

Abstract
MicroRNAs (miRNAs) regulate many physiological processes through post-transcriptional control of gene expression and are a major part of the small noncoding RNAs (snRNA). As hibernators can survive at low body temperatures (Tb) for many months without suffering tissue damage, understanding the mechanisms that enable them to do so are of medical interest. Because the brain integrates peripheral physiology and white adipose tissue (WAT) is the primary energy source during hibernation, we hypothesized that both of these organs play a crucial role in hibernation, and thus, their activity would be relatively increased during hibernation. We carried out the first genomic analysis of small RNAs, specifically miRNAs, in the brain and WAT of a hibernating bat (Myotis ricketti) by comparing deeply torpid with euthermic individual bats using high-throughput sequencing (Solexa) and qPCR validation of expression levels. A total of 196 miRNAs (including 77 novel bat-specific miRNAs) were identified, and of these, 49 miRNAs showed significant differences in expression during hibernation, including 33 in the brain and 25 in WAT (P≤0.01 &│logFC│≥1). Stem-loop qPCR confirmed the miRNA expression patterns identified by Solexa sequencing. Moreover, 31 miRNAs showed tissue- or state-specific expression, and six miRNAs with counts >100 were specifically expressed in the brain. Putative target gene prediction combined with KEGG pathway and GO annotation showed that many essential processes of both organs are significantly correlated with differentially expressed miRNAs during bat hibernation. This is especially evident with down-regulated miRNAs, indicating that many physiological pathways are altered during hibernation. Thus, our novel findings of miRNAs and Interspersed Elements in a hibernating bat suggest that brain and WAT are active with respect to the miRNA expression activity during hibernation.
Introduction
Hibernation (multiday torpor) and daily torpor in heterothermic mammals are characterized by substantial, temporal reductions of energy expenditure and body temperature (Tb). Torpor is often expressed when animals are exposed to low ambient temperature (Ta), and the decrease in energy use is crucial for survival during adverse conditions and food shortages [1]. Mammalian hibernation is an extreme example of hypometabolism with metabolic rate depression often to <5% of the euthermic rate, a minimum Tb of approximately 0°C, and cell preservation during prolonged dormancy periods [2, 3]. The physiological changes, such as the decrease and subsequent increase of metabolism and Tb during entry and arousal from torpor, are active and adaptive processes. They are often initiated in response to changes in environmental temperature and photoperiod and require the coordination of core and peripheral tissues by the brain [4, 5]. Importantly, the carbohydrate oxidation pathway shifts upon hormone activation towards lipid metabolism, and fat (mainly stored in white adipose tissue, WAT) becomes the primary energy source during the hibernation season [6]. Considering the brain’s important role in integrating peripheral tissues and WAT to provide energy during hibernation, we hypothesized that to adequately perform these tasks, both must be activated during hibernation, in contrast to other tissues and organs, where metabolism is generally suppressed [7].
A detailed understanding of the molecular mechanisms of this phenomenon would improve our understanding of hibernation at the subcellular level, but may also be useful in human medicine for improving organ transplantation, preventing skeletal muscle atrophy during long-term bed rest, and suppressing carcinogenesis [7]. In the past ten years, molecules that regulate hibernation and the molecular mechanisms underlying hibernation have been identified by transcriptome analysis [8–12]. Thousands of genes and proteins are differentially expressed in many species during hibernation [12–17]. However, the precise and timely control of gene expression during hibernation remains unclear.
Previous studies indicated that post-transcriptional regulation occurs during hibernation [18]. MicroRNAs (miRNAs) are short RNA molecules (18–25nt), and they are known to play key roles in directing post-transcriptional activity of genes involved in all cellular processes, including differentiation and development [19]. Since Morin and colleagues [20] first proved that miRNAs regulate ground squirrel [Ictidomys (Spermophilus) tridecemlineatus] hibernation, many detailed studies have analyzed miRNAs during torpor [18, 21–25] and found that miRNAs are involved in cold adaptation, epigenetic control and metabolic rate depression [26–28]. Differential miRNA expression, and thus their target genes, is an important component of the molecular adaptation required by organisms to respond to environmental changes, especially heterothermic mammals [18, 26–28].
There are more than 1100 species of bats (order Chiroptera), and they are among the most geographically widespread mammals [29]. Most bat families include species that enter some form of torpor, whereas multiday torpor (i.e., hibernation) occurs in at least five families [30]. The wide taxonomic diversity of heterothermy in bats suggests that bats are a good model to explore the evolutionary history of mammalian heterothermy [31]. Importantly, unlike ground squirrels, for which most molecular work on hibernation has been conducted, bats are not strongly seasonal in their torpor expression and many use multiday torpor throughout the year [32]. Therefore, a systematic study of genes or proteins involved in bat hibernation by comparing active and hibernating individuals will help us better understand the molecular mechanisms of hibernation and to explore the evolution of mammalian heterothermy [11, 31, 33, 34]. However, a systematic analysis of small RNAs, especially miRNAs, with regard to function and evolution of torpor in bats is currently lacking.
The latest 7× coverage genome sequence of Myotis lucifugus (http://www.ensembl.org/Myotis_lucifugus/Info/Index) provides us with the opportunity to survey miRNA expression in hibernating bats and to conduct comparative analyses with sequences from other metazoan species. Moreover, the RNA-seq sequencing-by-synthesis technique allows to sequence millions of short cDNAs and increases sensitivity by reducing personal error in library construction [35]. Based on the Illumina/Solexa sequencing platform and combined with qPCR validation, we performed the first in-depth miRNA analysis on two key tissues (brain and WAT) of the hibernating bat Myotis ricketti, which is congeneric with M. lucifugus. Our study aimed to discover bat-specific miRNAs, illustrate their potential role during bat hibernation, and contribute to the understanding of the evolution of mammalian heterothermy.
Methods
Ethics statement
All procedures involving animals were performed by strictly following the Guidelines and Regulations for the Administration of Laboratory Animals (Decree No. 2 of the State Science and Technology Commission of the People’s Republic of China on November 14, 1988) and were approved by the Animal Ethics Committee of East China Normal University (20080729).
Sample and RNA preparation
A total of six Myotis ricketti individuals with a body mass of 18–22 g were used in this study. Three active M. ricketti bats (Tb~36°C) were captured using mist nets in the Fangshan area (Beijing, China; 39°48′N, 115°42′E) on October 26, 2008 (Ta = 21°C). On March 6, 2009, three bats in deep hibernation (Tb~9°C) were captured in the same location (Ta = 7°C). The field studies did not involve endangered or protected species, and no specific permissions were required for M. ricketti in the Fangshan area. The bats were sacrificed humanely by decapitation immediately after measuring Tb and body mass. Whole brain and white intra-abdominal adipose tissue (WAT) of hibernating and euthermic bats were rapidly removed, immediately frozen in liquid nitrogen, and stored at -80°C until RNA extraction.
Total RNA was isolated from entire brains and adipose tissues (~0.2 g) of six bats using the RNAiso kit (TakaRa, Japan) according to the manufacturer’s instructions, and the concentration and RNA Integrity Number (RIN) was determined by an Agilent 2100 bioanalyzer. RNA samples were pooled to construct the transcriptome sequencing library as described previously [12, 21]. Briefly, total RNA from bat brains of the same stage were mixed in equal amounts (10 μg each) into two pooled samples, representing the hibernation and active state, hereafter referred to as HB (hibernating brain) and AB (active brain). Similarly, total WAT RNA from hibernating and active bats (10 μg each) were pooled and referred to as HA (hibernating WAT), or AA (active WAT). In total, four RNA pools (two tissues in two physiological states) were used to construct small RNA libraries. The RNA quality and quantity were as follows: HB: 480 ng/μl, RIN = 8.3, 28S/18S = 1.2; AB: 2210 ng/μl, RIN = 8.1, 28S/18S = 1.5; HA: 390 ng/μl, RIN = 8.4, 28S/18S = 1.4; AA: 1440 ng/μl, RIN = 7.5, 28S/18S = 1.5.
Construction and high-throughput sequencing of small RNA libraries
Small RNA cDNA libraries were constructed as described previously [36]. Briefly, small RNA (18~33 nt) was purified and enriched from each total RNA sample by polyacrylamide gel electrophoresis (PAGE), and 10 μg small RNA was ligated with the proprietary adapters used for cDNA synthesis and library construction, then Solexa sequencing-by-synthesis was performed (Illumina). All small RNA data series were submitted to NCBI Gene Expression Omnibus (GEO) with the accession number GSE29053.
Filter of small RNA reads
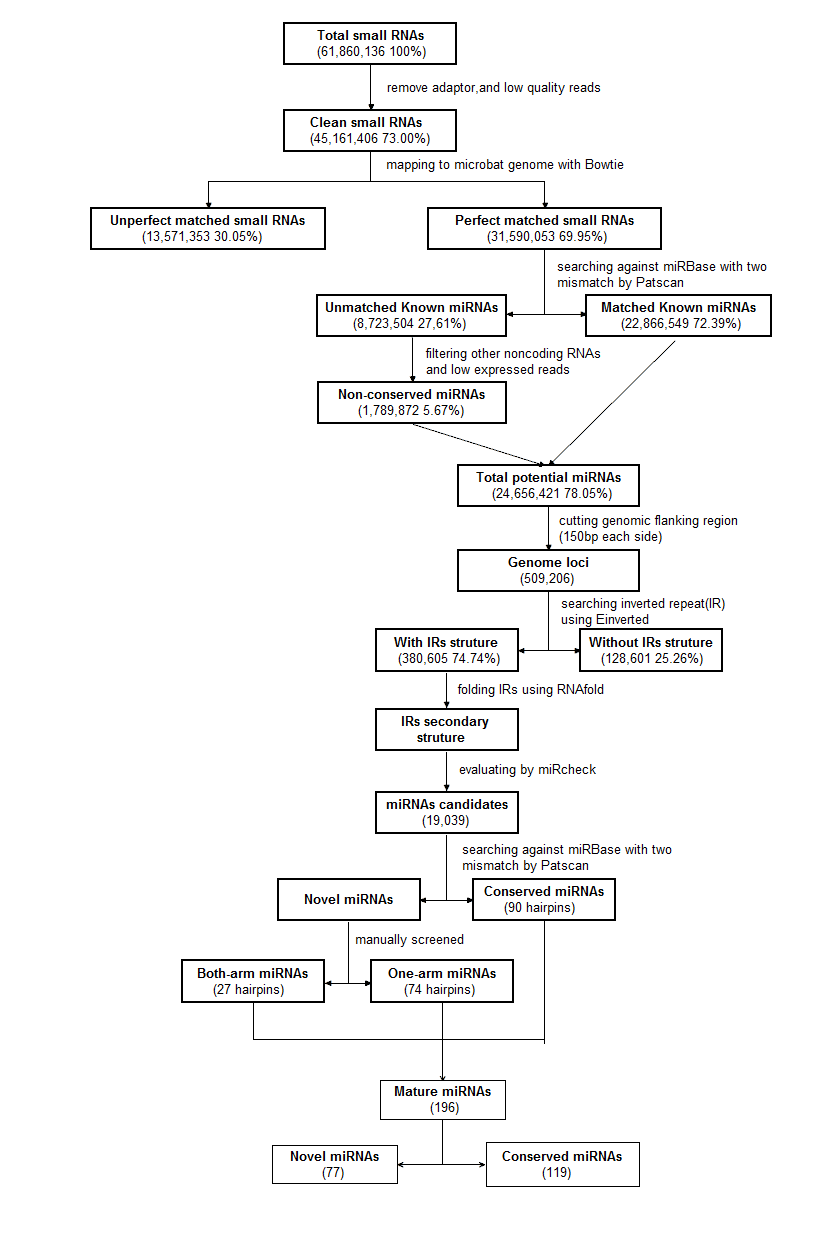
Individual sequence reads with base quality scores were produced by Illumina/Solexa. After removing contaminant reads (adapters, low quality and redundant reads), clean unique reads were mapped onto the Myotis lucifugus genome (http://asia.ensembl.org/Myotis_lucifugus/Info/Index) using the Bowtie program [37]. Perfectly mapped reads were scanned against the metazoan mature miRNA in the Sanger miRBase (Release 19) [38] to identify orthologs of known miRNAs using the Patscan program [39] with two mismatches allowed. Non-conserved unique reads were screened against Rfam databases (Release 10) [40] to filter the sequences of tRNA, rRNA, snoRNA, and other ncRNAs except miRNAs using Bowtie. Reads that matched to the genome more than 20 times were removed by miRDeep [41], and reads sequenced only one time were also removed. Finally, the remaining reads were considered potential miRNA reads and were used for miRNA identification.
MiRNA identification
MiRNA and its antisense strand can both be sequenced from the same precursor. Thus, to avoid repeated predictions and to reduce calculations, candidate reads whose distance in the reference genome were <200 nt were combined and examined as one genomic block. For each block, 150 nt of upstream and downstream extensions were extracted for secondary structure prediction. First, inverted repeats (IR) with stem-loop or hairpin structures were identified by Einverted of Emboss [42], with the following parameters: threshold = 30, match score = 3, mismatch score = 3, gap penalty = 6, and maximum repeat length = 240, as described previously [43]. The IR secondary structure was then predicted by RNAfold [44] with 10 nt upstream and downstream extensions and evaluated by MirCheck [43].
Predicted precursors that miRNA and antisense strand can be found in both arms were deemed miRNA candidates. Moreover, if the candidates have two or more unique reads resulted from the ±2 nt bias during cleavage and located at mature positions, they were deemed highly probable. When several length variants of the same miRNA were sequenced, only variants with the highest representation were considered. Finally, the miRNA candidates were submitted again to miRBase, and the miRNA precursors (hairpins) that passed MirCheck were manually inspected against the canonical miRNA structure to remove false predictions.
Repeat-derived siRNA detection
To screen repeat-derived small interfering RNA (siRNAs), the repeat sequences of the M. lucifugus genome were annotated by Repeatmasker (http://www.repeatmasker.org/) [45]. Reads that perfectly matched the M. lucifugus genome were aligned to repetitive elements using Bowtie, and reads that perfectly matched the repeats were considered genomic repeats-derived siRNAs.
Differential miRNA expression analysis and target prediction
Differentially expressed miRNAs that were statistically significant in relative abundance (reflected by Transcript Per Million, TPM) between hibernating and active states were identified by the edgeR function in Bioconductor [46]. Empirical Bayes estimation and exact tests based on the negative binomial distribution were used, and P≤0.01 & │logFC│≥1 considered statistically significant.
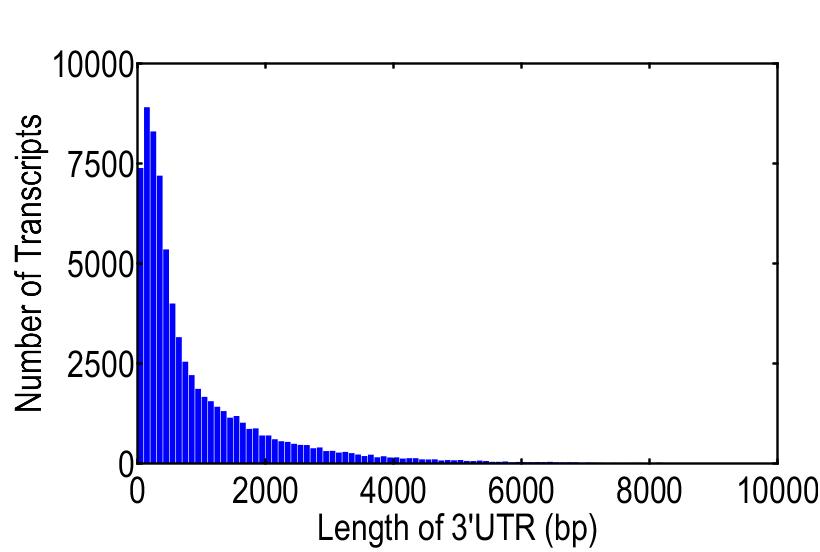
As there is no or little information about 3’ untranslated region (UTR) of M. lucifugus reference genes in database of Ensembl (http://www.ensembl.org/Myotis_lucifugus/Info/Index), to predict miRNA target genes, we first identified the orthologs of M. lucifugus genes by searching against Homo sapiens mRNA (http://www.ncbi.nlm.nih.gov/RefSeq/) with tblastx (e value ≤1e-10 and identity ≥60%) [47]. Orthologs with the best hits were kept. The H. sapiens mRNAs were used to analyze the 3’ UTR lengths, and 93.6% (67,594/72,204) of human mRNAs had a 3’ UTR less than 3 Kb (see S1 Fig), excluding 32,559 mRNAs that did not have a 3’ UTR. Thus, the 3 Kb region downstream of M. lucifugus reference gene coding sequences were extracted and aligned with the 3’UTR of H. sapiens mRNA orthologs by CLUSTALW [48] to trim the candidate sequences of 3’ UTR of M. lucifugus. For M. lucifugus reference genes that did not have a human ortholog, the 3 Kb region downstream of the coding sequence was considered the 3’UTR. The miRNA target was then predicted using the PITA program based on the interaction between miRNAs and their targets with the default criterion and ΔΔG≤−10 kcal/mol [49]. Finally, target sequence Gene Ontology (GO) annotation was performed by Interproscan [50] and target sequences were compared to the Kyoto Encyclopedia of Genes and Genomes database (KEGG, release 50) by BLASTX at E values ≤1e-10 [47, 51]. A Perl script was used to retrieve the KO (KEGG Orthology) information from BLAST results and to establish pathway associations between target genes and databases.
Quantitative miRNA expression by qPCR assay
Stem-loop reverse transcription (RT) qPCR was performed to quantify the expression level of 10 differentially expressed miRNAs obtained by Solexa sequencing in brains and WAT as described previously [52], and 5S rRNA was used as an endogenous control. Primers used in this study are listed in S1 Table. Each RT reaction contained 1 μg total RNA, 1× Reaction Buffer, dNTPs (5 mM each), 50 nM miRNA-specific stem-loop RT primer, 100 U RevertAid Premium Reverse Transcriptase and 0.5 U RiboLock RNase Inhibitor. RT reactions were incubated at 37°C for 30 min, 42°C for 60 min and 85°C for 5 min. PCR reactions were performed in a total of 20 μl, including 0.8 μl RT product, 10 μl 2× SYBR Green I Master Mix, 6 μM each of forward primer and reverse primer with the following program: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, 60°C for 30 s and 72°C for 15 s, then 95°C for 15 s, 60°C for 1 min and a final hold at 4°C. All reactions were performed on three biological replicates in each tissue and each physiological state, and each was run three times. Negative controls containing all reagents except template were included on each reaction plate. The 2−ΔΔCT method was used to calculate relative expression (fold change); and data were analyzed by One-Way ANOVA using SPSS 18.0 software.
Results
Small RNA sequencing and statistics
We isolated total RNA from hibernating and active Myotis ricketti brain and intra-abdominal adipose tissue, and the RNA concentration and RNA Integrity Number (RIN) met the requirements for Solexa small RNA library construction and sequencing.
After small RNAs were isolated and processed for deep sequencing on the Illumina/Solexa platform, we sequenced a total of 1.56×107, 1.54×107, 1.53×107 and 1.55×107 reads from the HB, AB, HA and AA libraries, respectively. We obtained a total of 4.51×107 high-quality small RNA reads after removing ambiguous reads. In these high quality reads, 6.3×105 (HB), 4.2×105 (AB), 1.2×106 (HA), 5.4×105 (AA) and 2.3×106 (in total) were clean unique reads, and the percentage of small RNA reads (>18 bp) in four libraries were above 60% (see S2 and S3 Tables). The redundancies of the four libraries were 94.82% (HB), 95.85% (AB), 89.92% (HA) and 95.31% (AA), and among these, the redundancy of co-expressed unique reads among libraries was 11.3% and that of state/tissue-specific reads was 88.7% (S3 Table).
Less than 5% of total small RNA reads from the bat libraries perfectly matched the Myotis lucifugus genome >20 times (S4 Table); thus, we removed these unique reads to avoid self-contradiction of miRNA prediction. Therefore, a total of 3.16×107 remained after mapping the sequences to the M. lucifugus genome (Table 1 and S2 Fig). The size distribution of perfectly matched small RNA reads is shown in Fig 1A. We identified a total of 2.47×107 potential miRNA reads, including 159,807 distinct reads, by searching against miRBase and filtering the ncRNAs and low-expressing reads (S5 Table). Subsequently, 1.51×107 (47.7% in total) miRNA reads met the fold-back structure (hairpin) and MirCheck criteria, and miRNA reads were the most abundant fraction (range from 32.7% in HA to 55.6% in AB, Table 1 and Fig 1B). Other small RNAs, such as ncRNAs, genomic repeats, transcript repeats, and unknown genomic regions, comprised 20.4%, 2.3%, 2.8% and 26.8%, respectively (Table 1 and Fig 1B). Upon further inspection of genomic repeat-derived siRNAs, we determined that Short INterspersed Element (SINE) and Long INterspersed Element (LINE) were the major contributors to the four libraries (Table 1 and Fig 2).
Table 1. Classification of unique small RNA reads.
| Classification | Total | HB | AB | HA | AA | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of unique reads | Percentage (%) | Total sequences | Percentage (%) | Number of unique reads | Percentage (%) | Total sequences | Percentage (%) | Number of unique reads | Percentage (%) | Total sequences | Percentage (%) | Number of unique reads | Percentage (%) | Total sequences | Percentage (%) | Number of unique reads | Percentage (%) | Total sequences | Percentage (%) | |
| Total Unique small RNA Reads | 2323170 | 100 | 45161406 | 100 | 629751 | 100 | 12160105 | 100 | 415452 | 100 | 10012888 | 100 | 1164049 | 100 | 11549959 | 100 | 536995 | 100 | 11438454 | 100 |
| Total Perfect Matched small RNA reads | 720862 | 31.0 | 31590053 | 69.9 | 201875 | 32.1 | 8682951 | 71.4 | 120695 | 29.1 | 7266543 | 72.6 | 373224 | 32.1 | 7502137 | 65.0 | 152674 | 28.4 | 8138422 | 71.1 |
| miRNAs a | 7530 | 1.0 | 15068478 | 47.7 | 3365 | 1.7 | 4451386 | 51.3 | 2684 | 2.2 | 4041090 | 55.6 | 3967 | 1.1 | 2454320 | 32.7 | 3164 | 2.1 | 4121682 | 50.6 |
| Non-Coding RNA b | 71131 | 9.9 | 6443708 | 20.4 | 39956 | 19.8 | 1556064 | 17.9 | 27417 | 22.7 | 709223 | 9.8 | 47132 | 12.6 | 2798792 | 37.3 | 29197 | 19.1 | 1379629 | 17.0 |
| rRNA | 39273 | 5.4 | 5495818 | 17.4 | 25585 | 12.7 | 1311512 | 15.1 | 18091 | 15.0 | 539528 | 7.4 | 31298 | 8.4 | 2452251 | 32.7 | 20826 | 13.6 | 1192527 | 14.7 |
| tRNA | 18189 | 2.5 | 244828 | 0.8 | 7063 | 3.5 | 54006 | 0.6 | 3977 | 3.3 | 36283 | 0.5 | 7954 | 2.1 | 112091 | 1.5 | 3540 | 2.3 | 42448 | 0.5 |
| snoRNA | 2360 | 0.3 | 36480 | 0.1 | 1030 | 0.5 | 9531 | 0.1 | 660 | 0.5 | 5882 | 0.1 | 1280 | 0.3 | 13861 | 0.2 | 729 | 0.5 | 7206 | 0.1 |
| snRNA | 454 | 0.1 | 7249 | 0.0 | 214 | 0.1 | 1107 | 0.0 | 130 | 0.1 | 614 | 0.0 | 309 | 0.1 | 4669 | 0.1 | 107 | 0.1 | 859 | 0.0 |
| other ncRNA | 10855 | 1.5 | 659333 | 2.1 | 6064 | 3.0 | 179908 | 2.1 | 4559 | 3.8 | 126916 | 1.7 | 6291 | 1.7 | 215920 | 2.9 | 3995 | 2.6 | 136589 | 1.7 |
| Transcript-derived reads | 289993 | 40.2 | 873840 | 2.8 | 52157 | 25.8 | 173709 | 2.0 | 31598 | 26.2 | 105720 | 1.5 | 173270 | 46.4 | 445779 | 5.9 | 52878 | 34.6 | 148632 | 1.8 |
| Genomic repeat-derived reads | 67509 | 9.4 | 732080 | 2.3 | 24669 | 12.2 | 66751 | 0.8 | 11222 | 9.3 | 47251 | 0.7 | 25197 | 6.8 | 91641 | 1.2 | 10348 | 6.8 | 526437 | 6.5 |
| LINE | 33783 | 50.0 | 124881 | 17.1 | 12312 | 49.9 | 36423 | 54.6 | 5312 | 47.3 | 26188 | 55.4 | 13129 | 52.1 | 32850 | 35.8 | 4622 | 44.7 | 29420 | 5.6 |
| SINE | 5681 | 8.4 | 512264 | 70.0 | 1830 | 7.4 | 8885 | 13.3 | 941 | 8.4 | 10160 | 21.5 | 2178 | 8.6 | 26692 | 29.1 | 1016 | 9.8 | 466527 | 88.6 |
| LTR | 10571 | 15.7 | 35643 | 4.9 | 3637 | 14.7 | 10730 | 16.1 | 1797 | 16.0 | 5286 | 11.2 | 4007 | 15.9 | 13374 | 14.6 | 1751 | 16.9 | 6253 | 1.2 |
| DNA | 8644 | 12.8 | 19283 | 2.6 | 2881 | 11.7 | 5062 | 7.6 | 1674 | 14.9 | 2877 | 6.1 | 3398 | 13.5 | 8261 | 9.0 | 1444 | 14.0 | 3083 | 0.6 |
| SSR | 2293 | 3.4 | 3002 | 0.4 | 1012 | 4.1 | 1216 | 1.8 | 372 | 3.3 | 427 | 0.9 | 658 | 2.6 | 868 | 0.9 | 316 | 3.1 | 491 | 0.1 |
| Low | 2524 | 3.7 | 3194 | 0.4 | 1895 | 7.7 | 2287 | 3.4 | 178 | 1.6 | 196 | 0.4 | 338 | 1.3 | 483 | 0.5 | 173 | 1.7 | 228 | 0.0 |
| Unknown Repeats | 4013 | 5.9 | 33813 | 4.6 | 1102 | 4.5 | 2148 | 3.2 | 948 | 8.4 | 2117 | 4.5 | 1489 | 5.9 | 9113 | 9.9 | 1026 | 9.9 | 20435 | 3.9 |
| Unknown | 284699 | 39.5 | 8471947 | 26.8 | 81728 | 40.5 | 2435041 | 28.0 | 47774 | 39.6 | 2363259 | 32.5 | 123658 | 33.1 | 1711605 | 22.8 | 57087 | 37.4 | 1962042 | 24.1 |
HB: hibernating state brain; AB: active state brain; HA: hibernating state adipose tissue; AA: active state adipose tissue. rRNA, ribosomal RNA; tRNA, transfer RNA; snoRNA, small nucleolar RNA; snRNA, small nuclear RNA; LINE, Long INterspersed Elements; SINE, Short INterspersed Elements; LTR, Transposable elements with Long Terminal Repeats; DNA, DNA transposons; SSR, Simple Sequence Repeats; Low, Low Complexity Sequences; unknown, derived from unannotated/intergenic regions.
a miRNA reads identified by miCheck, searched against the Sanger miRBase (Release 19) and manually inspected for canonical structure from 24656421 total potential reads (S2 Fig and S5 Table).
b Screening against non-coding RNA databases (Release 10) to filter ncRNAs (tRNA, rRNA, snoRNA and other ncRNA) by Bowtie.
Fig 1. Overview of small RNA gene expression in four bat libraries generated by Solexa deep sequencing.

(A) Length distribution of perfectly matched small RNA reads. The percentage of total or distinct (unique) small RNA reads that perfectly matched the reference genome are shown. (B-F) Breakdown of the proportions (in percentage) of various classes of small RNAs detected by sequencing of total/all combined, brain (HB, AB), and WAT (HA, AA). Various classes of small RNAs are shown by percentages. The miRNA family comprises the majority of small RNAs (47.7% in total). snoRNA, small nucleolar RNA; rRNA, ribosomal RNA; tRNA, transfer RNA; unknown, derived from unannotated/intergenic regions.
Fig 2. Illustration of genomic repeat-derived reads in four libraries.

LINE, Long INterspersed Elements; SINE, Short INterspersed Elements; LTR, Transposable elements with Long Terminal Repeats; DNA, DNA transposons; SSR, Simple Sequence Repeats; Low, Low Complexity Sequences; unknown, derived from unannotated/intergenic regions.
MiRNA identification
To better understand the length distribution of metazoan miRNA precursors, we analyzed known miRNA precursors in five out-grouped organisms (two vertebrates and three invertebrates, S3 Fig) in miRBase. The majority of pre-mature miRNAs from these five organisms were less than 150 nt with a mean length of approximately 90 nt (S3 Fig). We mapped the potential miRNA reads onto the reference genome and retrieved 509,206 block sequences with 150 nt upstream and downstream extensions; we then used these sequences for secondary structure prediction. From these, we used Einverted of Emboss [42] and identified 380,605 potential reads with inverted repeats. We identified 90 conserved hairpins in 28 families and 101 novel hairpins (S6 Table).
Finally, we obtained a total of 196 mature miRNAs, including 119 conserved miRNAs and 77 novel bat-specific miRNAs, and sequence raw counts varied from one to 1,946,308 (S7 Table). Further analysis showed that 12.2% (23/188, brain) and 4.6% (8/173, WAT) of the miRNA was expressed in a tissue-specific manner, whereas 10.1% (19/188, brain), and 10.9% (19/173, WAT) exhibited state-specific expression (Fig 3 and S7 Table). In the 31 state/tissue-specific miRNAs, six (miR-1298-5p, miR-124-5p, miR-153-5p, miR-153-3p, miR-551-3p and miR-1298-3p) had counts above 100, and they were all specifically expressed in the brain (S7 Table).
Fig 3. MiRNA expression patterns in four bat libraries.

The expression of miRNAs identified in this study were calculated by sequence counts (Transcript Per Million, TPM). Heat maps represent the clustering of miRNAs (up) and the Venn diagram shows the number of miRNAs in each library (down). (A) 119 conserved miRNAs match to the Metazoan mature miRNAs in Sanger miRBase; (B) 77 Novel miRNAs identified by manual screening. Details of differentially expressed miRNAs are shown in S7 Table. HB: hibernating state brain; AB: active state brain; HA: hibernating state adipose tissue; AA: active state adipose tissue.
Differential miRNA expression analysis and validation
A total 49 out of 196 miRNAs were differentially expressed (P≤0.01 & │logFC│≥1) between the hibernation vs. the active state, including 33 miRNAs in brain, 25 miRNAs in WAT and 9 miRNAs in both tissues (Table 2 and S7 Table). Of 33 miRNAs previously shown to be differentially expressed during torpor (S8 Table), ten (mir-1, mir-24, miR-29, miR-124a, mir-142, mir-181a, mir-181b mir-206, mir-378 and mir-486) were detected in our study (S7 Table). Moreover, we found that mir-378 was differentially expressed, and mir-142, mir-181b and mir-486 were also somewhat differentially expressed during hibernation (S7 Table).
Table 2. 49 differentially expressed miRNAs identified by Solexa sequencing in the brain or adipose tissues during hibernation.
| HA vs. AA | HB vs. AB | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Name | Total counts | logFC | logTPM | PValue | Mark | logFC | logTPM | PValue | Mark |
| miR-124b-3p | 101576 | 2.259908382 | 5.6190503 | 0.0018537 | UP | -0.536674 | 14.312553 | 0.4127694 | DOWN |
| miR-139-3p | 3183 | 0.37423932 | 8.5019562 | 0.5727299 | UP | -2.1306716 | 8.4057206 | 0.0019162 | DOWN |
| miR-200a-3p | 334 | 7.578561186 | 3.2895048 | 2.45E-06 | UP | -0.886215 | 5.923925 | 0.1986791 | DOWN |
| miR-200b-3p | 420 | 4.364567511 | 6.7026081 | 3.08E-08 | UP | -1.8932045 | 4.6037305 | 0.0113473 | DOWN |
| miR-222-3p | 12491 | 1.429407306 | 10.720804 | 0.0321579 | UP | -1.8021156 | 10.33994 | 0.0075425 | DOWN |
| miR-222-5p | 290 | 3.030812254 | 5.7697748 | 5.80E-05 | UP | 2.1583491 | 4.6655436 | 0.0044099 | UP |
| miR-3135-5p | 6682 | -2.637365825 | 9.938779 | 0.0001553 | DOWN | -0.1976903 | 8.1896251 | 0.7652108 | DOWN |
| miR-363-3p | 590 | 2.133071814 | 5.9211148 | 0.0028745 | UP | -0.0815074 | 6.3009552 | 0.9118204 | DOWN |
| miR-378-3p | 198036 | -2.818466705 | 15.121184 | 5.51E-05 | DOWN | -1.5232279 | 10.232562 | 0.0228152 | DOWN |
| miR-448-3p | 8783 | -2.143218357 | 7.3054357 | 0.0022535 | DOWN | 0.0288125 | 10.656037 | 0.9653039 | UP |
| miR-455-3p | 1010 | -1.921311717 | 6.8853723 | 0.0061558 | DOWN | 0.1968478 | 6.3795769 | 0.7761823 | UP |
| miR-486-3p | 809 | -0.156052775 | 6.9078207 | 0.825439 | DOWN | -1.9680416 | 5.8518332 | 0.0056228 | DOWN |
| miR-574a-5p | 400 | -2.521905714 | 6.1292436 | 0.0006325 | DOWN | -2.7422469 | 2.7923067 | 0.0039648 | DOWN |
| miR-574b-5p | 153 | -4.250113976 | 4.6476538 | 1.32E-05 | DOWN | -3.8191272 | 1.987474 | 0.0030428 | DOWN |
| miR-590-3p | 136 | 0.378106825 | 4.4405947 | 0.6392554 | UP | 2.6462719 | 3.3179244 | 0.0035346 | UP |
| miR-7c-5p | 32436 | 2.712605457 | 8.7286431 | 0.0001116 | UP | 1.1902664 | 12.649161 | 0.0723922 | UP |
| miR-7f-5p | 227183 | 0.390233519 | 13.83588 | 0.5510089 | UP | -1.8441761 | 15.019439 | 0.0062266 | DOWN |
| miR-92b-3p | 9041 | 2.884880248 | 5.7195942 | 0.0001081 | UP | -0.1793825 | 10.807021 | 0.7843292 | DOWN |
| Novel-10-3p | 111 | 3.016403333 | 12.848138 | 0.0001838 | UP | -4.4272759 | 12.275541 | 4.45E-06 | DOWN |
| Novel-14-3p | 97 | 0.936226836 | 12.677215 | 0.2187093 | UP | -4.3606505 | 11.683673 | 3.70E-05 | DOWN |
| Novel-15-3p | 1669 | -2.944335536 | 11.581472 | 0.0018222 | DOWN | 7.2036728 | 15.19745 | 3.14E-14 | UP |
| Novel-16-3p | 87 | -2.344769395 | 12.036684 | 0.0062019 | DOWN | -1.2012847 | 9.4737976 | 0.3901332 | DOWN |
| Novel-17-3p | 81 | -2.304954477 | 12.263073 | 0.0052596 | DOWN | -0.5052158 | 9.9593452 | 0.7250474 | DOWN |
| Novel-18a-3p | 1471 | -0.426177519 | 15.116184 | 0.5299252 | DOWN | 2.3688352 | 16.00558 | 0.0006634 | UP |
| Novel-18b-3p | 344 | -1.02311079 | 13.052043 | 0.1656782 | DOWN | 2.5486332 | 13.71619 | 0.0004685 | UP |
| Novel-1b-3p | 121 | -2.293820099 | 12.911157 | 0.0026867 | DOWN | -1.1555324 | 10.404784 | 0.2531532 | DOWN |
| Novel-21-3p | 1424 | 2.451014248 | 15.801168 | 0.0004914 | UP | -0.4894437 | 15.830676 | 0.4628932 | DOWN |
| Novel-22-3p | 71 | 0 | 0 | 1 | _ | 7.397751 | 11.114078 | 2.45E-06 | UP |
| Novel-25a-5p | 1088 | 1.581470973 | 15.986822 | 0.0200239 | UP | -3.2647271 | 15.214248 | 8.67E-06 | DOWN |
| Novel-27-3p | 52 | 0 | 0 | 1 | _ | 3.8813759 | 10.646132 | 0.0002438 | UP |
| Novel-28-5p | 50 | 2.607474245 | 11.636459 | 0.0034908 | UP | -3.7091105 | 11.077865 | 0.0008149 | DOWN |
| Novel-29-5p | 50 | 1.097898289 | 11.646378 | 0.1811178 | UP | -4.2594804 | 10.735381 | 0.0002438 | DOWN |
| Novel-2a-3p | 157 | 0.806179078 | 13.221511 | 0.2539667 | UP | -1.9817143 | 12.411679 | 0.0100787 | DOWN |
| Novel-2a-1-5p | 38 | 3.353470354 | 10.81473 | 0.002812 | UP | -0.7705951 | 10.858367 | 0.4453967 | DOWN |
| Novel-31-3p | 910 | -2.058174551 | 12.059167 | 0.0145875 | DOWN | 5.722207 | 14.947979 | 2.83E-11 | UP |
| Novel-32-3p | 649 | -2.308464878 | 13.31712 | 0.0020344 | DOWN | 3.1963163 | 13.574877 | 3.13E-05 | UP |
| Novel-34-3p | 329 | -0.707605215 | 12.444825 | 0.3636482 | DOWN | 3.301607 | 13.981698 | 1.33E-05 | UP |
| Novel-36-3p | 309 | 1.484842174 | 11.704508 | 0.070754 | UP | 2.9220594 | 13.854956 | 8.30E-05 | UP |
| Novel-37-3p | 3841 | -2.056735288 | 13.166302 | 0.0056955 | DOWN | 6.1069365 | 16.429287 | 4.25E-13 | UP |
| Novel-38-5p | 238 | -0.844950584 | 13.898719 | 0.2282207 | DOWN | -2.8099732 | 12.134971 | 0.0009641 | DOWN |
| Novel-3b-3p | 153 | -0.732186096 | 11.093626 | 0.4453967 | DOWN | 2.9094233 | 12.254351 | 0.0004231 | UP |
| Novel-3c-3p | 127 | -0.989181873 | 10.999282 | 0.2980811 | DOWN | 2.707911 | 11.810942 | 0.0012308 | UP |
| Novel-3f-3p | 66 | -1.301092394 | 10.228842 | 0.2836528 | DOWN | 2.9447601 | 11.027109 | 0.0019476 | UP |
| Novel-4a-3p | 148 | -1.243075915 | 13.337668 | 0.0862047 | DOWN | -3.8778764 | 11.233557 | 0.0003435 | DOWN |
| Novel-4b-3p | 1962 | -0.712484479 | 17.03775 | 0.2811898 | DOWN | -4.5924403 | 15.198042 | 8.55E-09 | DOWN |
| Novel-4d-3p | 93 | -1.074767845 | 12.588034 | 0.1491761 | DOWN | -3.2491131 | 10.65837 | 0.0047291 | DOWN |
| Novel-5-3p | 146 | 0 | 0 | 1 | _ | 4.8140123 | 11.573737 | 2.47E-06 | UP |
| Novel-6-3p | 139 | 2.426333279 | 12.19918 | 0.0032194 | UP | 0.5738677 | 12.756475 | 0.4367159 | UP |
| Novel-9-5p | 126 | 2.212281384 | 13.005661 | 0.0042355 | UP | -5.0103113 | 12.29918 | 1.27E-06 | DOWN |
To validate miRNA expression level, we tested 10 differentially expressed miRNAs by miRNA-specific stem-loop qPCR. According to the results of Solexa sequencing, five of these miRNAs (mir-222-5p, mir-574a-5p, Novel-9-5p, Novel-10-3p and Novel-37-3p) were differentially expressed during hibernation in both the brain and WAT; another five miRNAs, including three (mir-7f-5p, mir-139-3p and mir-222-3p) differentially expressed only in the brain and two (mir-124b-3p and mir-378-3p) only in WAT, were used as negative controls. PCR confirmed the Solexa sequencing expression patterns (Table 3). In WAT (HA/AA), six miRNAs (mir-124b-3p, mir-222-5p, mir-378-3p, mir-574a-5p, Novel-9-5p and Novel-10-3p) showed consistent expression patterns and all reached significance by both technologies (Table 3). In the brain (HB/AB), the differentially expressed patterns of four miRNAs (mir-222-5p, Novel-9-5p, Novel-10-3p and Novel-37-3p) were identical in both technologies. Moreover, two negative controls, mir-7f-5p in WAT and mir-378-3p in brain showed no significant change detected in sequencing data, were confirmed by qPCR.
Table 3. Comparison of ten miRNA expression pattern in four libraries, as detected by Solexa sequencing and qPCR.
| HA vs. AA | HB vs. AB | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Illumina | Real-time PCR | Illumina | Real-time PCR | |||||||||
| miRNA Name | logFC | P-value a | Mark | log2−ΔΔCT | P-value b | Mark | logFC | P-value a | Mark | log2−ΔΔCT | P-value b | Mark |
| miR-124b-3p | 2.26 | 1.85E-03 | ↑ * | 1.87 | 1.74E-03 | ↑ * | -0.542 | 0.41 | ↓ | -0.98 | 0.12 | ↓ |
| miR-139-3p | 0.37 | 0.57 | ↑ | -2.89 | 3.91E-04 | ↓ * | -2.13 | 1.92E-03 | ↓ * | -0.67 | 0.11 | ↓ |
| miR-222-3p | 1.43 | 0.03 | ↑ | 1.07 | 1.36E-05 | ↑ * | -1.8 | 7.54E-03 | ↓ * | -0.34 | 0.15 | ↓ |
| miR-222-5p | 3.03 | 5.79E-05 | ↑ * | 0.31 | 6.81E-03 | ↑ * | 2.16 | 4.41E-03 | ↑ * | 0.59 | 7.67E-03 | ↑ * |
| miR-378-3p | -2.82 | 5.51E-05 | ↓ * | -0.82 | 1.57E-06 | ↓ * | -1.52 | 0.02 | ↓ | -0.1 | 0.04 | ↓ * |
| miR-574a-5p | -2.52 | 6.32E-04 | ↓ * | -6.23 | 2.08E-07 | ↓ * | -2.74 | 3.96E-03 | ↓ * | -0.24 | 0.83 | ↓ |
| miR-7f-5p | 0.39 | 0.55 | ↑ | -0.05 | 0.12 | ↓ | -1.84 | 6.23E-03 | ↓ * | -0.796 | 0.24 | ↓ |
| Novel-10-3p | 3.02 | 1.84E-04 | ↑ * | 1.05 | 0.01 | ↑ * | -4.43 | 4.45E-06 | ↓ * | -0.67 | 8.52E-07 | ↓ * |
| Novel-37-3p | -2.06 | 5.69E-03 | ↓ * | -0.04 | 0.25 | ↓ | 6.11 | 4.20E-13 | ↑ * | 0.36 | 7.27E-03 | ↑ * |
| Novel-9-5p | 2.21 | 4.24E-03 | ↑ * | 35.07 | 1.69E-03 | ↑ * | -5.01 | 1.27E-06 | ↓ * | -1.86 | 3.82E-24 | ↓ * |
PCR was performed in three biological replicates of each state with triplicate wells for each individual sample.
↑, up-regulated
↓, down-regulated
*, P-valuea<0.01 or P-valueb<0.05.
Target prediction and annotation
To better understand the potential role and mechanism of miRNAs in hibernation, we predicted the miRNA target genes. Firstly, we obtained 22,432 M. lucifugus reference genes and 104,763 H. sapiens mRNAs and searched for orthologous genes by tblastx. A total of 20,282 M. lucifugus genes were orthologous to H. sapiens mRNAs with a BLAST e value ≤1e-10 and identity ≥60%. As 93.6% of human mRNAs have a 3’ UTR less than 3 Kb (S1 Fig), we extracted 3 Kb downstream of M. lucifugus coding sequences, including 2151 M. lucifugus genes with no human orthologs. Finally, we identified 87.8% (19,698 / 22,432) reference genes as the targets of 196 miRNAs. Both KEGG pathway and GO annotation analyses indicated that most physiological processes and cell functions are regulated by 196 miRNAs. We did not observe any significant differences in the number of target genes when we compared 119 conserved miRNAs vs. 196 miRNAs and 77 novel miRNAs vs. 196 miRNAs (S9 Table). In contrast, further analysis showed that many pathways in both tissues were markedly affected by the differentially expressed miRNAs, especially by the miRNAs down-regulated during hibernation (S9 Table).
We further examined the adipocytokine signaling pathway to determine the relationship between miRNAs and target mRNAs during hibernation, as adipocytokine signaling is co-regulated by both brain and adipose tissues. Moreover, we searched for mRNAs from our on-going project of digital gene expression (DGE) sequencing to enrich for target genes. DGE sequencing has been conducted on the same animals used here (unpublished data). A total of 64 mRNAs involved in adipocytokine signaling pathway were identified by DGE sequencing (S10 Table, DGE unpublished data). By combining the miRNA and target mRNA expression patterns, we found that 41 of 49 differentially expressed miRNAs regulated the expression of 51 genes, and more than half of the genes in each tissue were up-regulated during hibernation. Among these, the differential expression of three mRNAs (Insulin Receptor Substrate, IRS; Adenosine 5‘-monophosphate (AMP)-activated protein kinase, AMPK; Retinoid X receptor, RXR) was significant (P≤0.01), including IRS over-expression. The others were down-regulated during HA/AA (Fig 4 and S10 Table).
Fig 4. Co-regulation of miRNAs and target mRNAs in the adipocytokine signaling pathway.

(A) Expression pattern and relationship of differentially expressed miRNAs (left) and mRNAs involved in the adipocytokine signaling pathway (right, identified by DGE, unpublished data). The expression profile of a miRNA or gene in the brain (HB/AB) and adipose tissue (HA/AA) was calculated by sequence counts (TPM). The red and green colors represent high and low gene expression after comparing the hibernating state vs. the active state. Differentially expressed miRNAs and mRNAs (P≤0.01) during hibernation are linked with red lines; (B) Network of adipocytokine signaling pathway. Red box, predicted target genes of differentially expressed miRNAs that were also identified by DGE; Blue box, predicted target genes of differentially expressed miRNAs, but that were not identified by DGE; Green box, genes identified by DGE.
Discussion
Our study shows that in the hibernating bat Myotis ricketti, 33 brain and 25 white adipose tissue (WAT) miRNAs were differentially expressed between euthermia and hibernation. Consequently, they are likely involved in regulating functional processes and energy metabolism during hibernation.
Our findings were possible with the ultrahigh-throughput sequencing technique (RNA-seq) Illumina/Solexa sequencing, which provides a great platform to discover and analyze small RNAs. It has been previously used in non-model species, such as the Arctic ground squirrel [Urocitellus (Spermophilus) parryii] and the sea cucumber (Apostichopus japonicus) during hypometabolism, but these species’ genomic backgrounds and miRNA data are not available in miRbase [18, 21]. The clear advantages of Solexa sequencing are the generation of several million small RNA sequences in each small RNA library of one run, the ability to identify novel miRNAs, the potential to overcome the drawbacks of microarrays, and the generation of expression profiles in a greater and reproducible dynamic range [53]. However, this technology can be influenced by sequencing errors and raw data processing before miRNA identification [53]. In the present study, we performed Solexa high-throughput sequencing with stem-loop qPCR validation of expression levels to analyze miRNAs involved in hibernation. Moreover, we chose a widely distributed bat species in China, congeneric with Myotis lucifugus, as the animal model to explore miRNA expression in two vital organs (brain and WAT) during hibernation.
Our data show that the sequencing information from four libraries was saturated, and 88.7% unique reads were tissue/state-specifically expressed (S3 Table). These data indicate a high quality of small RNA library construction and screening and that the libraries meet the needs of small RNA annotation and analysis. After mapping the unique reads to the M. lucifugus genome database and removing the reads that matched genome ≥20, the small RNA annotation indicated that miRNAs (47.7% in total) are the most abundant fraction of small RNAs (see Table 1 and Fig 1). These results are similar to those of previous miRNA profiling of multi-animal species [18, 21, 36]. Furthermore, we found that SINE and LINE, which are retroposons widely used as phylogenetic markers [54, 55], were the major source of genomic repeat-derived siRNAs in all four libraries. The detailed information of SINE and LINE provided us with the ability to further explore the phylogenetic evolution of heterothermy in mammals, especially in bats.
Our data also revealed a large number of conserved and novel miRNAs. We identified a total of 196 mature miRNAs, including 119 conserved miRNAs and 77 novel miRNAs (S7 Table). Moreover, we found that 31 (22 conserved and nine novel) of 196 miRNAs were expressed in a tissue/state-specific manner, suggesting a complex mechanism of miRNA regulation during hibernation. Of those, six conserved miRNAs with expression counts >100 were specifically expressed in the brain, including miR-153, which has been shown to function in neuronal protection and tumorigenesis [56, 57], miR-124, which regulates metabolic and immune processes [58, 59], miR-551, which is implicated in the stress response [60], and miR-1298, which has been associated with neural and endocrinological disease [61]. However, in the absence of direct and detailed evidence between the molecular data and animal function, it is necessary to further investigate the roles of these tissue- and state-specific miRNAs during hibernation.
Of 196 miRNAs, 49 were differentially expressed (P≤0.01 & │logFC│≥1) (Table 2 and S7 Table), including ten miRNAs that had been shown to regulate mammalian hibernation in other species (S8 Table) [18, 20, 22–25]. Both microarray and qPCR have been used to validate miRNA expression in many studies, as they are more accurate than RNA-seq sequencing [18, 21]. In our study, qPCR validation of 10 out of 49 differentially expressed miRNAs provided consistent results with those of Solexa sequencing (Table 3). We conclude that our data are reliable. Although we validated a few differentially expressed miRNAs by qPCR in our study, the limitations of RNA-seq, and the value of sample pooling in high-throughput sequencing, we suggest that microarray hybridization should be performed in the future to further validate the significance of differentially expressed miRNAs obtained here. Microarray analysis could potentially compensate for the drawbacks of RNA-seq sequencing [53] and estimate the biological variability.
To gain insight into the potential broader functions of the miRNAs we identified, we predicted their putative targets and classified them by KEGG pathway assay and GO annotation. Comparing the targets of 196 miRNAs, we found that miRNAs were differentially expressed in both brain and adipose tissues between euthermia and hibernation states, and this is likely a reflection of the different activities of many essential processes during these states (e.g., lipid metabolism and signal transduction, S9 Table). We found that only 49 of 196 miRNAs regulated most target gene expression (12,909 of 20,370), and thus most physiological processes during hibernation, contributing to reduced energy expenditure during hypometabolism. Moreover, both KEGG and GO assays identified the same enriched functional groups (P<0.05), and all of them were affected by differentially expressed miRNAs, particularly by down-regulated miRNAs (S9 Table). By binding to target mRNAs, miRNAs can reversibly degrade their targets at the transcriptional level and/or inhibit their translation [19]. Thus, during torpor, brain and WAT physiological processes were globally affected by down-regulated miRNAs, suggesting that these processes were activated, supporting our hypothesis. Moreover, previous studies have shown that some specific brain regions such as the hypothalamus are involved in regulating mammalian hibernation [62–64]. Our study was limited to the use of entire brains, but future work on specific brain regions may identify the precise regulation of miRNA expression in important brain areas. Such studies could further expand our knowledge of hibernation regulation.
The brain plays an important role in controlling and synchronizing peripheral physiology to reduce energy expenditure and minimizing tissue damage and/or disease during hibernation [3, 4, 7]. In contrast, adipose tissue is the primary energy source during hibernation and is especially critical during periodic rewarming [6, 65]. Thus, specific molecular changes are required to accommodate the specific roles of brain and adipose tissue during hibernation. Because the potential network of miRNAs and targets is highly complicated, we chose to examine the adipocytokine signaling pathway, which plays an important role in signaling transduction and energy metabolism [66, 67]. In both tissues, more than half mRNAs were up-regulated (Fig 4A) in contrast to miRNA expression, indicating that this pathway was activated to some extent. According to KEGG analysis, the adipocytokine signaling pathway can be subdivided into three interrelated parts, TNFα (Tumor Necrosis Factor α)-related genes, LEP (Leptin)-related genes and ADIPO (Adiponectin)-related genes (Fig 4B). Andrews et al. [13] previously showed that energy metabolism of hibernators is shifted at the gene level from glycometabolism in the liver and/or pancreas to lipid metabolism in adipose tissue during hibernation. We found that the significant up-regulation of IRS in WAT could be indicative of glucose uptake inhibition, whereas a decrease in AMPK in WAT could promote gluconeogenesis. Our data help to illustrate how miRNAs and mRNAs control the metabolic switch by inhibiting the glycometabolic pathway to burning fat as the main energy source, supporting previous reports [65]. While the regulation of fat metabolism is crucial during hibernation, the regulation of food intake is important during euthermia. Negative AMPK expression or the inhibition of AMPK and AMPK-related gene activity in the hypothalamus result in reduced food intake [68, 69]. We hypothesize that down-regulation of AMPK and RXR in WAT lead to decreased food intake and increased energy expenditure. Network construction increases our understanding of molecular mechanisms mediated by miRNAs and target genes. It also provides clues to uncover how brain and adipose tissue cooperate to regulate hibernation by decreasing food intake, inhibiting glucose metabolism and increasing fatty acid metabolism to meet the physiological needs of hibernators.
Conclusions
We identified 196 miRNAs (77 novel miRNAs) in bats by in-depth analysis of the small RNAs in the hibernating species Myotis ricketti. Differential miRNA expression suggests that some physiological pathways in the brain and adipose tissue are activated during hibernation. In contrast to other peripheral tissues that are physiologically suppressed, our findings advance the understanding of the molecular mechanisms of hibernation. However, further study is required to validate the expression pattern during hibernation vs. euthermia, estimate biological variability, and elucidate the mechanisms of specific miRNAs and their target mRNAs in hibernation. Moreover, the Interspersed Elements (LINE and SINE) identified here may help further explore the phylogenetic evolution of heterothermy in bats. With the progress of the M. lucifugus genome sequencing project and the increased accuracy of gene annotation, more detailed information on miRNAs involved in hibernation will likely be uncovered from bat transcriptome libraries. These can be used to better understand heterothermy the physiology and evolution of heterothermy.
Supporting Information
Homo sapiens reference sequences were downloaded from the NCBI Reference Sequence database (http://www.ncbi.nlm.nih.gov/RefSeq/).
(JPG)
{kind=link}
(PNG)
{kind=link}
gga: Gallus gallus; has: Homo sapiens; cel: Caenorhabditis elegans; dme: Drosophila melanogaster; sme: Schmidtea mediterranea.
(JPG)
{kind=link}
(DOC)
The table summarizes the total number of small RNA reads by Solexa sequencing. After removing adapter contaminants, reads less than 18 nt and containing Ns, the clean reads from each library were used for the small RNA screen. HB: hibernating state brain; AB: active state brain; HA: hibernating state adipose tissue; AA: active state adipose tissue.
(DOC)
Redundancy among libraries was calculated to estimate the quality of the Solexa small RNA libraries. Moreover, the numbers of state/tissue-specific expressed reads were identified. HB: hibernating state brain; AB: active state brain; HA: hibernating state adipose tissue; AA: active state adipose tissue.
(DOC)
(DOC)
(DOC)
(A) 90 conserved miRNA hairpins. (B) 101 novel miRNA hairpins.
(XLSX)
(A) Expression details of 196 miRNAs (119 conserved miRNAs and 77 novel miRNAs). (B) List of 33 miRNAs differentially expressed in the brain (P≤0.01 & │logFC ≥1). (C) List of 25 miRNAs differentially expressed in adipose tissue (P≤0.01 & │logFC│≥1). (D) List of 9 miRNAs differentially expressed in both brain and adipose tissue (P≤0.01 & │logFC│≥1). (E) List of 31 tissue/state-specifically expressed miRNAs in brain (23) and adipose tissue (8).
(XLSX)
(XLSX)
KEGG pathway analysis (A) and GO annotation (B) of target genes regulated by 196 miRNAs, 119 conserved miRNAs, 8 conserved miRNAs differentially expressed in the brain, 13 conserved miRNAs differentially expressed in adipose tissue, 77 novel miRNAs, 25 novel miRNAs differentially expressed in the brain, and 12 novel miRNAs differentially expressed in adipose tissue. T-tests were performed to analyze the bio-activity of each physiological process in both tissues by comparing hibernation vs. active state.
(XLS)
Genes, only identified by DGE sequencing and not the targets of differentially expressed miRNAs which obtained in this study, are in green.
(XLS)
Acknowledgments
The authors wish to thank Junpeng Zhang and Jinshuo Zhang for their assistance collecting samples and Bronwyn McAllan for commenting on the manuscript.
Data Availability
All small RNA data series were submitted to NCBI Gene Expression Omnibus (GEO) with accession number GSE29053.
Funding Statement
This work was funded by grants awarded to SY Zhang under the Key Construction Program of the National "985" Project and to LH Yuan under the National Natural Science Fund of China (31301012). LH Yuan is also supported by the Guangzhou Pearl River Scientific and Technological New Star Project (2012J2200003), and Outstanding Youth Science and Technology Talent Fund of Guangdong Academy of Sciences (rcjj201402). JP Chen is supported by the Program for Innovative Research Team of Cross-discipline and cooperation from Chinese Academy of Sciences. The authors confirm that MininGene Biotechnology Co. Ltd provided support in the form of salaries for author (HBS), but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of these authors are articulated in the 'author contributions' section.
References
- 1. Geiser F (2013) Hibernation. Curr Biol 23, R188–193. 10.1016/j.cub.2013.01.062 [DOI] [PubMed] [Google Scholar]
- 2. Geiser F (2004) Metabolic rate and body temperature reduction during hibernation and daily torpor. Annu Rev Physiol 66, 239–274. [DOI] [PubMed] [Google Scholar]
- 3. Carey HV, Andrews MT,Martin SL (2003) Mammalian hibernation: cellular and molecular responses to depressed metabolism and low temperature. Physiol Rev 83, 1153–1181. [DOI] [PubMed] [Google Scholar]
- 4. Drew KL, Rice ME, Kuhn TB, Smith MA (2001) Neuroprotective adaptations in hibernation: therapeutic implications for ischemia-reperfusion, traumatic brain injury and neurodegenerative diseases. Free Radic Biol Med 31, 563–573. [DOI] [PubMed] [Google Scholar]
- 5. Frerichs KU (1999) Neuroprotective strategies in nature—novel clues for the treatment of stroke and trauma. Acta Neurochir Suppl 73, 57–61. [DOI] [PubMed] [Google Scholar]
- 6. Florant GL (1998) Lipid Metabolism in Hibernators: The Importance of Essential Fatty Acids. American Zoologist 38, 331–340. [Google Scholar]
- 7. Storey KB (2010) Out cold: biochemical regulation of mammalian hibernation—a mini-review. Gerontology 56, 220–230. 10.1159/000228829 [DOI] [PubMed] [Google Scholar]
- 8. Williams DR, Epperson LE, Li W, Hughes MA, Taylor R, Rogers J, et al. (2005) Seasonally hibernating phenotype assessed through transcript screening. Physiol Genomics 24, 13–22. [DOI] [PubMed] [Google Scholar]
- 9. Brauch KM, Dhruv ND, Hanse EA, Andrews MT (2005) Digital transcriptome analysis indicates adaptive mechanisms in the heart of a hibernating mammal. Physiol Genomics 23, 227–234. [DOI] [PubMed] [Google Scholar]
- 10. Yan J, Burman A, Nichols C, Alila L, Showe LC, Showe MK, et al. (2006) Detection of differential gene expression in brown adipose tissue of hibernating arctic ground squirrels with mouse microarrays. Physiol Genomics 25, 346–353. [DOI] [PubMed] [Google Scholar]
- 11. Chen J, Yuan L, Sun M, Zhang L, Zhang S (2008) Screening of hibernation-related genes in the brain of Rhinolophus ferrumequinum during hibernation. Comp Biochem Physiol B Biochem Mol Biol 149, 388–393. [DOI] [PubMed] [Google Scholar]
- 12. Lei M, Dong D, Mu S, Pan YH, Zhang S (2014) Comparison of brain transcriptome of the greater horseshoe bats (Rhinolophus ferrumequinum) in active and torpid episodes. PLoS One 9, e107746 10.1371/journal.pone.0107746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Andrews MT, Squire TL, Bowen CM, Rollins MB (1998) Low-temperature carbon utilization is regulated by novel gene activity in the heart of a hibernating mammal. Proc Natl Acad Sci U S A 95, 8392–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fahlman A, Storey JM, Storey KB (2000) Gene up-regulation in heart during mammalian hibernation. Cryobiology 40, 332–342. [DOI] [PubMed] [Google Scholar]
- 15. Hittel DS, Storey KB (2002) Differential expression of mitochondria-encoded genes in a hibernating mammal. J Exp Biol 205, 1625–1631. [DOI] [PubMed] [Google Scholar]
- 16. Yan J, Barnes BM, Kohl F, Marr TG (2008) Modulation of gene expression in hibernating arctic ground squirrels. Physiol Genomics 32, 170–181. [DOI] [PubMed] [Google Scholar]
- 17. Shao C, Liu Y, Ruan H, Li Y, Wang H, Kohl F, et al. (2010) Shotgun proteomics analysis of hibernating arctic ground squirrels. Mol Cell Proteomics 9, 313–326. 10.1074/mcp.M900260-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu Y, Hu W, Wang H, Lu M, Shao C, Menzel C, et al. (2010) Genomic analysis of miRNAs in an extreme mammalian hibernator, the Arctic ground squirrel. Physiol Genomics 42A, 39–51. 10.1152/physiolgenomics.00054.2010 [DOI] [PubMed] [Google Scholar]
- 19. Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. [DOI] [PubMed] [Google Scholar]
- 20. Morin P Jr, Dubuc A, Storey KB (2008) Differential expression of microRNA species in organs of hibernating ground squirrels: a role in translational suppression during torpor. Biochim Biophys Acta 1779, 628–633. 10.1016/j.bbagrm.2008.07.011 [DOI] [PubMed] [Google Scholar]
- 21. Chen M, Zhang X, Liu J, Storey KB (2013) High-throughput sequencing reveals differential expression of miRNAs in intestine from sea cucumber during aestivation. PLoS One 8, e76120 10.1371/journal.pone.0076120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kornfeld SF, Biggar KK, Storey KB (2012) Differential expression of mature microRNAs involved in muscle maintenance of hibernating little brown bats, Myotis lucifugus: a model of muscle atrophy resistance. Genomics Proteomics Bioinformatics 10, 295–301. 10.1016/j.gpb.2012.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maistrovski Y, Biggar KK, Storey KB (2012) HIF-1alpha regulation in mammalian hibernators: role of non-coding RNA in HIF-1alpha control during torpor in ground squirrels and bats. J Comp Physiol B 182, 849–859. 10.1007/s00360-012-0662-y [DOI] [PubMed] [Google Scholar]
- 24. Biggar KK, Storey KB (2014) Identification and expression of microRNA in the brain of hibernating bats, Myotis lucifugus . Gene 544, 67–74. 10.1016/j.gene.2014.04.048 [DOI] [PubMed] [Google Scholar]
- 25. Lang-Ouellette D (2014) Differential expression of miRNAs with metabolic implications in hibernating thirteen-lined ground squirrels, Ictidomys tridecemlineatus . Molecular and cellular biochemistry 394, 291–298. 10.1007/s11010-014-2105-4 [DOI] [PubMed] [Google Scholar]
- 26. Lyons PJ, Lang-Ouellette D, Morin P Jr (2013) CryomiRs: towards the identification of a cold-associated family of microRNAs. Comp Biochem Physiol Part D Genomics Proteomics 8, 358–364. 10.1016/j.cbd.2013.10.001 [DOI] [PubMed] [Google Scholar]
- 27. Biggar KK, Storey KB (2010) The emerging roles of microRNAs in the molecular responses of metabolic rate depression. Journal of molecular cell biology, mjq045. [DOI] [PubMed] [Google Scholar]
- 28. Morin P Jr, Storey KB (2009) Mammalian hibernation: differential gene expression and novel application of epigenetic controls. Int J Dev Biol 53, 433–442. 10.1387/ijdb.082643pm [DOI] [PubMed] [Google Scholar]
- 29. Jones KE, Purvis A, Maclarnon ANN, Bininda-Emonds OR, Simmons NB (2002) A phylogenetic supertree of the bats (Mammalia: Chiroptera) Biological Reviews 77, 223–259. [DOI] [PubMed] [Google Scholar]
- 30. Stawski C, Willis CKR, Geiser F (2014) The importance of temporal heterothermy in bats. Journal of Zoology 292, 86–100. [Google Scholar]
- 31. Yuan L, Zhao X, Lin B, Rossiter SJ, He L, Zuo X, et al. (2011) Adaptive evolution of Leptin in heterothermic bats. PLoS One 6, e27189 10.1371/journal.pone.0027189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Geiser F, Stawski C (2011) Hibernation and torpor in tropical and subtropical bats in relation to energetics, extinctions, and the evolution of endothermy. Integr Comp Biol 51, 337–348. 10.1093/icb/icr042 [DOI] [PubMed] [Google Scholar]
- 33. Pan YH, Zhang Y, Cui J, Liu Y, McAllan BM, Liao CC, et al. (2013) Adaptation of phenylalanine and tyrosine catabolic pathway to hibernation in bats. PLoS One 8, e62039 10.1371/journal.pone.0062039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lin B, Yuan L, Chen J (2012) Selection pressure drives the co-evolution of several lipid metabolism genes in mammals. Chinese Science Bulletin 57, 877–885. [Google Scholar]
- 35. Green CD, Simons JF, Taillon BE, Lewin DA (2001) Open systems: panoramic views of gene expression. Journal of immunological methods 250, 67–79. [DOI] [PubMed] [Google Scholar]
- 36. Morin RD, O'Connor MD, Griffith M, Kuchenbauer F, Delaney A, Prabhu AL, et al. (2008) Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res 18, 610–621. 10.1101/gr.7179508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ (2008) miRBase: tools for microRNA genomics. Nucleic Acids Res 36, D154–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dsouza M, Larsen N, Overbeek R (1997) Searching for patterns in genomic data. Trends Genet 13, 497–498. [DOI] [PubMed] [Google Scholar]
- 40. Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A (2005) Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 33, D121–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Friedlander MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, et al. (2008) Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol 26, 407–415. 10.1038/nbt1394 [DOI] [PubMed] [Google Scholar]
- 42. Rice P, Longden I, Bleasby A (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16, 276–277. [DOI] [PubMed] [Google Scholar]
- 43. Jones-Rhoades MW, Bartel DP (2004) Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol Cell 14, 787–799. [DOI] [PubMed] [Google Scholar]
- 44. Hofacker IL, WF, Stadler P F, Bonhoeffer L S, Tacker M, Schuster P (1994) Fast folding and comparison of RNA secondary structures Monatshefte für Chemie / Chemical Monthly Volume 125, 167–188. [Google Scholar]
- 45. Tarailo-Graovac M, Chen N (2009) Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics, 4.10. 11–14.10. 14. [DOI] [PubMed] [Google Scholar]
- 46. Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, et al. (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 31, 3497–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E (2007) The role of site accessibility in microRNA target recognition. Nat Genet 39, 1278–1284. [DOI] [PubMed] [Google Scholar]
- 50. Zdobnov EM, Apweiler R (2001) InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848. [DOI] [PubMed] [Google Scholar]
- 51. Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, et al. (2006) From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res 34, D354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Biggar KK, Kornfeld SF, Storey KB (2011) Amplification and sequencing of mature microRNAs in uncharacterized animal models using stem-loop reverse transcription-p olymerase chain reaction. Analytical biochemistry 416, 231–233. 10.1016/j.ab.2011.05.015 [DOI] [PubMed] [Google Scholar]
- 53. Git A, Dvinge H, Salmon-Divon M, Osborne M, Kutter C, Hadfield J, et al. (2010) Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression. Rna 16, 991–1006. 10.1261/rna.1947110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ohshima K (2013) RNA-Mediated Gene Duplication and Retroposons: Retrogenes, LINEs, SINEs, and Sequence Specificity. Int J Evol Biol 2013, 424726 10.1155/2013/424726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hartig G, Churakov G, Warren WC, Brosius J, Makalowski W, Schmitz J (2013) Retrophylogenomics place tarsiers on the evolutionary branch of anthropoids. Sci Rep 3, 1756 10.1038/srep01756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fragkouli A, Doxakis E (2014) miR-7 and miR-153 protect neurons against MPP(+)-induced cell death via upregulation of mTOR pathway. Front Cell Neurosci 8, 182 10.3389/fncel.2014.00182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang L, Pickard K, Jenei V, Bullock MD, Bruce A, Mitter R, et al. (2013) miR-153 supports colorectal cancer progression via pleiotropic effects that enhance invasion and chemotherapeutic resistance. Cancer Res 73, 6435–6447. 10.1158/0008-5472.CAN-12-3308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wei J, Wang F, Kong L-Y, Xu S, Doucette T, Ferguson SD, et al. (2013) MiR-124 inhibits STAT3 signaling to enhance T cell-mediated immune clearance of glioma. Cancer research 73, 3913–3926. 10.1158/0008-5472.CAN-12-4318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li M, Plecita L, McKeon B, Flockton A, Frid MG, Jezek P, et al. (2014) Mir-124 Regulates The Metabolic State Of Vascular Adventitial Fibroblasts In Pulmonary Hypertension Through The Rna Splicing Factor Ptbp1. Am J Respir Crit Care Med 189, A3986. [Google Scholar]
- 60.Hood L, Wang K, Lee I, Zhou Y, Cho JH, Ghosh D (2013) Markers and methods for detecting posttraumatic stress disorder (ptsd), Google Patents.
- 61. Kishore S, Stamm S (2011) Role of Alternative Splicing of the 5-HT2C Receptor in the Prader-Willi Syndrome In 5-HT2C Receptors in the Pathophysiology of CNS Disease. (Springer; ), pp. 413–427. [Google Scholar]
- 62. Pakhotin PI, Pakhotina ID, Belousov AB (1993) The study of brain slices from hibernating mammals in vitro and some approaches to the analysis of hibernation problems in vivo. Prog Neurobiol 40, 123–161. [DOI] [PubMed] [Google Scholar]
- 63. Zhu T, Yuan L, Jones G, Hua P, He G, Chen J, et al. (2014) OB-RL silencing inhibits the thermoregulatory ability of Great Roundleaf Bats (Hipposideros armiger) General and comparative endocrinology 204, 80–87. 10.1016/j.ygcen.2014.04.028 [DOI] [PubMed] [Google Scholar]
- 64. Dark J, Kilduff TS, Craig Heller H, Licht P, Zucker I (1990) Suprachiasmatic nuclei influence hibernation rhythms of golden-mantled ground squirrels. Brain research 509, 111–118. [DOI] [PubMed] [Google Scholar]
- 65. Andrews MT (2004) Genes controlling the metabolic switch in hibernating mammals. Biochem Soc Trans 32, 1021–1024. [DOI] [PubMed] [Google Scholar]
- 66. Chen J, Tan B, Karteris E, Zervou S, Digby J, Hillhouse EW, et al. (2006) Secretion of adiponectin by human placenta: differential modulation of adiponectin and its receptors by cytokines. Diabetologia 49, 1292–1302. [DOI] [PubMed] [Google Scholar]
- 67. Ogunyemi D, Xu J, Mahesan AM, Rad S, Kim E, Yano J, et al. (2013) Differentially expressed genes in adipocytokine signaling pathway of adipose tissue in pregnancy. Journal of Diabetes Mellitus 3, 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, et al. (2004) AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428, 569–574. [DOI] [PubMed] [Google Scholar]
- 69. Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, et al. (2004) AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem 279, 12005–12008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Homo sapiens reference sequences were downloaded from the NCBI Reference Sequence database (http://www.ncbi.nlm.nih.gov/RefSeq/).
(JPG)
(PNG)
gga: Gallus gallus; has: Homo sapiens; cel: Caenorhabditis elegans; dme: Drosophila melanogaster; sme: Schmidtea mediterranea.
(JPG)
(DOC)
The table summarizes the total number of small RNA reads by Solexa sequencing. After removing adapter contaminants, reads less than 18 nt and containing Ns, the clean reads from each library were used for the small RNA screen. HB: hibernating state brain; AB: active state brain; HA: hibernating state adipose tissue; AA: active state adipose tissue.
(DOC)
Redundancy among libraries was calculated to estimate the quality of the Solexa small RNA libraries. Moreover, the numbers of state/tissue-specific expressed reads were identified. HB: hibernating state brain; AB: active state brain; HA: hibernating state adipose tissue; AA: active state adipose tissue.
(DOC)
(DOC)
(DOC)
(A) 90 conserved miRNA hairpins. (B) 101 novel miRNA hairpins.
(XLSX)
(A) Expression details of 196 miRNAs (119 conserved miRNAs and 77 novel miRNAs). (B) List of 33 miRNAs differentially expressed in the brain (P≤0.01 & │logFC ≥1). (C) List of 25 miRNAs differentially expressed in adipose tissue (P≤0.01 & │logFC│≥1). (D) List of 9 miRNAs differentially expressed in both brain and adipose tissue (P≤0.01 & │logFC│≥1). (E) List of 31 tissue/state-specifically expressed miRNAs in brain (23) and adipose tissue (8).
(XLSX)
(XLSX)
KEGG pathway analysis (A) and GO annotation (B) of target genes regulated by 196 miRNAs, 119 conserved miRNAs, 8 conserved miRNAs differentially expressed in the brain, 13 conserved miRNAs differentially expressed in adipose tissue, 77 novel miRNAs, 25 novel miRNAs differentially expressed in the brain, and 12 novel miRNAs differentially expressed in adipose tissue. T-tests were performed to analyze the bio-activity of each physiological process in both tissues by comparing hibernation vs. active state.
(XLS)
Genes, only identified by DGE sequencing and not the targets of differentially expressed miRNAs which obtained in this study, are in green.
(XLS)
Data Availability Statement
All small RNA data series were submitted to NCBI Gene Expression Omnibus (GEO) with accession number GSE29053.