 246.5177 (base peak) (Fig 5B) corresponding to the loss of C5H12NO2S (Fig 6) in the LC-MS/MS spectrum of protonated M6, which is also a characteristic ion observed in the LC-MS/MS of protonated M2 (Fig 3B) (Fig 4), indicating the presence of Pt+(NH3)2(OH) moiety (two ammine groups, one hydroxy group and platinum atom) in the structure of M6. This was further supported by the MS/MS spectrum of deuterated M6 in which the m/z 246.5177 ion got shifted to m/z 247.5244 due to incorporation of one deuterium atom (Pt-OH→Pt-OD). Further, protonated M6 yields the m/z 378.1751 (C5H17N3O2PtS; -2.21 ppm) ion by the loss of H2O as evidenced by its HRMS data which clearly shows that H2O is eliminated from hydroxy group that is directly attached to platinum atom (Pt-OH) and not from the-COOH group of methionine moiety. This is also supported by the fact that protonated M5 loses HCl to form m/z 378.1751 (Fig 6). Similarly to M5, the formation of fragment ions at m/z 150.0587 (protonated methionine) (Fig 5B), m/z 154.0816 (deuterated methionine) and the neutral loss of C5H12NO2S (m/z 246.5177) (Fig 6) from the protonated molecular ion, reflects the presence of methionine moiety in the structure of M6. In addition, protonated M6 yields other product ions at m/z 350.1799 (loss of HCOOH), m/z 333.1390 (loss of NH3 from m/z 350.1799) and m/z 74.0231 (C2H4NO2 +), supports the presence of methionine moiety and hydroxy group in M6.
M7 ([M+H] + ; m/z 386.2316): The metabolite M7 at m/z 386.2316 ([M+H]+; C3H14N3O2PtSCl; 3.15 ppm) was eluted at 23.5 min. Similarly to protonated CP, M1, M4 and M5, the LC-MS/MS spectrum of protonated M7 displays the diagnostic product ion at m/z 264.4732 (base peak) corresponding to the loss of C3H8NO2S (Fig 7), authenticating the presence of Pt+(NH3)2(Cl) moiety in its structure. The protonated M7 yields diagnostic product ion at m/z 122.0275 corresponding to protonated cysteine, confirms the presence of cysteine moiety in its structure. This was further confirmed by the MS/MS of deuterated M7 in which m/z 122.0275 ion got shifted to m/z 127.0576 due to incorporation of five deuterium atoms (-NH2,-COOH,-SH and mobile proton). Further, the neutral loss fragments C3H8NO2S (m/z 264.4732) and HCl (m/z 350.1422) from the protonated molecular ion (Fig 7), substantiate the presence of cysteine moiety and chlorine atom, respectively, in M7. Besides, the spectrum also shows other ions at m/z 340.1688 (loss of HCOOH) and m/z 323.1365 (loss of NH3 from m/z 340.1688) which are consistent with the proposed structure. Based on these data, M7 was identified as a cysteine metabolite of CP.
Fig 7. Proposed fragmentation mechanism for metabolites M7-M10.

M8 ([M+H] + ; m/z 368.1261): The metabolite M8 at m/z 368.1261 ([M+H]+; C3H15N3O3PtS; -3.22 ppm) was detected at 18.5 min. The HRMS data revealed that M8 lacks a chlorine atom and inclusion of hydroxy group as compared to M7, suggesting that it is a hydroxylated cysteine metabolite of CP. Similarly to M2 and M6, the LC-MS/MS spectrum of protonated M8 shows significant product ion at m/z 246.5177 formed by the loss of C3H8NO2S (Fig 5C) (Fig 7), indicating the presence of Pt+(NH3)2(OH) moiety in its structure. This was further supported by the MS/MS spectrum of deuterated M8 in which the m/z 246.5177 ion got shifted to m/z 247.5244 due to incorporation of one deuterium atom (Pt-OH→Pt-OD). Further, the protonated M8 yields m/z 350.1422 (C3H13N3O2PtS; 2.12 ppm) ion by the loss of H2O as evidenced by its HRMS data which clearly shows that H2O is eliminated from hydroxy group that is directly attached to platinum atom (Pt-OH) (discussed in M6) and not from the-COOH group of cysteine moiety. This is also supported by the fact that protonated M7 loses HCl to form the base peak at m/z 350.1422 (Fig 7). Similarly to M7, the formation of diagnostic fragment ions at m/z 122.0275 (protonated cysteine) and m/z 127.0576 (deuterated cysteine), and the neutral loss of C3H8NO2S (m/z 246.5177) (Fig 7) from the protonated molecular ion, reflects the presence of cysteine moiety in the structure of M8. Besides, the LC-MS/MS spectrum of protonated M8 shows other product ions at m/z 322.1488 (loss of HCOOH) and m/z 305.1177 (loss of NH3 from m/z 322.1488) which are compatible with the proposed structure.
M9 ([M+H] + ; m/z 428.1711): The metabolite M9 at m/z 428.1711 ([M+H]+) with an elemental composition of C5H16N3O3PtSCl (3.66 ppm) was eluted at 24.2 min, suggesting an addition of acetyl group to M7. This can be seen from the LC-MS/MS spectrum of protonated M9 which clearly displays an abundant product ion at m/z 386.2316 (protonated M7) formed by the loss of C2H2O (Fig 7). Further, it can be seen from Fig 7 that the neutral loss of CH3CONH2 (m/z 369.1811) from the protonated molecular ion (m/z 428.1711), also confirms the presence of N-acetyl group in M9, as this is not the case for protonated M7 and M8 which eliminates NH3 due to the presence of-NH2 group. Similarly to protonated CP, M1, M4, M5 and M7, the appearance of significant product ion at m/z 264.4732 which involves the loss of C5H10NO3S (Fig 7), substantiates the presence of Pt+(NH3)2(Cl) moiety in the structure of M9. It can be noted that the formation of product ion at m/z 164.0544 (protonated acetylcysteine) and its corresponding deuterated species at m/z 168.0622 (-NH,-SH,-COOH and mobile proton), and the neutral loss of C5H10NO3S (m/z 264.4732) (Fig 7) from the protonated molecular ion, substantiates the presence of acetylcysteine moiety in the structure of M9. Other product ions at m/z 382.1311, m/z 323.1365 and m/z 392.1688, are also in line with the proposed structure. Based on these data, M9 was characterized as an acetylcysteine metabolite of CP.
M10 ([M+H] + ; m/z 410.1173): The metabolite M10 at m/z 410.1173 ([M+H]+) with an elemental composition of C5H17N3O4PtS (-4.09 ppm) was detected at 22.5 min, suggesting an addition of an acetyl group to M8. The HRMS data also revealed the lack of chlorine atom and inclusion of hydroxy group in M10 when compared to M9. The LC-MS/MS spectrum of protonated M10 gives an abundant peak at m/z 392.1688 (C5H15N3O3PtS; -4.09 ppm) (Fig 8A) by the loss of H2O, confirms the presence of Pt-OH in M10. This is also supported by the fact that protonated M9 loses HCl to form m/z 392.1688. The protonated M10 also yields m/z 368.1261 ion (Protonated M8) which is formed by the loss of C2H2O (Fig 7), confirms the presence of acetyl group in M10. Further, it can be seen from Fig 7 that the loss of CH3CONH2 (m/z 351.1388) from the protonated molecular ion (m/z 410.1173) of M10, also confirms the presence of N-acetyl group in its structure (Fig 7), as this is not the case for protonated M7 and M8 which eliminates NH3 due to the presence of-NH2 group (Fig 7). The LC-MS/MS spectrum of protonated M10 also shows other characteristic fragment ions at m/z 246.5177 which involves the loss of C5H10NO3S and m/z 164.0544 (protonated acetylcysteine) (Fig 7), authenticates the presence of Pt+(NH3)2(OH) moiety (discussed in M2, M6 and M8) and acetylcysteine moiety, respectively, in its structure. This was further supported by the MS/MS spectrum of deuterated M10 in which m/z 246.5177 and m/z 164.0544 ions got shifted to higher masses by one and four units, respectively, due to exchange of labile protons with deuterium atoms.
Fig 8. LC/ESI-MS/MS spectra of (a) Protonated M10 (b) Protonated M12 and (c) Protonated M17 at 28 eV.

M11 ([M+H] + ; m/z 572.0922): The metabolite M11 at m/z 572.0922 ([M+H]+; C10H24N5O6PtSCl; -2.11 ppm) was eluted at 8.2 min. The appearance of diagnostic product ion at m/z 264.4732 due to loss of C10H18N3O6S (Fig 9) in the LC-MS/MS spectrum of protonated M11, which is also a characteristic product ion observed in the LC-MS/MS of protonated CP, M1, M4, M5, M7 and M9, authenticates the presence of Pt+(NH3)2(Cl) moiety in its structure. The protonated M11 yields diagnostic product ion at m/z 308.0916 corresponding to protonated glutathione, confirms the presence of glutathione moiety in its structure. This was further confirmed by MS/MS of deuterated M11 in which the m/z 308.0916 ion got shifted to m/z 316.1426 due to incorporation of eight deuterium atoms (-NH2, two-NH, two-COOH,-SH and mobile proton). The LC-MS/MS spectrum of protonated M11 also shows other structure indicative fragment ions at m/z 526.1466 (loss of HCOOH), m/z 443.0811 (loss of C5H7NO3), m/z 323.1365 (simultaneous elimination of C3H5NO3 & C5H10N2O3) and m/z 130.0491 (C5H8NO3 +) which are consistent with the proposed structure for M11. Based on these data, M11 was identified as a glutathione metabolite of CP.
Fig 9. Proposed fragmentation mechanism for metabolites M11-M14.

M12 ([M+H] + ; m/z 554.1661): The metabolite M12 at m/z 554.1661 ([M+H]+; C10H25N5O7PtS; 4.41 ppm) was detected at 5.9 min. The HRMS data showed that M12 lacks a chlorine atom and inclusion of hydroxy group as compared to M11, suggesting that it is a hydroxylated glutathione CP metabolite. This has been confirmed by the appearance of the Pt+(NH3)2(OH) moiety consisting fragment ion at m/z 246.5177 (discussed in M2, M6, M8 and M10) which is formed by the neutral loss of C10H18N3O6S and m/z 308.0916 (protonated glutathione) (Fig 9), in the LC-MS/MS spectrum of protonated M12 (Fig 8B). This was also further confirmed by the MS/MS spectrum of deuterated M12 in which the above fragment ions got shifted to higher masses due to exchange of labile protons with deuterium i.e. m/z 246.5177→247.5244 and m/z 308.0916→316.1426. The protonated M12 also yields m/z 536.1528 ion (C10H23N5O6PtS; 2.51 ppm) by the loss of H2O, reflects the presence of hydroxy group (Pt-OH) in its structure which was further supported by the fact that protonated M11 loses HCl to form m/z 536.1528. In line with this, the MS/MS of deuterated M12 also showed the loss of D2O. Further support comes from other structure indicative product ions at m/z 508.1711 (loss of HCOOH), m/z 425.1344 (loss of C5H7NO3), m/z 305.1177 (simultaneous elimination of C3H5NO3 & C5H10N2O3) and m/z 130.0491 (C5H8NO3 +) observed in the LC-MS/MS of protonated M12.
M13 ([M+H] + ; m/z 327.1397): The metabolite M13 at m/z 327.1397 ([M+H]+) with an elemental composition of C2H13N2PtSCl (-2.88 ppm) was detected at 29.4 min. Similarly to protonated CP, M1, M4, M5, M7, M9 and M11, the LC-MS/MS spectrum of protonated M13 shows the characteristic fragment ion at m/z 264.4732 involves the loss of C2H7S, confirms the presence of Pt+(NH3)2(Cl) moiety (Fig 9) in its structure. From Fig 9, it is notable that the neutral loss fragments C2H7S (m/z 264.4732) and HCl (m/z 291.1165) from the protonated molecular ion, designate the presence of thioether moiety and chlorine atom, respectively, in M13. Based on these data, M13 was identified as a thioether metabolite of CP.
M14 ([M+H] + ; m/z 309.1141): The metabolite M14 at m/z 309.1141 ([M+H]+; C2H14N2OPtS; 1.92 ppm) was detected at 29.1 min. Its deuterated molecular at m/z 311.1171 (C2H12N2OPtSD2), suggests the presence of two exchangeable hydrogens in its structure (Pt-OH group and mobile proton). These HRMS data clearly showed that M14 lacks a chlorine atom and inclusion of hydroxy group as compared to M13, suggesting that it is a hydroxylated thioether CP metabolite. This has been confirmed by the appearance of Pt+(NH3)2(OH) moiety consisting fragment ion at m/z 246.5177 (discussed in M2, M6, M8, M10 and M12) formed by the loss of C2H7S and m/z 291.1165 ion which involves the loss of H2O from Pt-OH group (discussed in M6, M8 and M10) (Fig 9), in the LC-MS/MS spectrum of protonated M14. Further, this has been substantiated by the MS/MS spectrum of deuterated M14 in which the m/z 246.5177 ion got shifted to m/z 247.5244 ion due to exchange of one labile proton with deuterium atom (Pt-OH)→(Pt-OD) and the deuterated molecular ion (m/z 311.1171) yields the m/z 291.1165 ion by the loss of D2O (from Pt-OD and mobile proton).
M15 ([M+H] + ; m/z 528.4816): The second most abundant metabolite M15 at m/z 528.4816 ([M+H]+) with an elemental composition of C10H29N4O4PtS2 (3.96 ppm) was eluted at 14.6 min. The HRMS data suggests the lack of one chlorine atom and presence of an additional methionine moiety in M15 as compared to M5. This can be seen from the LC-MS/MS spectrum of protonated M15 which shows abundant product ions at m/z 378.4711 and m/z 228.4812 corresponding to a probable loss of one and two methionine moieties, respectively, and m/z 150.0587 (protonated methionine) (Fig 10). It can be noted that the peaks at m/z 264.4732 (Pt+(NH3)2Cl) and m/z 246.5177 (Pt+(NH3)2OH) would have been present and the sequential losses of two neutral species of methionine would have been absent in case of one methionine moiety in protonated M15 as discussed in M5 and M6. Further support comes from the appearance of moderately abundant product ions at m/z 482.4733 and m/z 436.4822 which were formed by the neutral loss of one and two formic acids, respectively, from protonated M15. Similarly to M5 and M6, the appearance of m/z 150.0587 ion (protonated methionine) in the MS/MS of protonated M15 and m/z 154.0816 ion (deuterated methionine) in the MS/MS of deuterated M15, reflects the presence of methionine moiety in the structure of M15. Based on these data, the structure of M15 can be assigned as a di-methionine metabolite of CP.
Fig 10. Proposed fragmentation mechanism for metabolite M15.
M16 ([M+H] + ; m/z 472.4933): The third most abundant metabolite M16 at m/z 472.4933 ([M+H]+) with an elemental composition of C6H21N4O4PtS2 (-2.96 ppm) was eluted at 12.9 min. The HRMS data suggests the lack of one chlorine atom and inclusion of an additional cysteine moiety in M16 as compared to M7. This can be seen from the LC-MS/MS spectrum of protonated M16 which shows abundant product ions at m/z 350.4819 and m/z 228.4812 corresponding to a probable loss of one and two cysteine moieties, respectively, and m/z 122.0275 (protonated cysteine). The peaks at m/z 264.4732 (Pt+(NH3)2Cl) and m/z 246.5177 (Pt+(NH3)2OH) would have been present and the sequential losses of two neutral species of cysteine would have been absent in case of one cysteine moiety in M16 as discussed in M7 and M8. The moderately abundant product ions at m/z 426.4871 (loss of HCOOH) and m/z 380.4762 (loss of two HCOOH), also designate the presence of two cysteine moieties in M16 (Fig 11). Similarly to M7 and M8, the appearance of m/z 122.0275 (protonated cysteine) and m/z 127.0576 (deuterated cysteine) in the MS/MS of protonated and deuterated M16, respectively, substantiates the presence of cysteine moiety in the structure of M16. Based on these data, M16 was characterized to be a di-cysteine metabolite of CP.
Fig 11. Proposed fragmentation mechanism for metabolites M16 and M17.

M17 ([M+H] + ; m/z 556.5014): The protonated metabolite M17 at m/z 556.5014 with an elemental composition of C10H25N4O6PtS2 (-3.88 ppm) was eluted at 15.7 min. The HRMS data suggests the lack of one chlorine atom and the inclusion of an additional acetylcysteine moiety in M17 as compared to M9. This can be seen from the LC-MS/MS spectrum of protonated M17 (Fig 8C) which shows abundant product ions at m/z 392.5017 and m/z 228.4812 corresponding to a probable loss of one and two acetylcysteine moieties, respectively and m/z 164.0544 (protonated acetylcysteine). Further support comes from the appearance of significant product ions at m/z 514.5149 and m/z 472.4933 & m/z 497.5116 and m/z 438.4933 & m/z 510.5144 and m/z 464.5011 (Fig 8C) which were formed by the neutral loss fragments of one and two C2H2O & CH3CONH2 & HCOOH, respectively, from protonated M17 (Fig 11). Similarly to M15 and M16, the peaks at m/z 264.4732 ((Pt+(NH3)2Cl)) and m/z 246.5177 ((Pt+(NH3)2OH)) would have been present and the sequential losses of two neutral species of acetylcysteine would have been absent in case of one acetylcysteine moiety in M17 as discussed in M9 and M10. Similarly to M9 and M10, the appearance of m/z 164.0544 (protonated acetylcysteine) in the MS/MS of protonated M17 (Fig 8C) and m/z 168.0622 (deuterated acetylcysteine) in the MS/MS of deuterated M17, confirms the presence of acetylcysteine moiety in the structure of M17. Based on these data, M17 was identified as a di-acetylcysteine metabolite of CP.
M18 (m/z 414.0922): The metabolite M18 at m/z 414.0922 with an elemental composition of C5H11NO2PtSCl2 (4.09 ppm) was eluted at 21.6 min. The HRMS data suggests the presence of two chlorine atoms and methionine moiety in the structure of M18. The LC-MS/MS spectrum of M18 shows abundant fragment ions at m/z 378.1412 and m/z 342.1655 due to loss of one and two HCl molecules, respectively, from the molecular ion, confirms the presence of two intact chlorine atoms in its structure. The absence of peak corresponds to loss of HCOOH, which is a major fragmentation observed in the LC-MS/MS of methionine containing metabolites (M5, M6 and M15), clearly eliminates the possibility of straight chain methionine moiety in the structure of M18. On the contrary, the appearance of diagnostic fragment ion at m/z 370.1542 corresponding to the loss of CO2 from the molecular ion, confirms the presence of chelated methionine ring in M18. Further, it can be noted that the ion at m/z 228.4812 (discussed in protonated CP and M1-M17) would have been present in case of Pt+N2H5 moiety (if the chelated methionine ring does not present in M18). Based on these data, M18 was characterized to be a chelated methionine CP metabolite.
M19 (m/z 396.1387): The metabolite M19 at m/z 396.1387 with an elemental composition of C5H12NO3PtSCl (3.66 ppm) was eluted at 15.7 min. The HRMS data suggests the lack of one chlorine atom and presence of hydroxy group in M19 as compared to M18. This has been confirmed from the LC-MS/MS spectrum of M19 which shows abundant fragment ion at m/z 378.1412 (C5H10ClNO2PtS; 1.56 ppm) involves the loss of H2O. The m/z 378.1412 ion loses HCl to form m/z 342.1655 ion which confirms the presence of intact chlorine atom in M19 as discussed in M18. Similarly to M18, the absence of peak corresponds to loss of HCOOH and appearance of diagnostic fragment ion at m/z 352.1173 formed by the loss of CO2 from molecular ion, clearly eliminates the possibility of straight chain methionine moiety and confirms the presence of chelated methionine ring in the structure of M19. Further, the absence of m/z 228.4812 (Pt+N2H5) (discussed in protonated CP and M1-M17), substantiates the presence of the chelated methionine ring in M19 as similar to M18. Based on these data, M19 was characterized to be a hydroxylated chelated methionine CP metabolite.
M20 (m/z 396.4976): The metabolite M20 at m/z 396.4976 with an elemental composition of C5H14N2O2PtSCl (-3.55 ppm) was eluted at 27.8 min. The elemental composition data indicates the lack of one chlorine atom and presence of ammine group in M20 as compared to M18. This can be seen from the LC-MS/MS spectrum of M20 which shows significant ion at m/z 379.1012 formed by the loss of NH3, has been evidenced by HRMS data (C5H11ClNO2PtS; -4.05 ppm). Further, the m/z 379.1012 ion loses HCl to form m/z 343.1181 ion which confirms the presence of intact chlorine atom in the structure of M20 as discussed in M18 and M19. Similarly to M18 and M19, the absence of peak corresponds to loss of HCOOH and appearance of diagnostic fragment ion at m/z 352.1157 corresponding to loss of CO2 from M20, clearly eliminates the possibility of straight chain methionine moiety and confirms the presence of chelated methionine ring in M20. Similarly to M18 and M19, the absence of m/z 228.4812 ion (Pt+N2H5) (discussed in protonated CP and M1-M14), supports presence of the chelated methionine ring in M20.
M21 (m/z 386.1451): The metabolite M21 at m/z 386.1451 with an elemental composition of C3H7NO2PtS Cl2 (2.88 ppm) was eluted at 22.3 min. The HRMS data suggests the presence of two chlorine atoms and cysteine moiety in the structure of M21. The LC-MS/MS spectrum of M21 shows abundant fragment ions at m/z 350.1556 and m/z 314.1277 due to loss of one and two HCl molecules, respectively, from the molecular ion, confirms the presence of two intact chlorine atoms in M21. The absence of peak corresponds to loss of HCOOH and the appearance of diagnostic fragment ion at m/z 342.1175 formed by the loss of CO2 from molecular ion, clearly eliminates the possibility of straight chain cysteine moiety and substantiates the presence of chelated cysteine ring in M21. The absence of m/z 228.4812 ion (Pt+N2H5) (discussed in protonated CP and M1-M17), also supports the presence of chelated cysteine ring in M21. Based on these data, M21 was characterized to be a chelated cysteine CP metabolite.
M22 (m/z 368): The metabolite M22 at m/z 368.1911 with an elemental composition of C3H8NO3PtSCl (4.03 ppm) was eluted at 17.8 min. The HRMS data suggests the lack of one chlorine atom and presence of hydroxy group in M22 as compared to M21. This can be seen from the LC-MS/MS spectrum of M22 which shows abundant fragment ion at m/z 350.1556 (C3H6ClNO2PtS; 2.93 ppm) formed by the loss of H2O. Further, the m/z 350.1556 ion loses HCl to form m/z 314.1277 ion which confirms the presence of intact chlorine atom in the structure of M22 as discussed in M21. Similarly to M21, the absence of peak corresponds to loss of HCOOH and the appearance of diagnostic fragment ion at m/z 324.1866 corresponding to the loss of CO2 from M22, clearly eliminates the possibility of straight chain cysteine moiety and confirms the presence of chelated methionine ring in M22. Similarly to M18-M21, the absence of m/z 228.4812 ion (Pt+N2H5) (discussed in protonated CP and M1-M17), supports the presence of chelated cysteine ring in M21. Based on these data, M22 was characterized to be a hydroxylated chelated cysteine CP metabolite.
M23 (m/z 368.5646): The metabolite M23 at m/z 368.5646 with an elemental composition of C3H10N2O2PtSCl (-4.77 ppm) was eluted at 28.7 min. The accurate mass measurements data indicates the lack of one chlorine atom and presence of ammine group in M23 as compared to M21. This can be seen from the LC-MS/MS spectrum of M23 which shows diagnostic fragment ion at m/z 351.1566 formed by the loss of NH3 (C3H7ClNO2PtS; -3.32 ppm). Further, the m/z 351.1566 ion loses HCl to form m/z 315.4811 ion which confirms the presence of intact chlorine atom in the structure of M23 as discussed in M21 and M22. Similarly to M21 and M22, the absence of the peak corresponds to the loss of HCOOH and the appearance of m/z 324.4622 ion due to the loss of CO2 from molecular ion, clearly eliminates the possibility of straight chain cysteine moiety and confirms the presence of chelated cysteine ring in M23.
M24 ([M+H] + ; m/z 529.0496): The metabolite M24 at m/z 529.0496 ([M+H]+) with an elemental composition of C10H23N2O4PtS2Cl (4.55 ppm) was eluted at 9.1 min. The LC-MS/MS spectrum of protonated M24 displays abundant product ions at m/z 379.0733 (loss of C5H12NO2S), m/z 483.0687 (loss of HCOOH) and m/z 150.0587 (protonated methionine) (Fig 12), indicates the presence of straight chain methionine in its structure. The formation significant ion at m/z 485.0583 which involves the loss of CO2 and absence of the m/z 228.4812 ion (Pt+N2H5) (discussed in protonated CP and M1-M17), substantiate the presence of one chelated methionine ring in M24. Another moderately abundant ion at m/z 493.2145 formed by the loss of HCl (Fig 12), reflects the presence of intact chlorine atom in M24.
Fig 12. Proposed fragmentation mechanism for metabolites M24 and M25.
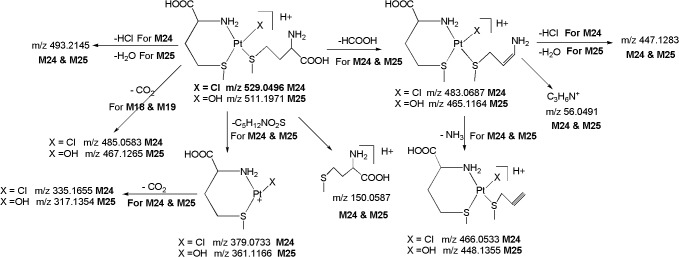
M25 ([M+H] + ; m/z 511.1971): The metabolite M25 at m/z 511.1971 ([M+H]+) with an elemental composition of C10H24N2O5PtS2 (4.38 ppm) was eluted at 6.6 min. The elemental composition data indicates the lack of one chlorine atom and inclusion of hydroxy group in M25 as compared to M24. This can be seen from the LC-MS/MS spectrum of protonated M25 which displays an abundant product ion at m/z 493.2145 (C10H22N2O4PtS2; 3.36 ppm) formed by the loss of H2O. This was further supported by the fact that protonated M24 loses HCl to form the base peak m/z 493.2145. This clearly indicates that H2O is eliminated from Pt-OH group and not from the-COOH group of methionine group. In line with this the MS/MS of deuterated M25 also showed the loss of D2O. The protonated M25 yields abundant product ions at m/z 361.1166 (loss of C5H12NO2S), m/z 465.1164 (loss of HCOOH) and m/z 150.0587 (protonated methionine), indicates the presence of straight chain methionine in its structure. Further, the formation an abundant product ion at m/z 467.1265 (Fig 12) which involves the loss of CO2 and absence of the m/z 228.4812 ion (Pt+N2H5) (discussed in protonated CP and M1-M17), supports the presence of one chelated methionine ring in M25.
M26 ([M+H] + ; m/z 473.1263): The metabolite M26 at m/z 473.1263 ([M+H]+; C6H15N2O4PtS2Cl; 4.11 ppm) was detected at 10.2 min. The LC-MS/MS spectrum of protonated M26 displays abundant product ions at m/z 351.1167 (loss of C3H8NO2S), m/z 427.1153 (loss of HCOOH) and m/z 122.0275 (protonated cysteine) (Fig 13), demonstrating the presence of strain chain cysteine in M26. Further, the formation an abundant ion at m/z 429.1165 which involves the loss of CO2 and the absence of m/z 228.4812 ion (Pt+N2H5) (discussed in protonated CP and M1-M17), confirms the presence of chelated cysteine ring in M26. It is notable that the m/z 437.1877 ion which is formed by the loss of HCl, reflects the presence of intact chlorine atom in M26.
Fig 13. Proposed fragmentation mechanism for metabolites M26 and M27.
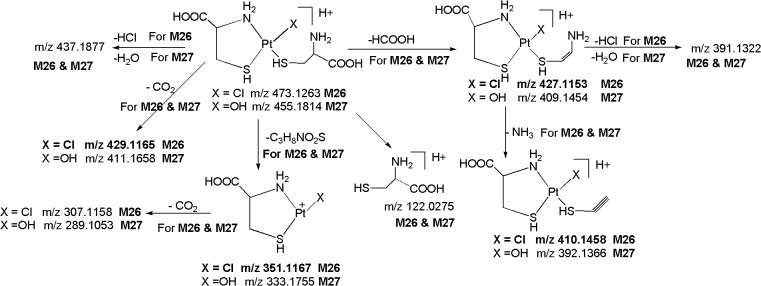
M27 ([M+H] + ; m/z 455.1814): The metabolite M27 at m/z 455.1814 ([M+H]+) with an elemental composition of C6H16N2O5PtS2 (2.66 ppm) was eluted at 7.6 min. The HRMS data suggests the lack of one chlorine atom and inclusion of hydroxy group in M27 as compared to M26. This can be seen from the LC-MS/MS spectrum of protonated M27 which displays an abundant ion at m/z 437.1877 (C6H14N2O4PtS2; 2.66 ppm) formed by the loss of H2O. This has been confirmed by the MS/MS of deuterated M27 which showed the loss of D2O. Similarly to M26, the LC-MS/MS spectrum of protonated M27 shows abundant product ions at m/z 333.1755 (C3H8NO2S), m/z 409.1454 (loss of HCOOH) and m/z 122.0275 (protonated cysteine) (Fig 13), represents the presence of strain chain methionine in its structure. Further, the formation an abundant product ion at m/z 411.1658 which involves the loss of CO2 and absence of the m/z 228.4812 ion (Pt+N2H5) (discussed in protonated CP and M1-M17), authenticates the presence of chelated cysteine ring in M27.
M28 ([M+H] + ; m/z 379.0851), M29 ([M+H] + ; m/z 494.5511), M30 ([M+H] + ; m/z 351.1822) and M31 ([M+H] + ; m/z 438.4955): The metabolites M28 at m/z 379.0851 (([M+H]+); C5H18N3O2PtS; 3.77 ppm), M29 at m/z 494.5511 (([M+H]+); C10H23N2O4PtS2; -3.88 ppm), M30 at m/z 351.1822 (([M+H]+); C3H14N3O2PtS; 1.88 ppm) and M31 at m/z 438.4955 (([M+H]+); C6H15N2O4PtS2; -4.55 ppm) were eluted at 25.9, 10.6, 26.8 and 11.8 min, respectively. The LC-MS/MS spectra of protonated M28 and M30 display abundant product ions at m/z 335.1655 (C4H18N3PtS; 2.87 ppm) and m/z 307.1344 (C2H14N3PtS; 2.61 ppm) which were formed by the elimination of CO2 from the protonated molecular ion, clearly indicates the presence of chelated monomethionine and chelated monocysteine rings in M28 and M30, respectively. It can be noted that the appearance of diagnostic product ion at m/z 228.4812 in the LC-MS/MS spectrum of protonated M28 and M30, which is also a characteristic ion observed in the LC-MS/MS spectra of protonated CP and metabolites M1-M17, confirms the presence of Pt+N2H5 group. Based on these data, M28 and M30 were characterized to be chelated monomethionine and monocysteine CP metabolites, respectively.
Similarly, the LC-MS/MS spectra of protonated M29 and M31 display abundant product ions at m/z 450.4981 (C9H23N2O2PtS2; 4.11 ppm) & m/z 406.5011 (C8H23N2PtS2; 2.69 ppm) and m/z 394.5011 (C5H15N2O2PtS2; -4.07 ppm) & m/z 350.4968 (C4H15N2PtS2; -3.56 ppm), respectively, formed by the sequential elimination of one & two CO2 molecules, respectively, clearly indicates the presence of bismethionine and biscysteine chelated rings in M29 and M31, respectively. Further, the absence of m/z 228.4812 ion in the LC-MS/MS spectra of M29 and M31, which is diagnostic for the presence of Pt+N2H5 group (discussed in protonated CP and M1-M17), substantiates the presence of bismethionine and biscysteine rings in the structures of M29 and M31, respectively. Based on these data, M29 and M31 were identified as bismethionine and biscysteine CP metabolites, respectively.
Conclusions
A total of thirty one in vivo metabolites of CP formed in rat kidney tissue homogenate samples, have been identified and characterized by using liquid chromatography positive ion electrospray ionization high resolution tandem mass spectrometry (LC/ESI-HR-MS/MS). The structures of identified metabolites have been elucidated based on fragmentation pattern, accurate mass measurements combined with online HDX LC-MS/MS experiments. This study describes the utility of online deuterated experiments in the identification and structural characterization of drug metabolites. The dissociation of one of the chlorine atoms from CP results in a positive charge on the platinum that will attract the negatively charged sulfur present on the active thio group containing molecules. The results showed that CP undergoes a series of ligand exchange biotransformation reactions with water and active thio group containing nucleophiles like methionine, cysteine, acetylcysteine, glutathione and thioether to form various toxic metabolites. The major metabolites of CP are, mono aqua CP, monohydroxy CP, dihydroxy CP, hydroxy aqua CP, mono methionine CP, mono hydroxylated methionine CP, bismethionine CP, mono cysteine CP, mono hydroxylated cysteine CP, biscysteine CP, mono acetylcysteine CP, mono hydroxylated acetylcysteine CP, mono glutathione CP, mono hydroxylated glutathione CP, mono thioether CP, mono hydroxylated thioether CP. The newly identified metabolites have a scope to be assessed for their efficacy and toxicity. The structural identification of these metabolites might provide essential information for further pharmacological and clinical studies of CP, and may also be useful to develop various effective new anticancer agents. We further focus on the functions and toxicology studies of CP and its metabolites including nephrotoxicity in our future study. However, from the present it is difficult to comment on the toxic studies of CP and its metabolites in detail.
Acknowledgments
This work was supported by Proteogenomic Research Program, and the Bio- and Medical Technology Development Program (Project No. 2012M3A9B6055305) through the National Research Foundation of Korea funded by the Korean Ministry of Education, Science and Technology (to K.P.K.), Republic of Korea.
Data Availability
All relevant data are within the paper.
Funding Statement
This work was supported by Proteogenomic Research Program, and the Bio- and Medical Technology Development Program (Project No. 2012M3A9B6055305) through the National Research Foundation of Korea funded by the Korean Ministry of Education, Science and Technology (to K.P.K.), Republic of Korea.
References
- 1. Matabosch X, Pozo OJ, Perez-Mana C, Farre M, Marcos J, Segura J, Ventura R. Identification of budesonide metabolites in human urine after oral administration. Anal. Bioanal. Chem. 2012; 404: 325–340. 10.1007/s00216-012-6037-0 [DOI] [PubMed] [Google Scholar]
- 2. Liu ZY, Huang LL, Chen DM, Dai MH, Tao YF, Wang YL, Yuan ZH. Application of electrospray ionization hybrid ion trap/time-of-flight mass spectrometry in the rapid characterization of quinocetone metabolites formed in vitro. Anal. Bioanal. Chem. 2010; 396: 1259–1271. 10.1007/s00216-009-3245-3 [DOI] [PubMed] [Google Scholar]
- 3. Peterman SM, Duczak N, Kalgutkar AS, Lame ME, Soglia JR. Application of a linear ion trap/orbitrap mass spectrometer in metabolite characterization studies: Examination of the human liver microsomal metabolism of the non-tricyclic anti-depressant nefazodone using data-dependent accurate mass measurements. J. Am. Soc. Mass Spectrom. 2006; 17: 363–375. 10.1016/j.jasms.2005.11.014 [DOI] [PubMed] [Google Scholar]
- 4. Strom T, Shokati T, Klawitter J, Hoffman K, Schiebel HM, Christiansa U. Structural identification of SAR-943 metabolites generated by human liver microsomes in vitro using mass spectrometry in combination with analysis of fragmentation patterns. J. Mass Spectrom. 2011; 46: 615–624. 10.1002/jms.1930 [DOI] [PubMed] [Google Scholar]
- 5. Fan Y, Han H, He C, Yang L, Wang Z. Identification of the metabolites of gigantol in rat urine by ultra-performance liquid chromatography combined with electrospray ionization quadrupole time-of-flight tandem mass spectrometry. Biomed. Chromatogr. 2014; 28: 1808–1815. 10.1002/bmc.3224 [DOI] [PubMed] [Google Scholar]
- 6. Liu D, Jiang J, Zhang L, Tan F, Wang Y, Hu P. Metabolite characterization of a novel anti‐cancer agent, icotinib, in humans through liquid chromatography/quadrupole time‐of‐flight tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2011; 25: 2131–2140. 10.1002/rcm.5061 [DOI] [PubMed] [Google Scholar]
- 7. Jirasko R, Holcapek M, Nobilis M. Identification of phase I and phase II metabolites of benfluron and dimefluron in rat urine using high performance liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2011; 25: 2153–2162. 10.1002/rcm.5097 [DOI] [PubMed] [Google Scholar]
- 8. Jiaa Y, Shenb J, Li X, Xie H, Wangc J, Luoa J, et al. Identification and analysis of gastrodin and its five metabolites using ultra-fast liquid chromatography electrospray ionization tandem mass spectrometry to investigate influence of multiple-dose and food. J. Chromatogr. A. 2014; 1358: 110–116. 10.1016/j.chroma.2014.06.080 [DOI] [PubMed] [Google Scholar]
- 9. Michopoulos F, Whalleya N, Theodoridis G, Wilson ID, Dunkleya TPJ, Critchlowa SE. Targeted profiling of polar intracellular metabolites using ion-pair-high performance liquid chromatography and ultra-high performance liquid chromatography coupled to tandem mass spectrometry: Applications to serum, urine and tissue extracts. J. Chromatogr. A. 2014; 1349: 60–68. 10.1016/j.chroma.2014.05.019 [DOI] [PubMed] [Google Scholar]
- 10. Wanga Y, Armando AM, Quehenberger O, Yana C, Dennis EA. Comprehensive ultra-performance liquid chromatographic separation and mass spectrometric analysis of eicosanoid metabolites in human samples. J. Chromatogr. A. 2014; 1359: 60–69. 10.1016/j.chroma.2014.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu DQ, Hop CE. Strategies for characterization of drug metabolites using liquid chromatography tandem mass spectrometry in conjunction with chemical derivatization and on-line H/D exchange approaches. J. Pharm. Biomed. Anal. 2005; 37: 1–18. 10.1016/j.jpba.2004.09.003 [DOI] [PubMed] [Google Scholar]
- 12. Beuck S, Schanzer W, Thevis M. Investigation of the in vitro metabolism of the emerging drug candidates S107 for doping-preventive purposes. J. Mass Spectrom. 2011; 46: 112–130 . 10.1002/jms.1878 [DOI] [PubMed] [Google Scholar]
- 13. Lam W, Ramanathan R. In electrospray ionization source hydrogen/deuterium exchange LC-MS and LC-MS/MS for characterization of metabolites. J. Am. Soc. Mass Spectrom. 2002; 13: 345–353. 10.1016/S1044-0305(02)00346-X [DOI] [PubMed] [Google Scholar]
- 14. Giaccone G. Clinical perspectives on platinum resistance. Drugs. 2000; 59: 9–17. 10.2165/00003495-200059004-00002 [DOI] [PubMed] [Google Scholar]
- 15. Hartmann JT, Lipp HP. Toxicity of platinum compounds. Expert Opin. Pharmacother. 2003; 4: 889–901. 10.1517/14656566.4.6.889 [DOI] [PubMed] [Google Scholar]
- 16. Yachnin JR, Wallin I, Lewensohn R, Sirzen F, Ehrsson H. The kinetics and cytotoxicity of cisplatin and its monohydrated complex. Cancer Lett. 1998; 132: 175–180. 10.1016/S0304-3835(98)00175-X [DOI] [PubMed] [Google Scholar]
- 17. Hall MD, Okabe M, Shen DW, Liang XJ, Gottesman MM. The role of cellular accumulation in determining sensitivity to platinum-based chemotherapy. Annu. Rev. Pharmacol. Toxicol. 2008; 48: 495–535. 10.1146/annurev.pharmtox.48.080907.180426 [DOI] [PubMed] [Google Scholar]
- 18. Ekborn A, Lindberg A, Laurell G, Wallin I, Eksborg S, Ehrsson H. Ototoxicity, nephrotoxicity and pharmacokinetics of cisplatin and its monohydrated complex in the guinea pig. Cancer Chemother. Pharmacol. 2003; 51: 36–42. 10.1007/s00280-002-0540-5 [DOI] [PubMed] [Google Scholar]
- 19. Miller RP, Tadagavadi RK, Ramesh G, Reeves WG. Mechanisms of cisplatin nephrotoxicity. Toxins (Basel). 2010; 2: 2490–2518. 10.3390/toxins2112490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pinedo HM, Schornagel JH. Platinum and other metal coordination compounds in cancer chemotherapy 2. Plenum; New York; 1996. pp 1–357. [Google Scholar]
- 21. Daley-Yates PT, McBrien DCH. Cisplatin metabolites in plasma, a study of their pharmacokinetics and importance in the nephrotoxic and antitumour activity of cisplatin. Biochem. Pharmacol. 1984; 33: 3063–3070. 10.1016/0006-2952(84)90610-5 [DOI] [PubMed] [Google Scholar]
- 22. Jones MM, Basinger MA, Beaty JA, Holscher MA. The relative nephrotoxicity of cisplatin, cis-[Pt(NH3)2(guanosine)2]2+, and the hydrolysis product of cisplatin in the rat. Cancer Chemother. Pharmacol. 1991; 29: 29–32. 10.1007/BF00686332 [DOI] [PubMed] [Google Scholar]
- 23. Madias NE, Harrington JT. Platinum nephrotoxicity. Am. J. Med. 1978; 65: 307–314. 10.1016/0002-9343(78)90825-2 [DOI] [PubMed] [Google Scholar]
- 24. Litterst CL, LeRoy AF, Guarino AM. Disposition and distribution of platinum following parenteral administration of cis-dichlorodiammineplatinum(II) to animals. Cancer Treat. Rep. 1979; 63: 1485–1492. [PubMed] [Google Scholar]
- 25. Litterst CL, Gram TE, Dedrick RL, Leroy AF, Guarino AM. Distribution and disposition of platinum following intravenous administration of cis-diamminedichloroplatinum(II) (NSC 119875) to dogs. Cancer Res. 1976; 36: 2340–2344. [PubMed] [Google Scholar]
- 26. Stewart DH, Mikhael NZ, Nanji AA, Noir RC, Kacew S, Howard K, et al. Renal and hepatic concentrations of platinum: relationship to cisplatin time, dose, and nephrotoxicity. J. Clin. Oncol. 1985; 3: 1251–1256. [DOI] [PubMed] [Google Scholar]
- 27. Mistry P, Lee C, McBrien DC. Intracellular metabolites of cisplatin in rat kidney. Cancer Chernother. Pharmacol. 1989; 24: 73–79. 10.1007/BF00263124 [DOI] [PubMed] [Google Scholar]
- 28. Daley-Yates PT, McBrien DC. The renal fractional clearance of platinum antitumour compounds in relation to nephrotoxicity. Biochem Pharmacol. 1985; 34: 1423–1428. 10.1016/0006-2952(85)90679-3 [DOI] [PubMed] [Google Scholar]
- 29. Daley-Yates PT, McBrien DC. Cisplatin metabolites: a method for their separation and for measurement of their renal clearance in vivo. Biochem Pharmacol. 1983; 32: 181–184. 10.1016/0006-2952(83)90677-9 [DOI] [PubMed] [Google Scholar]
- 30. Reece PA, Stafford I, Davy M, Freeman S. Disposition of unchanged cisplatin in patients with ovarian cancer. Clin. Pharmacol. Ther. 1987; 42: 320–325. [DOI] [PubMed] [Google Scholar]
- 31. Cole WC, Wolf W. Renal toxicity studies of protein bound platinum (Cis). Chem. biol. Interact. 1981; 35: 341–348. 10.1016/0009-2797(81)90009-0 [DOI] [PubMed] [Google Scholar]
- 32. Yao X, Panichpisal K, Kurtzman N, Nuqent K. Cisplatin nephrotoxicity: a review. Am. J. Med. Sci. 2007; 334: 115–124. 10.1097/MAJ.0b013e31812dfe1e [DOI] [PubMed] [Google Scholar]
- 33. Hamilton CR, Bliss JM, Horwich A. The late effects of cis-platinum on renal function. Eur. J. Cancer Clin. Oncol. 1989; 25: 185–189. 10.1016/0277-5379(89)90006-0 [DOI] [PubMed] [Google Scholar]
- 34. Litterst CL. Alterations in toxicity of cis-dichlorodiammineplatinum-II and in tissue localization of platinum as a function of NaCl concentration in the vehicle of administration. Toxicol. Appl. Pharmacol. 1981; 61: 99–108. 10.1016/0041-008X(81)90011-9 [DOI] [PubMed] [Google Scholar]
- 35. Bannister SJ, Sternson LA, Repta AJ. Evaluation of reductive amperometric detection in the liquid chromatographic determination of antineoplastic platinum complexes. J. Chromatogr. 1983; 273: 301–318. 10.1016/S0378-4347(00)80951-2 [DOI] [PubMed] [Google Scholar]
- 36. Safirstein R, Daye M, Guttenplan JB. Mutagenic activity and identification of excreted platinum in human and rat urine and rat plasma after administration of cisplatin. Cancer Lett. 1983; 18: 329–338. 10.1016/0304-3835(83)90244-6 [DOI] [PubMed] [Google Scholar]
- 37. Mauidin SK, Fichard FA, Plescia M, Wynck SD, Saucer A, Chaney SG. High-performance liquid chromatographic separation of platinum complexes containing the cis-1,2-diaminocyclohexane carrier ligand. Anal. Biochem. 1986; 157: 129–143. 10.1016/0003-2697(86)90206-X [DOI] [PubMed] [Google Scholar]
- 38. Kizu R, Hayakawa K, Miyazaki M. HPLC determination of cis-diamminedichloroplatinum(II) in plasma and urine with UV detection and column-switching. Biomed. Chromatogr. 1989; 3: 14–19. 10.1002/bmc.1130030105 [DOI] [PubMed] [Google Scholar]
- 39. Kinoshita M, Yoshimura N, Ogata H, Tsujino D, Takahashi T, Takahashi S, et al. High-performance liquid chromatographic analysis of unchanged cis-diamminedichloroplatinum (cisplatin) in plasma and urine with post-column derivatization. J. Chromatogr. 1990; 529: 462–467. 10.1016/S0378-4347(00)83855-4 [DOI] [PubMed] [Google Scholar]
- 40. Zhao Z, Tepperman K, Dorsey JG, Elder RC. Determination of cisplatin and some possible metabolites by ion-pairing chromatography with inductively coupled plasma mass spectrometric detection. J. Chromatogr. 1993; 615: 83–89. 10.1016/0378-4347(93)80293-D [DOI] [PubMed] [Google Scholar]
- 41. Andersson A, Ehrsson H. Determination of cisplatin and cis-diammineaquachloroplatinum(II) ion by liquid chromatography using post-column derivatization with diethyldithiocarbamate. J. Chromatogr. B. 1994; 652: 203–210. 10.1016/0378-4347(93)E0403-D [DOI] [PubMed] [Google Scholar]
- 42. Baumann RA, Gooijer C, Velthorst NH, Frei RW, Klein I, Van der Vijgh WJF. Quantitative determination of cisplatin in body fluids by liquid chromatography with quenched phosphorescence detection. J. Pharm. Biomed. Anal. 1987; 5: 165–170. 10.1016/0731-7085(87)80019-5 [DOI] [PubMed] [Google Scholar]
- 43. Maessen FJMJ, Seeverens PJH, Kreuning G. Analytical aspects of organic solvent load reduction in normal-power ICPs by aerosol thermostatting at low temperatures Spectrochim. Acta, Part B. 1984; 39: 1171–1180. 10.1016/0584-8547(84)80203-7 [DOI] [Google Scholar]
- 44. Marsh K, Sternson LA, Repta AJ. Postcolumn reaction detector for platinum (II) antineoplastic agents. Anal. Chem. 1984; 56: 491–497. 10.1021/ac00267a045 [DOI] [PubMed] [Google Scholar]
- 45. Farrish HH, Hsyu PH, Pritchard JF, Brouwer KR, Jarrett J. Validation of a liquid chromatography post-column derivatization assay for the determination of cisplatin in plasma. J. Pharm. Biomed. Anal. 1994; 12: 265–271. 10.1016/0731-7085(94)90038-8 [DOI] [PubMed] [Google Scholar]
- 46. EI-Khateeb M, Appleton TG, Charles BG, Gahan LR. Development of HPLC conditions for valid determination of hydrolysis products of cisplatin. J. Pharm. Sci. 88: 1999; 319–326. 10.1021/js980287m [DOI] [PubMed] [Google Scholar]
- 47. Ehrsson HC, Wallin LB, Andemson AS, Edlund PO. Cisplatin, transplatin, and their hydrated complexes: Separation and identification using porous graphitic carbon and electrospray ionization mass spectrometry. Anal. Chem. 1995; 67: 3608–3611. 10.1021/ac00115a035 [DOI] [Google Scholar]
- 48. Du Y, Zhang N, Cui M, Liu Z, Liu S. Investigation on the hydrolysis of the anticancer drug cisplatin by Fourier transform ion cyclotron resonance mass spectrometry. Rapid Commun Mass Spectrom. 2012; 26: 2832–6. 10.1002/rcm.6408 [DOI] [PubMed] [Google Scholar]
- 49. Rosenberg B. Platinum complex-DNA interactions and anticancer activity. Biochimie. 1978; 60: 859–867. 10.1016/S0300-9084(78)80570-7 [DOI] [Google Scholar]
- 50. Lim MC, Martin RB, The nature of cis amine Pd(II) and antitumor cis amine Pt(II) complexes in aqueous solutions. J. Inorg, Nucl. Chem. 1976; 38: 1911–1914. 10.1016/0022-1902(76)80121-2 [DOI] [Google Scholar]
- 51. Cleare MJ, Hydes PC, Hepburn DR. Malerbi BW. In cisplatin, current Status and new developments (Eds. Prestayko A. W., Crooke S. T. and Carter S.K.); Academic Press; New York: 1980. p 149. [Google Scholar]
- 52. Zunno F, Pratesi G, Micheloni A, Cavalletti E, Sala F, Tofanetti O. Protective effect of reduced glutathione against cisplatin-induced renal and systemic toxicity and its influence on the therapeutic activity of the antitumor drug. Chem. Biol. Interact. 1989; 70: 89–101. 10.1016/0009-2797(89)90065-3 [DOI] [PubMed] [Google Scholar]
- 53. Jones MM, Basinger MA. Thiol and thioether suppression of cis-platinum-induced nephrotoxicity in rats bearing the Walker 256 carcinosarcoma. Anticancer Res. 1989; 9: 1937–1941. [PubMed] [Google Scholar]
- 54. Melvik JE, Pettersen E. Reduction of cis-Dichlorodiammineplatinum-induced cell inactivation by methionine. Inorg. Chim. Acta, 1987; 137: 115–118. 10.1016/S0020-1693(00)87128-5 [DOI] [Google Scholar]
- 55. Miller SE, House DA. The hydrolysis products of cisdichlorodiammineplatinum (II). hydrolysis kinetics at physiological pH. Inorg. Chim. Acta. 1990; 173: 53–60. 10.1016/S0020-1693(00)91054-5 [DOI] [Google Scholar]
- 56. Gullo JJ, Litterst CL, Maguire PJ, Sikic BI, Hoth DF, Woolley PV. Pharmacokinetics and protein binding of cis-dichlorodiammine platinum (II) administered as a one hour or as a twenty hour infusion. Cancer Chemother. Pharmacol. 1980; 5: 21–26. 10.1007/BF00578558 [DOI] [PubMed] [Google Scholar]
- 57. Dedon PC, Borch RF. Characterization of the reactions of platinum antitumor agents with biologic and nonbiologic sulfur-containing nucleophiles. Biochem. Pharmacol. 1987; 36: 1955–1964. 10.1016/0006-2952(87)90494-1 [DOI] [PubMed] [Google Scholar]
- 58. Townsend DM, Tew KD, He L, King JB, Hanigan MH. Role of glutathione S-transferase Pi in cisplatin-induced nephrotoxicity. Biomed. Pharmacother. 2009; 63:79–85. 10.1097/01.ASN.0000042803.28024.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sadzuka Y, Shimizu Y, Takino Y, Hirota S. Protection against cisplatin-induced nephrotoxicity in the rat by inducers and an inhibitor of glutathione S-transferase. Biochem. Pharmacol. 1994; 48: 453–459. 10.1016/0006-2952(94)90274-7 [DOI] [PubMed] [Google Scholar]
- 60. Townsend DM, Deng M, Zhang L, Lapus MG, Hanigan MH. Metabolism of cisplatin to a nephrotoxin in proximal tubule cells. J. Am. Soc. Nephrol. 2003; 14: 1–10. doi: 1046-6673/1401-0001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Townsend DM, Hanigan MH. Inhibition of gamma-glutamyl transpeptidase or cysteine S-conjugate beta-lyase activity blocks the nephrotoxicity of cisplatin in mice. J. Pharmacol. Exp. Ther. 2002; 300: 142–148. doi: 0022-3565/02/3001-142-148$3.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang L, Hanigan MH. Role of cysteine S-conjugate β-lyase in the metabolism of cisplatin. J. Pharmacol. Exp. Ther. 2003; 306: 988–994. 10.1124/jpet.103.052225 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.