

Abstract
Patients with chronic obstructive pulmonary disease (COPD) exhibit increases in lung volume due to expiratory airflow limitation. Increases in lung volumes may affect upper airway patency and compensatory responses to inspiratory flow limitation (IFL) during sleep. We hypothesized that COPD patients have less collapsible airways inversely proportional to their lung volumes, and that the presence of expiratory airflow limitation limits duty cycle responses to defend ventilation in the presence of IFL. We enrolled 18 COPD patients and 18 controls, matched by age, body mass index, sex, and obstructive sleep apnea disease severity. Sleep studies, including quantitative assessment of airflow at various nasal pressure levels, were conducted to determine upper airway mechanical properties [passive critical closing pressure (Pcrit)] and for quantifying respiratory timing responses to experimentally induced IFL. COPD patients had lower passive Pcrit than their matched controls (COPD: −2.8 ± 0.9 cmH2O; controls: −0.5 ± 0.5 cmH2O, P = 0.03), and there was an inverse relationship of subject's functional residual capacity and passive Pcrit (−1.7 cmH2O/l increase in functional residual capacity, r2 = 0.27, P = 0.002). In response to IFL, inspiratory duty cycle increased more (P = 0.03) in COPD patients (0.40 to 0.54) than in controls (0.41 to 0.51) and led to a marked reduction in expiratory time from 2.5 to 1.5 s (P < 0.01). COPD patients have a less collapsible airway and a greater, not reduced, compensatory timing response during upper airway obstruction. While these timing responses may reduce hypoventilation, it may also increase the risk for developing dynamic hyperinflation due to a marked reduction in expiratory time.
Keywords: upper airway obstruction, COPD, obstructive sleep apnea, inspiratory flow limitation, lung volume
chronic obstructive pulmonary disease (COPD) stands as a leading cause of death, disability, and reduced quality of life. While dyspnea on exertion remains the major reason for impaired quality of life, disturbed sleep and morning fatigue represent the second most common complaints in these patients (3, 18). While obstructive sleep apnea (OSA) is a well-recognized source for sleep disturbances and morning fatigue in patients with COPD, other markers of COPD, such as lung hyperinflation (20), increased respiratory efforts (9, 12, 26), alterations in gas exchange (6, 15, 21, 27, 34), and inspiratory flow limitation, have also been associated with sleep disturbances. Understanding how these factors produce sleep disturbances may allow investigators to develop specific strategies for preventing and treating sleep disturbances in patients with COPD.
Inspiratory flow limitation is a hallmark of upper airway obstruction, which occurs in individuals with impaired mechanical properties of the upper airways (10, 25). The mechanical properties of the upper airway is best quantified by determining the critical closing pressure (Pcrit) during a neural passive condition (passive Pcrit) (19, 25). Our laboratory has recently shown that the passive Pcrit predicts sleep apnea disease susceptibility and rises with increasing body mass index (BMI) and age (19). Moreover, our laboratory and others recently demonstrated that increases in lung volume reduce the passive Pcrit in normal individuals and in patients with sleep apnea (23, 31, 32). The effect of increasing lung volumes in patients with COPD on mechanical properties of the upper airway has not been shown.
Upper airway obstruction elicits compensatory responses that help to prevent hypoventilation. Specifically, the inspiratory duty cycle [proportion of inspiratory time (Ti) to respiratory cycle length] increases in response to upper airway obstruction, and this rise in Ti is at the expense of the expiratory time (Te) (5, 14, 28). Patients with COPD, however, often exhibit long Te to cope with expiratory flow limitation and intrinsic positive end-expiratory pressure (11, 13). It is possible that the presence of expiratory flow limitation impairs duty cycle response to upper airway obstruction in patients with COPD. In the present study, we recruited COPD patients and BMI, age, sex, and apnea hypopnea index (AHI) matched individuals to determine 1) passive Pcrit and 2) respiratory timing responses to experimentally induced upper airway obstruction. We hypothesize that 1) COPD patients have a lower passive Pcrit (and thus lower risk for upper airway obstruction) in association with elevations in lung volume; and that 2) compensatory timing responses to upper airway obstruction are reduced in COPD patients compared with normal individuals; and 3) that the reduction in timing responses are in proportion to the degree of expiratory flow limitation [forced expiratory volume in 1 s (FEV1)].
METHODS
Subjects
Subjects were recruited from a pool of individuals participating in a trial examining the effect of sleep on COPD outcomes. A total of 96 COPD patients and 41 non-COPD smokers received a previous sleep study and were available for our study. From this group, a total of 36 subjects were recruited (Table 1). Both men (n = 12) and women (n = 6) who were diagnosed with COPD [FEV1/forced vital capacity (FVC) < 70%] were individually matched for age, sex, BMI and AHI with eighteen subjects without COPD. In addition, subset analysis was done on 12 COPD patients and 12 controls, matched by their mechanical upper airway properties (Passive Pcrit) and BMI. Subjects using respiratory depressants, sedatives or oral steroids were excluded. The subjects were also screened for unstable cardiovascular disease, uncontrolled hypertension, recent oropharyngeal surgery, neurological disorders, renal and hepatic insufficiency, current pregnancy, bleeding disorders, psychiatric issues, a mean resting O2 hemoglobin saturation of <88% and allergies to lidocaine. Written informed consent was obtained from each participant for this study, which was approved by the Johns Hopkins Medical Institution Human Investigations Review Board.
Table 1.
General demographics
| Variable | Controls | COPD Patients | P Value |
|---|---|---|---|
| n | 18 | 18 | |
| Anthropometry | |||
| Age, yr | 50.3 ± 1.6 | 57.1 ± 1.9 | <0.05 |
| Height, cm | 170.0 ± 2.7 | 174.2 ± 2.1 | NS |
| Weight, kg | 97.9 ± 6.6 | 86.5 ± 4.1 | NS |
| BMI, kg/m2 | 31.6 ± 1.6 | 28.6 ± 1.2 | NS |
| Sex | 12M; 6 F | 12M; 6 F | NS |
| Lung volumes | |||
| FEV1, liters | 3.1 ± 0.2 | 1.9 ± 0.2 | < 0.05 |
| FEV1/FVC, % | 79 ± 1.2 | 54 ± 2.6 | <0.05 |
| TLC, liters | 5.9 ± 0.3 | 6.7 ± 0.2 | < 0.05 |
| RV, liters | 1.9 ± 0.1 | 3.0 ± 0.2 | <0.05 |
| FRC, liters | 2.8 ± 0.2 | 4.0 ± 0.2 | < 0.05 |
| RV/TLC, % | 33.8 ± 1.8 | 45.5 ± 2.4 | <0.05 |
| Polysomnography | |||
| TST, min | 365.6 ± 10.7 | 353.7 ± 15.9 | NS |
| Sleep efficiency, %TST | 86.9 ± 2.0 | 79.4 ± 3.0 | <0.05 |
| NREM, %TST | 84.7 ± 1.5 | 83.9 ± 1.9 | NS |
| Stage 1 | 19.2 ± 2.4 | 27.1 ± 5.2 | NS |
| Stage 2 | 60.2 ± 2.5 | 50.8 ± 4.3 | NS |
| Stage 3 | 5.2 ± 1.8 | 6.0 ± 2.3 | NS |
| REM, %TST | 15.3 ± 1.5 | 16.1 ± 1.9 | NS |
| Total AHI, events/h | 28.9 ± 8.1 | 28.9 ± 8.6 | NS |
| NREM AHI, events/h | 27.8 ± 8.1 | 28.2 ± 8.6 | NS |
| REM AHI, events/h | 36.1 ± 7.7 | 33.3 ± 10.0 | NS |
| %Hypopnea NREM | 76.9 ± 6.1 | 75.9 ± 6.7 | NS |
| %Hypopnea REM | 73.3 ± 7.3 | 62.2 ± 8.6 | NS |
| Arterial SaO2, % | |||
| Average baseline value | 96.4 ± 0.4 | 93.9 ± 0.5 | <0.05 |
| Average low value | 91.5 ± 0.7 | 90.2 ± 0.6 | NS |
Values are averages ± SE of main demographic parameters for both groups; n, no. of subjects. COPD, chronic obstructive pulmonary disease; BMI, body mass index; M, male; F, female; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; TLC, total lung capacity; RV, residual volume; FRC, functional residual capacity; TST, total sleep time; NREM, non-rapid eye movement; REM, rapid eye movement; AHI, apnea hypopnea index; SaO2, arterial O2 saturation; NS, nonsignificant.
Experimental Techniques
Pulmonary function testing.
Standard techniques for pulmonary function testing (CPL Lung Function Analyzer; W.E. Collins, Braintree MA.) were conducted the morning after the diagnostic sleep study (2). The FEV1 and the FVC were determined using spirometry. Functional residual capacity (FRC) was measured using multiple-breath closed-circuit helium dilution, with slow vital capacity and expiratory reserve volume determined at the end of the procedure. Residual volume (RV) was calculated by subtracting the expiratory reserve volume from FRC. Total lung capacity could then be found by adding slow vital capacity to RV.
Polysomnography.
Standard diagnostic techniques were employed to determine baseline sleep characteristics. To monitor and stage sleep, electroencephalograph electrodes were placed at F3-A2, C3-A2, and O1-A2, in addition to submental electromyogram electrodes and left and right electroculogram electrodes. Respiratory events were scored using impedance thoracoabdominal belts to gauge respiratory effort, a nasal cannula was used to quantify airflow, and pulse oximetry was used to monitor hemoglobin oxygen saturation. Apneas were defined as cessation of breathing for 10 s, while hypopneas were scored when a 30% reduction in airflow was accompanied by ≥4% oxygen desaturation. Standard polysomnographic methods and scoring rules were utilized to stage sleep and classify sleep-disordered breathing events, according to the 2007 recommended scoring guidelines set forth by the American Academy of Sleep Medicine (1).
Respiratory physiology setup.
In addition to standard polysomnographic techniques, the subject was fitted with a gel mask attached to a custom designed continuous positive airway pressure unit capable of delivering pressures between −20 and +20 cmH2O (Resmed, Bella Vista, NSW, Australia). Airflow was measured with a pneumotachograph (model 4830, 0-400 l/min; Hans Rudolph, Kansas City, MO). Flow and pressure were acquired at a sampling rate of 100 Hz. All physiological signals were acquired and monitored on a computer station using Somnologica software (Medcare, Buffalo, NY).
Study Protocol
Initially, subjects underwent a diagnostic sleep study for determining severity of sleep-disordered breathing, which was required for matching individuals by AHI and anthropometric data (see Table 1). In addition, pulmonary function tests were performed the day after the sleep study to determine total lung volumes and disease status. We then performed a follow-up sleep study within a 2- to 4-wk period to determine the patients' mechanical upper airway properties (passive Pcrit) and the compensatory timing responses to upper airway obstruction. The procedures for Pcrit determination have been detailed earlier (24) and are illustrated in Fig. 1 and briefly described below.
Fig. 1.

Experimental protocol. Schematic diagram of the experimental protocols (top) and polysomnographic responses (bottom) during non-rapid eye movement sleep in an apneic male subject. Baseline (left): nasal pressure was adjusted to effective continuous positive airway pressure and held for at least 3 min (holding pressure) to establish a stable non-flow-limited breathing pattern (see flow trace, bottom left). Passive upper airway obstruction (middle left): a series of brief (5 breaths) drops in nasal pressure from holding pressure were performed without concomitant activation of EMG. During these drops, upper airway obstruction ensued, as indicated by inspiratory flow limitation as in the flow trace below. SaO2, arterial O2 saturation; Vimax, maximal inspiratory flow.
Passive condition: brief periods of upper airway obstruction.
We used the passive Pcrit to determine upper airway properties under a low, hypotonic neuromuscular activity. During stable non-rapid eye movement sleep, the nasal pressure was incrementally raised until non-flow-limited nasal breathing was achieved. The nasal pressure was then dropped in stepwise fashion in 1- to 2-cmH2O increments, for five breaths at a time, until the subject's airway occluded. A pressure drop would be aborted and repeated if an arousal or an increase in EMG activity without an arousal occurred. Periods of breathing were excluded if there were arousals from stable sleep.
Upper airway collapsibility.
Pressure-flow (maximal inspiratory flow at flow-limited conditions) relationships were generated for each subject by plotting the maximal airflow from breaths 3–5 of each pressure drop, as previously described (30). Linear regression was used to determine the nasal pressure at which there was zero flow, indicating that the upper airway had occluded (passive Pcrit), as demonstrated in Fig. 1.
Respiratory timing responses.
Ti, Te, the total time of each breath (Ttot) and respiratory rate were all determined. Inspiratory duty cycle (Ti/Ttot) was reported in the non-flow-limited condition, as well as during breaths with acute moderate to severe flow limitation, defined as a mean peak inspiratory airflow rate of 50–150 ml/s (Fig. 1) (28). The change in timing parameters from baseline to upper airway obstruction condition was used to determine individual strength in respiratory timing responses to upper airway obstruction.
Statistical Analysis
Individual values were averaged and compared for both the COPD and control groups using Student's unpaired t-test. Further subgroup analysis was performed in those with and without OSA. The changes in duty cycle response were compared between COPD and control subject groups with unpaired t-test. Comparisons pre- and postinduction of inspiratory flow limitation were performed using a paired t-test. Regression analysis between FRC and passive Pcrit was performed to determine the association between lung volume and upper airway collapsibility. A P value of 0.05 was considered statistically significant. All data are presented as means ± SE, unless otherwise stated.
RESULTS
Subject Characteristics
Demographic, anthropometric, polysomnographic, and pulmonary function statistics for all 18 COPD participants and 18 controls matched by the AHI are displayed in Table 1. Comparison of the anthropometric variables for the subjects individually matched for age (57.1 ± 1.9 vs. 50.3 ± 1.6 yr), sex (12 men; 6 women), BMI (28.8 ± 1.3 vs. 31.6 ± 1.6 kg/m2), and AHI (29 ± 8 vs. 29 ± 9 events/h) are shown in Table 1. One-half of the subjects (7 men and 2 women from each group) had sleep-disordered breathing with an event rate >10 events/h. COPD subjects, compared with the control subjects, had an FEV1 of 1.9 ± 0.2 vs. 3.1 ± 0.2 liters, and FEV1/FVC of 54 ± 0.0 vs. 79 ± 1.2%. Similarly, lung volumes were elevated in COPD patients vs. controls: total lung capacity was 6.7 ± 0.2 vs. 5.9 ± 0.3 liters, RV was 3.0 ± 0.2 vs. 1.9 ± 0.1 liters, and FRC was 4.0 ± 0.2 vs. 2.8 ± 0.2 liters.
Acute (Passive) Mechanical Upper Airway Properties
COPD subjects had a mean passive Pcrit of −2.8 cmH2O compared with that of −0.5 cmH2O for their matched controls (P = 0.03; unpaired t-test; Fig. 2). For the subgroup without OSA, COPD subjects had a mean passive Pcrit of −4.6 cmH2O compared with that of −0.8 cmH2O for their matched controls (P = 0.02; unpaired t-test). For the subgroup with OSA, COPD subjects had a mean passive Pcrit of −1.0 cmH2O compared with that of −0.2 cmH2O for their matched controls (P = 0.55). There was a significant association between FRC and passive Pcrit (r2 = 0.27, P = 0.002), as illustrated in Fig. 3, indicating that patients with higher lung volumes had lower passive Pcrit values.
Fig. 2.
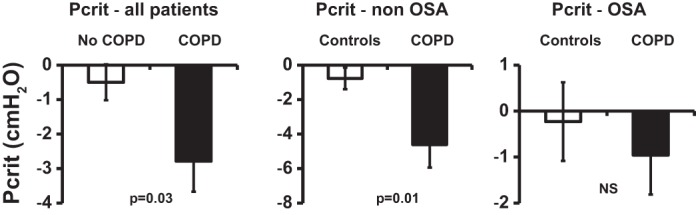
Passive critical closing pressure (Pcrit) measurements in chronic obstructive pulmonary disease (COPD) patients and controls. The graph represents average passive Pcrit values for COPD patients and matched controls. Left: all patients. Middle: non-obstructive sleep apnea (OSA) patients. Right: OSA patients. Values are means ± SE. Upper airway collapsibility (passive Pcrit) is lower in patients with COPD compared with subjects matched for age, sex, body mass index, and respiratory disturbance index (P = 0.03). This difference seems greater in nonapneic patients, but can no longer be noticed in apneic patients. NS, nonsignificant.
Fig. 3.
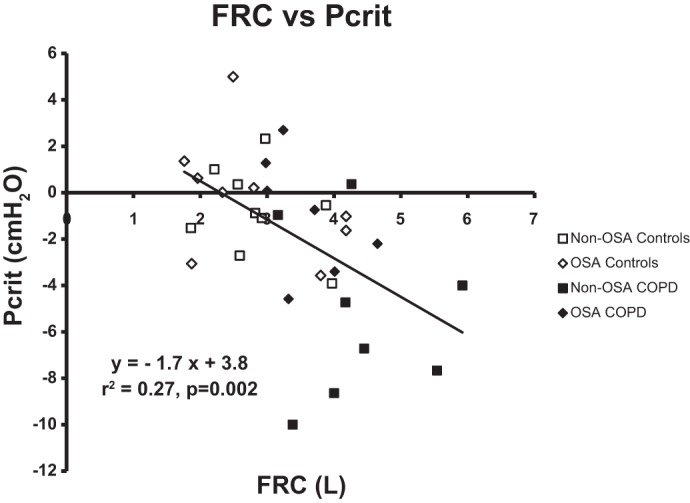
Passive Pcrit vs. functional residual capacity (FRC) relationship. The graph represents passive Pcrit and FRC relationship, with a linear regression using all subject values. □, Non-OSA controls; ◇, OSA controls; ■, non-OSA COPD; ⧫ OSA COPD. There was a statistically significant association between FRC and upper airway collapsibility (passive Pcrit) in all subjects (P = 0.002, r2 = 0.27). Each 1-liter increase in FRC was associated with an ∼2-cmH2O reduction in passive Pcrit, which was similar to the relationship observed previously in normal individuals and patients with sleep apnea (31, 32).
In the univariate analysis, there was statistically significant associations between passive Pcrit values and both FRC (P = 0.002) and BMI (P = 0.003). In the multivariate analysis, both factors remained determinants for passive Pcrit level (FRC, P = 0.021; BMI, P = 0.041; Table 2).
Table 2.
Passive Pcrit relationship with BMI and FRC
|
Model 1 |
Model 2 |
Model 3 |
||||
|---|---|---|---|---|---|---|
| β-coefficient | P value | β-coefficient | P value | β-coefficient | P value | |
| FRC, cmH2O/l | −1.66 ± 0.50 | 0.002 | −1.25 ± 0.51 | 0.021 | ||
| BMI, kg/m2 | 0.25 ± 0.08 | 0.003 | 0.17 ± 0.08 | 0.042 | ||
Values are means ± SE for β-coefficients. Multivariate models of passive critical closing pressure (Pcrit) with BMI and/or FRC as the independent variables are given. The β-coefficient of the passive Pcrit vs. FRC association decreases from −1.66 ± 0.50 to −1.24 ± 0.51 cmH2O/l when BMI is added to the regression model.
Acute Respiratory Timing Responses
In the subgroup of 12 patients paired for the analysis of the timing responses, the control group had an AHI of 29.8 ± 7.5 events/h and the CODP group had an AHI of 31.2 ± 6.8 events/h. The average holding pressure was 8.8 ± 0.8 cmH2O in controls and 8.1 ± 1.0 cmH2O in patients with COPD. The average delta pressure required for inducing severe inspiratory flow limitation was 6.4 ± 0.8 cmH2O for controls and 7.0 ± 0.9 cmH2O for patients with COPD.
Respiratory timing responses were assessed by comparing non-flow-limited condition (baseline) with periods of severe upper airway obstruction. The peak inspiratory airflow at baseline and severe upper airway obstruction were similar in both groups, declining from 383 ± 49 to 105 ± 11 ml/s in controls and from 387 ± 47 to 111 ± 12 ml/s in COPD patients. Respiratory timing parameters during baseline, non-flow-limited breathing, and in response to severe degree of inspiratory flow limitation are summarized in Table 3. In both non-flow-limited and flow-limited conditions, COPD patients and controls had similar timing indexes. While the control group had an ∼25% increase in the duty cycle from 0.41 to 0.51, the COPD patients had an ∼40% increase from 0.40 to 0.55 (P = 0.03 for the comparison on the increase in the duty cycle). Te decreased on average from 2.5 to 1.5 s in the COPD group (P < 0.01) and in the matched controls from 2.4 to 1.8 s (P < 0.01). Ttot decreased in COPD from 4.1 to 3.3 s (P < 0.05) and in the controls from 3.9 to 3.6 s (nonsignificant). Ti increased from 1.5 to 1.8 s (P < 0.01) in patients with COPD and from 1.6 to 1.8 s in controls (P < 0.05). None of the inspiratory or expiratory timing responses were correlated to the level of obstruction (FEV1) in patients with COPD.
Table 3.
Timing responses to induced flow limitation
| Inspiratory Time, s |
Expiratory Time, s |
Total Time, s |
Duty Cycle |
Respiratory Rate, breaths/min |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Control | COPD | Control | COPD | Control | COPD | Control | COPD | Control | COPD | |
| Baseline | 1.6 ± 0.1 | 1.5 ± 0.1 | 2.4 ± 0.2 | 2.5 ± 0.3 | 3.9 ± 0.2 | 4.1 ± 0.3 | 0.41 ± 0.02 | 0.40 ± 0.02 | 16.0 ± 0.9 | 15.5 ± 0.9 |
| IFL | 1.8 ± 0.1* | 1.8 ± 0.1* | 1.8 ± 0.2* | 1.5 ± 0.1* | 3.6 ± 0.3 | 3.3 ± 0.2* | 0.51 ± 0.02* | 0.55 ± 0.02*† | 18.1 ± 1.6 | 18.9 ± 1.1* |
Values are averages ± SE. Timing responses are shown for 12 COPD patients and 12 controls matched by sex, BMI (30.6 ± 1.3 vs. 29.8 ± 2.0 kg/m2), and passive Pcrit (−0.7 vs. −0.7 cmH2O). IFL, inspiratory flow limitation.
P < 0.05, comparison between flow-limited condition and non-flow-limited condition.
P < 0.05, comparison between response to flow limitation in patients with COPD and matched controls.
DISCUSSION
The main findings in the present study are that COPD patients have a less collapsible upper airway than age, sex, BMI, and AHI matched control subjects, and that the passive Pcrit inversely correlates with residual lung volume. Airway patency was increased in patients with COPD compared with matched controls, but this difference was much smaller and nonsignificant in the subgroup of patients with sleep apnea, indicating that sleep apnea disease status influences the mechanisms that mediate the lung volume and passive Pcrit relationship. Second, in contrast to our hypothesis, COPD patients demonstrated increased rather than a decreased duty cycle response to upper airway obstruction compared with matched controls. This rise in duty cycle was associated with an increased respiratory rate and a marked reduction in Te, both of which are known risk factors for dynamic hyperinflation. Taken together, our data suggest that, while elevations in lung volume may lower OSA susceptibility in patients with COPD, timing responses to upper airway obstruction may promote dynamic hyperinflation.
One of the main clinical features of COPD is a progressive elevation of lung volume. Our patient population had a moderate degree of COPD with FEV1/FVC ranging from 50–70% predicted and a mean increase in FRC of 1.2 liters compared with controls. Increases in lung volume decreases upper airway collapsibility in normal individuals and patients with OSA (23, 31, 32). The proposed mechanism for this association is an increase in caudal tracheal traction during inflation of the lungs producing a stiffer, less collapsible upper airway (16). We have now extended these findings to COPD patients with increased lung volumes and found a similar relationship between FRC and upper collapsibility of ∼2 cmH2O decrease in passive Pcrit per liter increase in FRC. A lower passive Pcrit is associated with a lower risk for developing OSA. Thus it appears that COPD patients with increased lung volumes have lower susceptibility to develop OSA.
We observed a lower passive Pcrit in nonapneic COPD patients compared with their matched nonapneic controls. One could suspect this difference to be due to an abnormally high passive Pcrit in the control group and not due to a decrease in passive Pcrit in the COPD group. Our laboratory has previously demonstrated that the passive Pcrit can be similar in non-OSA and OSA individuals, while the active Pcrit is lower in normal individuals compared with apneic patients, even when matched by passive Pcrit (19, 25). Passive Pcrit reveals mechanical airway properties in a nonneural activated state and approaches the Pcrit measurements under anesthesia. In fact, some reports during anesthesia have also shown Pcrit values close to atmospheric, particularly when studying older patients (7, 8). We, therefore, suggest that the difference in passive Pcrit between nonapneic COPD patients and controls is not due to an abnormally high Pcrit in the normal controls, but rather due to a decreased passive Pcrit in nonapneic COPD patients.
Although there was a difference in passive Pcrit between nonapneic COPD patients and their matched controls, we no longer observed this difference when comparing apneic COPD patients to their controls. We propose several mechanisms to explain this differential response. First, several investigators demonstrated that passive Pcrit decreases with elevations in lung volume, which is attributed to a caudal traction of the upper airway (31, 33, 35). Apneic individuals have larger tongues (29) and often narrow, high arched hard palate (17), both of which may have blunted the stretching effect provided by increases in lung volumes. Second, it is also possible that elevations in lung volume lower passive Pcrit through neural stimulation of intra- or extrapulmonary stretch receptors. Apneic individuals may have an attenuated neural response to elevations in lung volume similar to a diminished neural control of the upper airway in response to upper airway obstruction (22, 25). Alternatively, similar to the Hering Breuer reflex, which requires large inhaled volumes for eliciting the reflex response, passive Pcrit responses may also be dependent on the degree of hyperinflation. In some apneic COPD patients, increases in lung volumes may not have been large enough to reduce passive Pcrit. To determine why nonapneic COPD patients have a lower passive Pcrit compared with controls, while apneic COPD patients have similar passive Pcrit than apneic controls, mechanistic experiments involving neural and muscular activity assessment are required.
In the presence of upper airway obstruction, increasing the duty cycle is one of the main responses to sustain ventilation. The larger the increase in the duty cycle, the better individuals maintain their minute ventilation (5, 28, 36). In our present work, we observed that patients with COPD, compared with matched controls, have a larger duty cycle increase when challenged by upper airway obstruction. Thus COPD patients appear to be better protected from developing hypoventilation compared with matched controls when facing upper airway obstruction. In contrast, patients with COPD need very long Te to prevent dynamic lung hyperinflation. In the present work, we observed that COPD patients, as well as controls, decreased Te when challenged by inspiratory flow limitation. While mild reductions in Te usually do not affect breathing mechanics, the observed ∼50% reduction in Te may be enough to elicit dynamic hyperinflation. In fact, a similar reduction of Te by using a metronome during wakefulness induced dynamic hyperinflation in patients with moderate COPD (4). Subjects with normal lung function might well tolerate the Te reduction. However, in subjects with COPD inspiratory flow limitation, the observed reduction in Te may produce dynamic hyperinflation during sleep.
There are several strengths and limitations to the present study. The first strength is our approach for matching COPD patients to controls for age, sleep apnea disease severity (AHI), sex, and BMI, which are known risk factors for upper airway collapsibility. When we analyzed only the patients without OSA, the differences in upper airway collapsibility are even greater than for the whole group comparison, suggesting that we may have underestimated the effect of elevated lung volume on upper airway patency. The second strength is our experimental design for the determination of the timing parameters in response to upper airway obstruction. In previous studies, our laboratory has demonstrated that duty cycle increases with the severity of upper airway obstruction in a dose-dependent fashion. We, therefore, imposed similar severity of upper airway obstruction across our study participants. This experimental design allowed us to reduce the variability in timing responses while observing the differential responses between COPD patients and controls. In contrast, despite best matching of COPD patients and controls by disease status (AHI), age, and sex, we have only cross-sectional data on passive Pcrit in our study participants. It is possible that we may have underestimated the effect of lung volume on upper airway collapsibility due to a selection bias. We also did not measure intrinsic positive end-expiratory pressure, which is probably present in many of our COPD patients and could be altered by the experimentally induced flow limitation. We acknowledge that our COPD patients were significantly older than controls. Increased age is associated with either no change or increase in passive Pcrit (19). We, therefore, believe that the age difference cannot account for the lower passive Pcrit in the COPD group. Finally, it is possible that we did not achieve a similar neuromuscular passivity in the apneic controls vs. apneic COPD individuals. Patients with COPD have higher respiratory loads and, consequently, higher effort, which can translate into stronger neurological input to both respiratory and upper airway muscles. Although passive Pcrit measurements are designed to describe airway properties in a passive state, we cannot exclude that some of the observed upper airway differences reflect different upper airway muscle activation states.
Implications
The present work has several clinical implications. First, we show that elevations in lung volume are associated with a lower Pcrit (passive Pcrit) of the upper airway. Similarly, changes in lung volume may also affect passive Pcrit in COPD patients. Lung volume reduction surgery (LVRS) is one treatment strategy for severe COPD. If LVRS would increase passive Pcrit, treatment efficacy of LVRS may be impeded by the emergence of OSA. Second, in COPD patients, brief periods of severe upper airway obstruction elicited marked reductions in Te to levels comparable to findings of previous studies examining sources of dynamic hyperinflation. Dynamic hyperinflation is known to increase work of breathing and reduce sleep efficiency, both of which could be attributed to the morning fatigue commonly observed in COPD patients.
GRANTS
This study was funded by National Heart, Lung, and Blood Institute Grant RO1-HL-105546 and the Conselho Nacional de Desenvolvimento Científico e Tecnológico-Brasil.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: P.J.C.B., P.R.G., J.P.K., S.P.P., P.L.S., A.R.S., and H.S. analyzed data; P.J.C.B., P.R.G., J.P.K., S.P.P., P.L.S., A.R.S., and H.S. interpreted results of experiments; P.J.C.B., P.R.G., and H.S. prepared figures; P.J.C.B., P.R.G., A.R.S., and H.S. drafted manuscript; P.J.C.B., J.P.K., S.P.P., P.L.S., A.R.S., and H.S. edited and revised manuscript; P.J.C.B. and H.S. approved final version of manuscript; P.R.G., J.P.K., S.P.P., P.L.S., A.R.S., and H.S. conception and design of research; P.R.G., J.P.K., and H.S. performed experiments.
REFERENCES
- 1.American Academy of Sleep Medicine. The AASM Manual for the Scoring of Sleep and Associated Events: Rules, Terminology and Technical Specification. Westchester, IL: American Academy of Sleep Medicine, 2007. [PMC free article] [PubMed] [Google Scholar]
- 2.American Thoracic Society. Standardization of spirometry, 1994 update. American Thoracic Society. Am J Respir Crit Care Med 152: 1107–1136, 1995. [DOI] [PubMed] [Google Scholar]
- 3.Breslin E, van der Schans C, Breukink S, Meek P, Mercer K, Volz W, Louie S. Perception of fatigue and quality of life in patients with COPD. Chest 114: 958–964, 1998. [DOI] [PubMed] [Google Scholar]
- 4.Calligaro GL, Raine RI, Bateman ME, Bateman ED, Cooper CB. Comparing dynamic hyperinflation and associated dyspnea induced by metronome-paced tachypnea versus incremental exercise. COPD 11: 105–112, 2014. [DOI] [PubMed] [Google Scholar]
- 5.Chin CH, Kirkness JP, Patil SP, McGinley BM, Smith PL, Schwartz AR, Schneider H. Compensatory responses to upper airway obstruction in obese apneic men and women. J Appl Physiol 112: 403–410, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Douglas NJ, Calverley PMA, Leggett RJE, Brash DC, Flenley DC, Brezinova V. Transient hypoxaemia during sleep in chronic bronchitis and emphysema. Lancet 1: 1–4, 1979. [DOI] [PubMed] [Google Scholar]
- 7.Eastwood PR, Platt PR, Shepherd K, Maddison K, Hillman DR. Collapsibility of the upper airway at different concentrations of propofol anesthesia. Anesthesiology 103: 470–477, 2005. [DOI] [PubMed] [Google Scholar]
- 8.Eastwood PR, Szollosi I, Platt PR, Hillman DR. Comparison of upper airway collapse during general anesthesia and sleep. Lancet 359: 1207–1209, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Gold AR, Gold MS, Harris KW, Espeleta VJ, Amin MM, Broderick JE. Hypersomnolence, insomnia and the pathophysiology of upper airway resistance syndrome. Sleep Med 9: 675–683, 2008. [DOI] [PubMed] [Google Scholar]
- 10.Gold AR, Schwartz AR. The pharyngeal critical pressure. The whys and hows of using nasal continuous positive airway pressure diagnostically. Chest 110: 1077–1088, 1996. [DOI] [PubMed] [Google Scholar]
- 11.Gorini M, Misuri G, Corrado A, Duranti R, Iandelli I, De Paola E, Scano G. Breathing pattern and carbon dioxide retention in severe chronic obstructive pulmonary disease. Thorax 51: 677–683, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guilleminault C, Stoohs R, Clerk A, Cetel M, Maistros P. A cause of excessive daytime sleepiness. The upper airway resistance syndrome. Chest 104: 781–787, 1993. [DOI] [PubMed] [Google Scholar]
- 13.Haluszka J, Chartrand DA, Grassino AE, Milic-Emili J. Intrinsic PEEP and arterial Pco2 in stable patients with chronic obstructive pulmonary disease. Am Rev Respir Dis 141: 1194–1197, 1990. [DOI] [PubMed] [Google Scholar]
- 14.Hoshino Y, Ayuse T, Kurata S, Ayuse T, Schneider H, Kirkness JP, Patil SP, Schwartz AR, Oi K. The compensatory responses to upper airway obstruction in normal subjects under propofol anesthesia. Respir Physiol Neurobiol 166: 24–31, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hudgel DW, Martin RJ, Capehart M, Johnson B, Hill P. Contribution of hypoventilation to sleep oxygen desaturation in chronic obstructive pulmonary disease. J Appl Physiol 55: 669–677, 1983. [DOI] [PubMed] [Google Scholar]
- 16.Kairaitis K, Byth K, Parikh R, Stavrinou R, Wheatley JR, Amis TC. Tracheal traction effects on upper airway patency in rabbits: the role of tissue pressure. Sleep 30: 179–186, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Kim JH, Guilleminault C. The nasomaxillary complex, the mandibule, and sleep-disordered breathing. Sleep Breath 15: 185–193, 2011. [DOI] [PubMed] [Google Scholar]
- 18.Kinsman RA, Yaroush RA, Fernandez E, Dirks JF, Schocket M, Fukuhara J. Symptoms and experiences in chronic bronchitis and emphysema. Chest 83: 755–761, 1983. [DOI] [PubMed] [Google Scholar]
- 19.Kirkness JP, Schwartz AR, Schneider H, Punjabi NM, Maly JJ, Laffan AM, McGinley BM, Magnuson T, Schweitzer M, Smith PL, Patil SP. Contribution of male sex, age, and obesity to mechanical instability of upper airway during sleep. J Appl Physiol 104: 1618–1624, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krachman SL, Chatila W, Martin UJ, Nugent T, Crocetti J, Gaughan J, Criner GJ. Effects of lung volume reduction surgery on sleep quality and nocturnal gas exchange in patients with severe emphysema. Chest 128: 3221–3228, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Leitch AG, Gloney LJ, Leggett RJE. Arterial blood gas tensions, hydrogen ion, and electroencephalogram during sleep in patients with chronic ventilatory failure. Thorax 31: 730–735, 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGinley BM, Schwartz AR, Schneider H, Kirkness JP, Smith PL, Patil SP. Upper airway neuromuscular compensation during sleep is defective in obstructive sleep apnea. J Appl Physiol 105: 197–205, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Owens RL, Malhotra A, Eckert DJ, White DP, Jordan AS. The influence of end-expiratory lung volume on measurements of pharyngeal collapsibility. J Appl Physiol 108: 455–451, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patil SP, Punjabi NM, Schneider H, O'Donnell CP, Smith PL, Schwartz AR. A simplified method for measuring critical pressures during sleep in the clinical setting. Am J Respir Crit Care Med 170: 86–93, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Patil SP, Schneider H, Marx JJ, Gladmon E, Schwartz AR, Smith PL. Neuromechanical control of upper airway patency during sleep. J Appl Physiol 102: 547–556, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Powers CR, Frey WC. Maintenance of wakefulness test in military personnel with upper airway resistance syndrome and mild to moderate sleep apnea. Sleep Breath 13: 253–258, 2009. [DOI] [PubMed] [Google Scholar]
- 27.Sanders MH, Newman AB, Haggerty CL, Redline S, Lebowitz M, Samet J, O'Connor GT, Punjabi NM, Shahar E. Sleep and Sleep-disordered breathing in adults with predominantly mild obstructive airway disease. Am J Respir Crit Care Med 167: 7–14, 2003. [DOI] [PubMed] [Google Scholar]
- 28.Schneider H, Krishnan V, Pichard LE, Patil SP, Smith PL, Schwartz AR. Inspiratory duty cycle responses to flow limitation predict nocturnal hypoventilation. Eur Respir J 33: 1068–1076, 2009. [DOI] [PubMed] [Google Scholar]
- 29.Schwab RJ, Pasirstein M, Pierson R, Mackley A, Hachadoorian R, Arens R, Maislin G, Pack AI. Identification of upper airway anatomic risk factors for obstructive sleep apnea with volumetric magnetic resonance imaging. Am J Respir Crit Care Med 168: 522–530, 2003. [DOI] [PubMed] [Google Scholar]
- 30.Schwartz AR, O'Donnel CP, Baron J, Schubert N, Alam D, Samadi SD, Smith PL. The hypotonic upper airway in obstructive sleep apnea. Role of structures and neuromuscular activity. Am J Respir Crit Care Med 157: 1051–1057, 1998. [DOI] [PubMed] [Google Scholar]
- 31.Squier SB, Patil SP, Schneider H, Kirkness JP, Smith PL, Schwartz AR. Effect of end-expiratory lung volume on upper airway collapsibility in sleeping men and women. J Appl Physiol 109: 977–985, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tagaito Y, Isono S, Remmers JE, Tanaka A, Nishino T. Lung volume and collapsibility of the passive pharynx in patients with sleep-disordered breathing. J Appl Physiol 103: 1379–1385, 2007. [DOI] [PubMed] [Google Scholar]
- 33.Thut DC, Schwartz AR, Roach D, Wise RA, Permutt S, Smith PL. Tracheal and neck position influence upper airway airflow dynamics by altering airway length. J Appl Physiol 75: 2084–2090, 1993. [DOI] [PubMed] [Google Scholar]
- 34.Trask CH, Gress EM. Oximeter studies on patients with chronic pulmonary emphysema, awake and asleep. N Engl J Med 266: 639–642, 1962. [DOI] [PubMed] [Google Scholar]
- 35.Van de Graaff WB. Thoracic traction on the trachea: mechanisms and magnitude. J Appl Physiol 70: 1328–1336, 1991. [DOI] [PubMed] [Google Scholar]
- 36.Younes M. Role of respiratory control mechanisms in the pathogenesis of obstructive sleep disorders. J Appl Physiol 105: 1389–1405, 2008. [DOI] [PubMed] [Google Scholar]