

ABSTRACT
RNA interference (RNAi) screens intended to identify host factors that restrict virus replication may fail if the virus already counteracts host defense mechanisms. To overcome this limitation, we are investigating the use of viral host range mutants that exhibit impaired replication in nonpermissive cells. A vaccinia virus (VACV) mutant with a deletion of both the C7L and K1L genes, K1L−C7L−, which abrogates replication in human cells at a step prior to late gene expression, was chosen for this strategy. We carried out a human genome-wide small interfering RNA (siRNA) screen in HeLa cells infected with a VACV K1L−C7L− mutant that expresses the green fluorescent protein regulated by a late promoter. This positive-selection screen had remarkably low background levels and resulted in the identification of a few cellular genes, notably SAMD9 and WDR6, from approximately 20,000 tested that dramatically enhanced green fluorescent protein expression. Replication of the mutant virus was enabled by multiple siRNAs to SAMD9 or WDR6. Moreover, SAMD9 and WDR6 clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 knockout HeLa cell lines were permissive for replication of the K1L−C7L− mutant, in agreement with the siRNA data. Expression of exogenous SAMD9 or interferon regulatory factor 1 restricted replication of the K1L−C7L− mutant in the SAMD9−/− cells. Independent interactions of SAMD9 with the K1 and C7 proteins were suggested by immunoprecipitation. Knockout of WDR6 did not reduce the levels of SAMD9 and interactions of WDR6 with SAMD9, C7, and K1 proteins were not detected, suggesting that these restriction factors act independently but possibly in the same innate defense pathway.
IMPORTANCE
The coevolution of microbial pathogens with cells has led to an arms race in which the invader and host continuously struggle to gain the advantage. For this reason, traditional siRNA screens may fail to uncover important immune mechanisms if the pathogens have already developed effective responses. However, host-restricted viral mutants have lost one or more defense genes needed for their replication in nonpermissive cells. By screening human genome libraries of short RNAs that inhibit the expression of individual host genes in nonpermissive cells, we identified SAMD9 and WDR6 as major restriction factors that prevented replication of a vaccinia virus mutant and suggest that host range screening can be generally useful for the investigation of host-pathogen interactions.
INTRODUCTION
The coevolution of microbial pathogens with cells has led to an arms race in which the invader and host continuously struggle to gain the advantage. In principle, human genome-wide small interfering RNA (siRNA) screening of infected cells has the potential to reveal novel immune mechanisms. However, knocking down expression of a host defense gene may have little effect if the pathogen has already developed an effective counterresponse. Theoretically, this limitation could be overcome by using a microbial mutant that has lost the ability to effectively respond to a specific immune mechanism. Since cells vary in the extent to which they express innate defenses, such microbial mutants often exhibit a host range phenotype. Consequently, one strategy would be to screen siRNA libraries in nonpermissive cells infected with host range mutants and monitor rescue of infection. An attractive feature of such a screen is that knocking down mRNA expression would enable replication of the mutant and therefore elicit a positive response, which is likely to minimize nonrelevant indirect effects. The present study demonstrates the power of this approach using a poxvirus host range mutant.
Poxviruses are large DNA viruses that reproduce in the cytoplasm and encode numerous proteins involved in host interactions and replicative functions (1). The best known poxvirus species belong to the orthopoxvirus genus and include variola virus, the vanquished agent of smallpox; vaccinia virus (VACV), the live vaccine that eradicated smallpox; monkeypox virus, the cause of a smallpox-like zoonosis; and cowpox virus, the agent of a zoonosis causing mainly localized skin lesions. Approximately half of the 200 genes of VACV, the most intensively studied orthopoxvirus, are conserved in all chordopoxviruses (2) and most of these genes are essential for replication. The remaining genes are mainly involved in virus-cell interactions, and some determine host range and virulence (3, 4). Although host range defects may be associated with loss of a single gene, the loss of both C7L and K1L is necessary to restrict VACV replication in mammalian cell lines (5–7). The requirement for both C7L and K1L is intriguing, because these two complementary genes are unrelated in sequence. Moreover, a third unrelated gene from cowpox virus that is absent from VACV is also able to functionally complement the absence of C7L and K1L genes (6). Without these host range genes, the replication block is manifested at the level of viral gene expression (8–12).
One or more homologs of the C7 protein are encoded by most poxvirus genera, although they are only 20 to 30% identical and share no recognizable motif (13). Myxoma virus carries genes encoding three tandem C7 homologs, but only MO62 could substitute for VACV C7 in overcoming host range restriction (14). Further studies indicated that the MO62 protein binds to the host SAMD9 (sterile alpha motif domain-containing 9) protein and that MO63, a second myxoma C7 homolog, facilitates the latter interaction (15). Importantly, knockdown of SAMD9 expression partially relieved the host range restriction of the MO62 null mutant. However, the VACV C7 protein was reported not to bind SAMD9 in the context of a myxoma virus infection, leaving open the cellular antagonist of C7 (13, 15). The K1 protein, which is encoded by some but not all members of the orthopoxvirus genus, has no nonpoxvirus homologs and is comprised almost entirely of ankyrin repeats that are important for function (16–18). K1 has been reported to inhibit NF-κB activation by preventing IκBα degradation (19) and to inhibit protein kinase R phosphorylation (20). A common property of C7 and K1 proteins is their ability to antagonize antiviral activities induced by type 1 interferon and interferon-regulated factor 1 (IRF1) (21). Nevertheless, the K1L−C7L− mutant is unable to replicate in IRF1-negative mouse embryo fibroblasts, suggesting that the latter is not the primary restriction factor in mouse cells.
We considered it likely that VACV C7 and K1 target the same host defense pathway and decided to conduct a human genome-wide siRNA screen to rescue the replication defect. In this communication, we report our finding that both SAMD9 and WDR6 inhibit replication of the VACV K1L−C7L− mutant specifically. Inhibition of the K1L−C7L− mutant by SAMD9 has also been reported by Liu and McFadden (22).
RESULTS
Genes that restrict the VACV K1L−C7L− host range mutant identified by a human genome-wide siRNA screen.
We employed the VACV mutant vK1L−C7L−/GFP+ with a deletion of the K1L gene and replacement of the C7L gene with an open reading frame (ORF) encoding the green fluorescent protein (GFP) regulated by a VACV late promoter (7). GFP fluorescence was a sensitive and appropriate readout, since late genes are not expressed in nonpermissive cells. The ability of vK1L−C7L−/GFP+ to replicate in African green monkey BS-C-1 cells, but not in human HeLa cells, is shown by plaque formation in Fig. 1A. To prepare for the siRNA screen, we calibrated conditions by parallel infections of permissive BS-C-1 and restrictive HeLa cell lines in a 384-well plate with serial dilution of the mutant virus. GFP-positive (GFP+) cells were scored using automated fluorescence microscopy. Even at the highest multiplicity of infection, there were few GFP+ HeLa cells (Fig. 1B). We empirically chose to use a multiplicity of 0.1 PFU of the mutant virus per cell, which resulted in the spread of virus to ~50% of the permissive cells after 18 h but close to zero in the nonpermissive cells (Fig. 1B). These conditions provided an extremely robust screen capable of detecting even minute changes in virus replication.
FIG 1 .

Rescue of vK1L−C7L−/GFP+ by siRNAs. (A) Replication of vK1L−C7L−/GFP+ in monkey BS-C-1 cells, but not in human HeLa cells. Cells were infected with wild-type VACV expressing GFP (vWT) or vK1L−C7L−/GFP+ and incubated for 48 h at 37°C with a methylcellulose overlay. The images were taken with a fluorescence microscope. (B) Effect of virus multiplicity on virus spread. HeLa and BS-C-1 cells were seeded into 384-well plates, and 48 wells of each cell type were infected with each serial dilution of vK1L−C7L−/GFP+. After 18 h, the cells were fixed with 2% paraformaldehyde. The nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) and imaged by automated fluorescence microscopy. GFP-positive cells were scored and plotted against a logarithmic scale of the multiplicity of infection (MOI). Values are means ± standard deviations (error bars). Values that are significantly different (P ≤ 0.0001) as calculated by two-way ANOVA and Bonferroni test after ANOVA comparing each dilution in permissive versus nonpermissive cells are indicated by an asterisk. Calculations were made using PRISM by GraphPad. (C) SAMD9 siRNAs restore replication of vK1L−C7L−/GFP+ in HeLa cells. Images of the GFP channel (top panels) and DAPI stain (bottom panels) were taken from the genome-wide siRNA screens. Three separate SAMD9 siRNAs and a control siRNA (siControl) were tested. The percentages of GFP-positive cells are indicated.
The primary high-throughput screen was performed with the Silencer Select siRNA library from Ambion, which consists of three different siRNAs targeting each of 21,566 human genes in individual wells. A secondary high-throughput siRNA screen was carried out with the OnTargetPlus genome-wide siRNA library from Dharmacon, which consists of four pooled siRNAs to each of 17,320 human genes. The complete data sets are provided in Tables S1.1 to S1.3 and S2 in the supplemental material. The redundant siRNA analysis tool (RSA) was used to minimize the impact of off-target activities (23). Table 1 shows the 30 RSA top-ranked siRNA targets. By far, the strongest siRNA hit was to SAMD9, which increased the number of GFP+ cells to 27 to 46% for each of the three siRNAs in the primary screen (Fig. 1C and Table 1) and was also the strongest hit in the secondary screen (Table 1). WDR6 and FTSJ1 were ranked number 2 and 3, respectively, by RSA and were the only other hits in which the GFP-positive cells exceeded 3% for each of the Ambion siRNAs and the pooled siRNAs (Table 1). SAMD9, WDR6, FTSJ1, and one lower ranked target, CDC37, were selected for additional specific testing. None of these prioritized targets were significant hits in a previous human genome-wide screen carried out with wild-type vaccinia virus (24).
TABLE 1 .
Priority hits of primary and secondary human genome-wide siRNA screensa
| Gene symbol | GeneID | % GFP-positive cells |
No. of siRNAs with >3% GFP+ cells | RSA logP score |
|||
|---|---|---|---|---|---|---|---|
| Ambion_1 | Ambion_2 | Ambion_3 | Dharmacon | ||||
| SAMD9 | 54809 | 46.49 | 34.10 | 26.98 | 16.10 | 4 | −12.7 |
| WDR6 | 11180 | 10.44 | 5.31 | 4.43 | 4.40 | 4 | −7.6 |
| FTSJ1 | 24140 | 5.31 | 3.77 | 3.41 | 6.18 | 4 | −6.7 |
| MAPK14 | 1432 | 3.68 | 3.54 | 3.22 | 0.29 | 3 | −6.5 |
| CDC37 | 11140 | 7.87 | 5.03 | 2.97 | 1.67 | 2 | −6.3 |
| GNPDA2 | 132789 | 5.68 | 5.15 | 2.97 | 0.36 | 2 | −6.3 |
| DPAGT1 | 1798 | 7.53 | 5.71 | 0.61 | 0.00 | 2 | −5.6 |
| CRHBP | 1393 | 10.15 | 3.36 | 2.38 | 0.38 | 2 | −5.3 |
| IARS | 3376 | 7.66 | 3.68 | 2.19 | 0.06 | 2 | −5.2 |
| ALPK2 | 115701 | 4.72 | 3.33 | 2.11 | 0.48 | 2 | −5.2 |
| SORCS3 | 22986 | 12.66 | 4.87 | 2.08 | NA | 2 | −5.1 |
| PPP1R11 | 6992 | 5.95 | 3.68 | 2.04 | 0.28 | 2 | −5.1 |
| DPAGT1 | 1798 | 7.53 | 5.71 | 0.61 | 0.00 | 2 | −4.7 |
| CKAP5 | 9793 | 4.80 | 4.80 | 0.00 | 0.00 | 2 | −4.6 |
| CDS1 | 1040 | 9.93 | 4.53 | 0.76 | 0.12 | 2 | −4.5 |
| PAQR5 | 54852 | 9.48 | 4.33 | 1.23 | 0.28 | 2 | −4.5 |
| PDCL | 5082 | 4.19 | 4.18 | 1.64 | 0.89 | 2 | −4.5 |
| ARPC1A | 10552 | 4.79 | 3.34 | 1.61 | 0.00 | 2 | −4.4 |
| UTP11L | 51118 | 5.66 | 4.09 | 0.68 | 0.56 | 2 | −4.3 |
| PPP3CB | 5532 | 8.97 | 3.87 | 0.19 | 0.39 | 2 | −4.3 |
| PDE4B | 5142 | 6.47 | 3.84 | 0.94 | 0.10 | 2 | −4.1 |
| AMACR | 23600 | 3.97 | 3.63 | 0.53 | 0.35 | 2 | −4.1 |
| AURKB | 9212 | 6.93 | 3.57 | 0.20 | 0.00 | 2 | −4.0 |
| PROCA1 | 147011 | 4.07 | 3.45 | 0.65 | 0.16 | 2 | −3.9 |
| PTPN11 | 5781 | 3.48 | 3.26 | 1.03 | 0.08 | 2 | −3.9 |
| POLD2 | 5425 | 3.41 | 3.24 | 0.48 | 0.03 | 2 | −3.9 |
| KLK7 | 5650 | 6.55 | 3.12 | 0.60 | 0.00 | 2 | −3.8 |
| MSTN | 2660 | 12.83 | 3.02 | 0.12 | 0.13 | 2 | −3.7 |
| ABHD4 | 63874 | 6.70 | 3.01 | 0.33 | 0.05 | 2 | −2.5 |
| KIF11 | 3832 | 5.03 | 1.63 | 0.62 | 6.28 | 2 | −2.5 |
The primary screen was performed with three individual siRNAs targeting each gene from the Ambion Silencer Select siRNA library. The secondary screen was performed with four pooled siRNAs targeting each gene from the Dharmacon OnTargetPlus siRNA library. The genes are ordered according to their RSA logP scores.
Ambion siRNAs targeting the selected genes and control siRNA were transfected individually into HeLa cells, which were subsequently infected with 0.01 PFU of vK1L−C7L−/GFP+ per cell and analyzed for GFP fluorescence by flow cytometry. The siRNAs to WDR6, although less effective than the siRNA to SAMD9, allowed the host range mutant to spread to about 15% of the cells (Fig. 2A). While still lower in efficacy, the siRNAs to FTJ1 and CDC37 increased spread above that of the control siRNA with a P value of <0.01 for the former (Fig. 2A). In the present study, we focused on SAMD9 and to a lesser extent on WDR6 as major human cell restriction factors for the K1L−C7L− host range mutant.
FIG 2 .
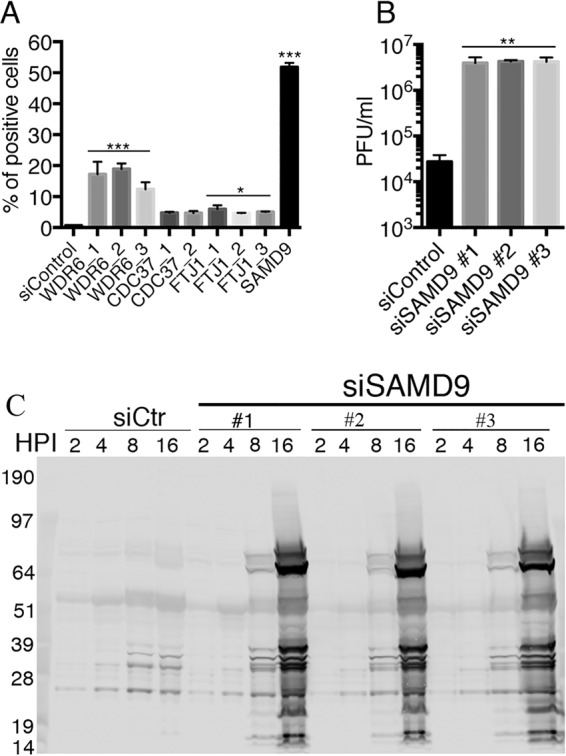
Validation of SAMD9 as a major host restriction factor for vK1L−C7L−/GFP+. (A) Comparison of positive-hit siRNAs. The indicated siRNAs were individually transfected into HeLa cells in a 24-well format. After 48 h, the cells were infected with 0.01 PFU of vK1L−C7L−/GFP+ per cell and incubated for 18 h. GFP-positive cells were scored by flow cytometry. Data from three experiments each carried out in triplicate were combined. Values are means plus standard deviation (error bars). Values that are significantly different from the siControl value calculated by Bonferroni test after the one-way ANOVA test using PRISM GraphPad software are indicated by asterisks as follows: ***, P ≤ 0.001; *, P ≤ 0.05. (B) Replication of vK1L−C7L−/GFP+ in HeLa cells transfected with SAMD9 siRNA. Three different SAMD9 siRNAs and control siRNA were transfected into HeLa cells in a 24-well format. After 48 h, the cells were infected with 0.01 PFU of vK1L−C7L−/GFP+ and incubated for 24 h. Cells were lysed by freezing and thawing, and infectious virus titers were determined by plaque assay on permissive BS-C-1 cells. Data from two experiments each carried out in triplicate were combined. Values are means plus standard deviation (error bars). Values that are significantly different (P ≤ 0.005) from the siControl value calculated by Bonferroni test after the one-way ANOVA test using PRISM GraphPad software are indicated (**). (C) Synthesis of viral proteins in HeLa cells transfected with SAMD9 siRNA and infected with vK1L−C7L−/GFP+. HeLa cells were transfected with control (siCtr) or three different SAMD9-specific siRNAs for 48 h and then infected with 3 PFU of vK1L−C7L−/GFP+ per cell for the indicated hours postinfection (HPI). The cells were lysed, and the proteins were resolved by SDS-PAGE and Western blotting with broadly reactive antibodies to VACV proteins. The electrophoretic positions of molecular mass markers (in kilodaltons) are indicated to the left of the gel.
SAMD9 knockdown restores the full replication cycle of the K1L−C7L− mutant.
Analyzing GFP expression in a virus spread assay was a convenient quantitative measure of virus replication. Nevertheless, we wanted to confirm the role of SAMD9 by conventional methods. HeLa cells were transfected with individual siRNAs specific for SAMD9 or with a control siRNA and then infected with 1 PFU of vK1L−C7L−/GFP per cell. After 24 h, the virus yields were determined by plaque assay in permissive BS-C-1 cells. There was a >150-fold increase in virus production after SAMD9 knockdown compared to the control (Fig. 2B). Since replication of the host range mutant is arrested at the stage of viral late protein synthesis, we also analyzed lysates of infected cells by Western blotting using antibody to VACV proteins. Between 8 and 16 h, there was a marked increase in synthesis of the major viral proteins in cells that had been transfected with each SAMD9 siRNA (Fig. 2C).
Interaction of VACV C7 and K1 with SAMD9.
In view of the evidence that SAMD9 is a major restriction factor for replication of the K1L−C7L− mutant in HeLa cells, we designed an experiment to determine whether the two proteins interact directly or indirectly with SAMD9. The parental VACV used to construct vK1L−C7L−/GFP+ was vTF7-3 (25), which carries the gene encoding the bacteriophage T7 RNA polymerase regulated by a VACV early/late promoter and has been used extensively in transfection assays for expression of genes preceded by a T7 promoter. Accordingly, we constructed plasmids with genes encoding V5 and FLAG epitope-tagged C7 and K1 proteins, respectively, with T7 promoters. Both proteins were expressed following transfection of permissive and nonpermissive cells that had been infected with vK1L−C7L−/GFP+ (Fig. 3A). Furthermore, expression of either protein alone rescued replication of the host range mutant, demonstrating their biological activity (Fig. 3B). Next, we carried out binding experiments in HeLa cells infected with vK1L−C7L−/GFP+. Antibodies to V5 and FLAG were used to capture C7 and K1, respectively. Western blotting showed that SAMD9 was pulled down with either viral protein (Fig. 3C).
FIG 3 .
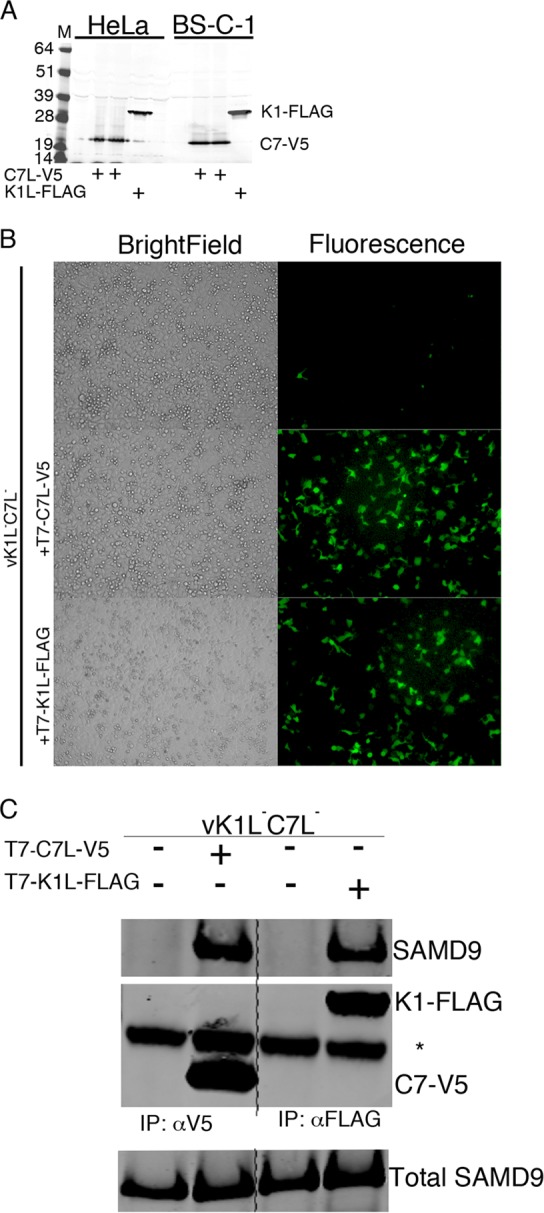
C7L and K1L proteins physically interact with SAMD9. (A) Expression of C7L-V5 and K1L-FLAG following transfection with bacteriophage T7 promoter plasmids. HeLa cells were infected with vK1L−C7L−/GFP+, which expresses the T7 RNA polymerase, and transfected 1 h later with plasmids T7-C7L-V5 and T7-K1L-FLAG encoding C7L-V5 (two clones) or K1L-FLAG regulated by T7 promoters, respectively. After 14 h, the cells were lysed, and their proteins were resolved by SDS-PAGE and Western blotting with antibodies to the V5 and FLAG epitope tags. The positions of molecular mass markers (M) (in kilodaltons) are indicated to the left of the gel. (B) Rescue of vK1L−C7L−/GFP+ by expression of C7 or K1 protein. HeLa cells were infected with vK1L−C7L−/GFP+ and mock transfected (upper panel) or transfected with T7-C7L-V5 or T7-K1L-FLAG for 16 h. Cells were imaged by GFP fluorescence microscopy and bright-field microscopy. (C) Association of C7 and K1 proteins with SAMD9. HeLa cells were infected with vK1L−C7L−/GFP+ and mock transfected (−) or transfected (+) with T7-C7L-V5 or T7-K1L-FLAG for 16 h. The cells were lysed and incubated with antibodies to the V5 or FLAG epitope tag and captured with magnetic beads conjugated to protein G. Input and eluted proteins were resolved by SDS-PAGE following Western blotting with antibodies to endogenous SAMD9 and to V5 and FLAG tags. The position of the heavy chain of the antibody is indicated by an asterisk to the right of the gel. The experiment was repeated three times with similar results. Abbreviations: IP, immunopurification; αV5, antibody to V5 epitope; αFLAG, antibody to FLAG epitope.
The above data could be explained by direct or indirect binding of C7 and K1 proteins to SAMD9. To eliminate the possibility of another viral protein mediating the association, we expressed C7 and K1 with different epitope tags using a cytomegalovirus (CMV) promoter in uninfected HeLa cells. SAMD9 was captured in association with hemagglutinin (HA) epitope-tagged C7 protein (C7-HA) and myc epitope-tagged K1 protein (K1-myc) and detected by Western blotting (Fig. 4A). These data eliminated the possibility that the binding of C7 and K1 to SAMD9 is mediated by another VACV protein but did not exclude the participation of another cellular protein.
FIG 4 .
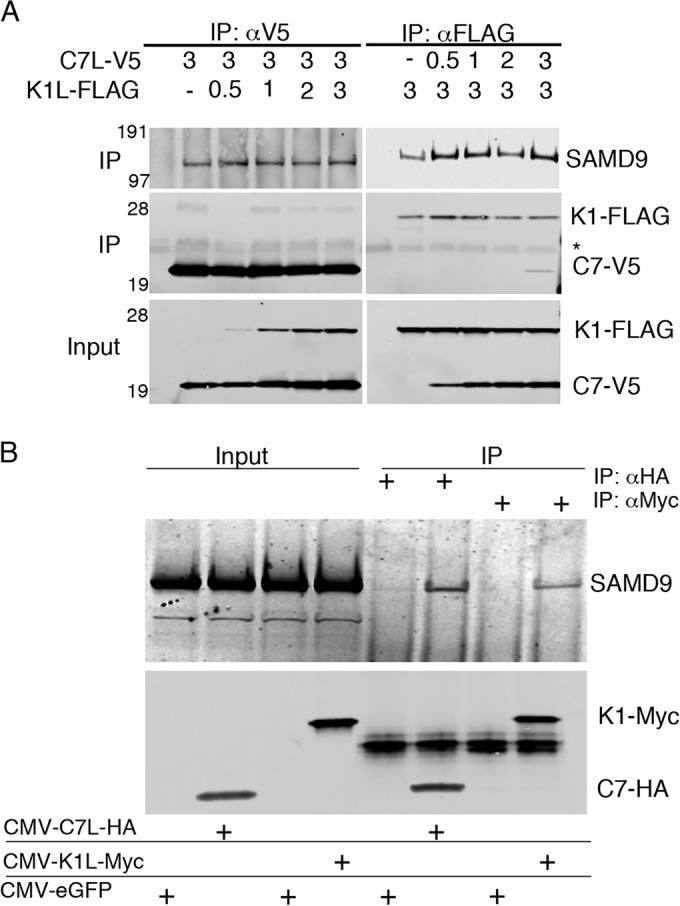
C7L and K1L interact with SAMD9 independently and in the absence of other viral proteins. (A) HeLa cells were infected with vK1L−C7L−/GFP+ and transfected with 3 µg of T7-C7L-V5 and increasing amounts of T7-K1L-FLAG or with 3 µg of T7-K1L-FLAG and increasing amounts of T7-C7L-V5. The amounts of T7-C7L-V5 and T7-K1L-FLAG (in micrograms) are given above the gels (−, none). After 16 h, the cells were lysed and incubated with antibodies for the V5 or FLAG epitope tag. Input and proteins captured by magnetic beads conjugated to protein G were resolved by SDS-PAGE and Western blotting with antibodies to endogenous SAMD9 and V5 or FLAG epitope tag. The positions of mass markers (in kilodaltons) are shown to the left of the gels. The positions of tagged proteins are shown to the right of the gels. The position of the antibody heavy chain is indicated by an asterisk. Abbreviations: IP, immunopurification; αV5, antibody to V5 epitope; αFLAG, antibody to FLAG epitope. (B) Uninfected HeLa cells were transfected with plasmids that express C7L-HA, K1L-myc, or enhanced GFP (eGFP) regulated by CMV promoters. After 24 h, the cells were lysed and incubated with antibodies to the HA or myc epitope tag. Input and eluted proteins were analyzed by SDS-PAGE and Western blotting to detect C7L-HA, K1L-myc, and endogenous SAMD9.
Assuming that C7 and K1 proteins bind directly to SAMD9, they could bind to the same or different sites. We carried out an additional experiment to determine whether binding of SAMD9 to C7 and K1 was enhanced or inhibited by the other. HeLa cells were infected with vK1L−C7L−/GFP+ and transfected with different ratios of the plasmids expressing V5 epitope-tagged C7 protein (C7-V5) and FLAG epitope-tagged K1 protein (K1-FLAG) regulated by T7 promoters. There was no obvious difference in the capture of SAMD9 by C7 in the presence of K1 and vice versa (Fig. 4B). In addition, there was no evidence for interaction of C7-V5 with K1-FLAG.
Replication of the K1L−C7L− mutant in SAMD9 knockout HeLa cells and reversal by expression of exogenous SAMD9 and IRF1.
Our evidence for the major role of SAMD9 in restricting replication of vK1L−C7L−/GFP+ was derived using siRNAs. To further investigate the host range defect of K1L−C7L− mutants, clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 technology was used to disrupt the SAMD9 ORF in HeLa cells. Only a trace of SAMD9 was detected by Western blotting in one of the three clonally derived cell lines tested (Fig. 5A). Using the latter, the replication of vK1L−C7L−/GFP+ was evident from the significant virus spread (Fig. 5B). Moreover, transfection of a SAMD9-expressing plasmid into the SAMD9 knockout cells prevented replication of the host range mutant (Fig. 5B).
FIG 5 .
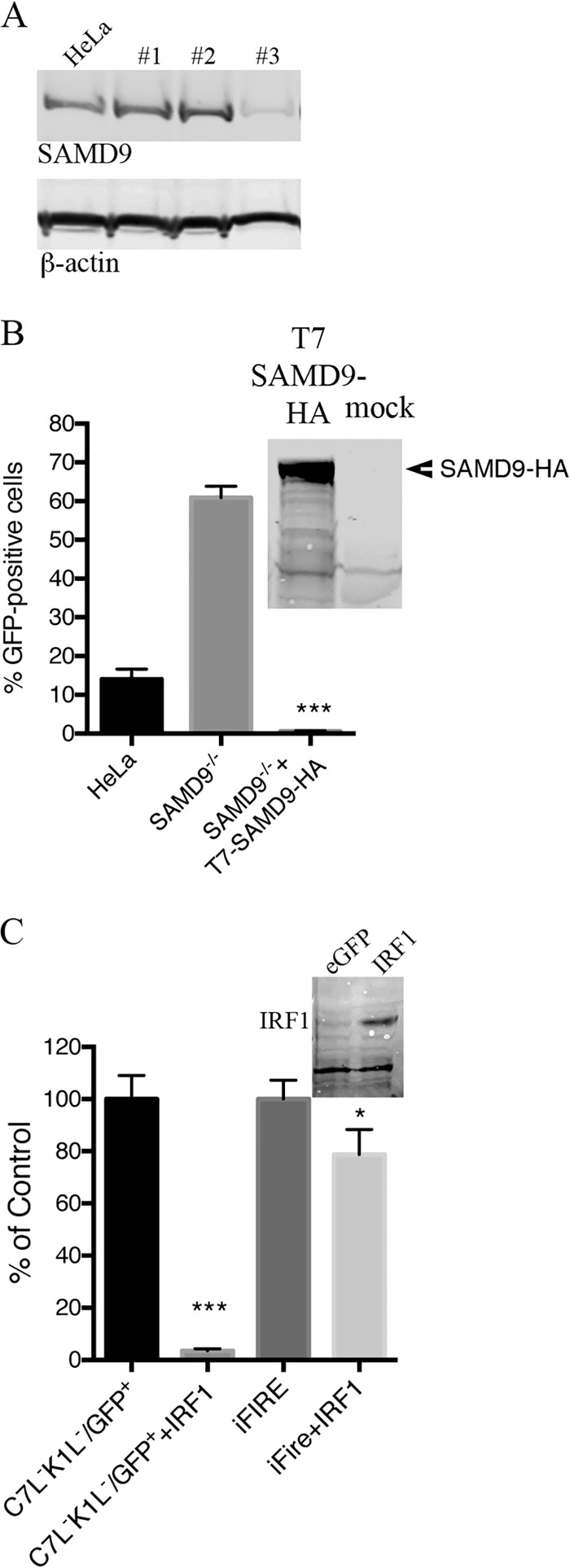
Rescue of vK1L−C7L−/GFP+ by inactivation of SAMD9 and inhibition of replication by IRF1. (A) Generation of SAMD9-deficient cells by CRISPR/CAS9 technology. HeLa cells were transfected with the CRISPR/Cas9 components as described in Materials and Methods. Colonies were lysed, and their proteins were resolved by SDS-PAGE and analyzed by Western blotting to detect endogenous SAMD9 and β-actin as a loading control. Colony 3 was chosen for further experiments and labeled as SAMD9−/− HeLa cells. (B) Functional validation of SAMD9−/− HeLa cells. Normal HeLa cells and SAMD9−/− HeLa cells were infected with 0.01 PFU of vK1L−C7L−/GFP+. One set of infected SAMD9−/− cells were transfected with T7-SAMD9-HA. After 18 h, GFP-positive cells were scored by flow cytometry. (Inset) Western blot demonstrating expression of SAMD9 by T7-SAMD9-HA. Data from two separate experiments each performed in triplicate were combined. Values are means plus standard deviations (error bars). The value that was significantly different (P ≤ 0.001) from the value for the untransfected control, calculated by Bonferroni test after one-way ANOVA using PRISM GraphPad software is indicated (***). (C) Overexpression of IRF1 prevents spread of vK1L−C7L−/GFP+ in SAMD9−/− HeLa cells. SAMD9−/− cells were mock transfected or transfected with plasmid expressing IRF1 regulated by the CMV promoter. At 30 h after transfection, the cells were infected with 0.01 PFU of vK1L−C7L−/GFP+. After an additional 18-h incubation, GFP-positive cells were scored by flow cytometry. (Inset) Western blot showing IRF1 expression. Data from three separate experiments performed in triplicate were combined. Values are means plus standard deviations (error bars). The values that were significantly different calculated by Bonferroni test after one-way ANOVA using PRISM GraphPad software are indicated by asterisks as follows: ***, P ≤ 0.001 relative to the value for C7L−K1L−/GFP+; *, P ≤ 0.05 relative to the value for iFire.
IRF1 induces a large number of interferon-stimulated genes (26) and was shown to inhibit replication of the VACV K1L−C7L− mutant in permissive human Huh7 cells (21). It was therefore of interest to see whether expression of IRF1 would inhibit replication of K1L−C7L− mutant in SAMD9 knockout cells. The latter cells were transfected with an IRF1 expression plasmid and infected with vK1L−C7L−/GFP+ or a control virus (iFire) expressing K1 and C7 proteins as well as GFP and luciferase (not relevant to this study). GFP expression was drastically reduced in the transfected cells that were infected with the K1L−C7L− virus but only modestly reduced in the control virus (Fig. 5C).
Replication of vK1L−C7L−/GFP+ in permissive human Huh-7.5.1 cells.
Meng and coworkers (21) had reported that the replication of the VACV K1L−C7L− mutant was restricted by type 1 interferons in permissive Huh-7 cells. Therefore, we wanted to determine whether these cells normally expressed SAMD9 and whether SAMD9 was induced by beta interferon (IFN-β). In contrast to HeLa cells, SAMD9 was not detected in Huh7.5.1 cells by Western blotting (Fig. 6A and B). However, IFN-β, but not IRF1, increased SAMD9 expression in Huh-7.5.1 cells (Fig. 6A and B). SAMD9 siRNA partially reduced SAMD9 expression induced by interferon (Fig. 6A and B) and enhanced vK1L−C7L−/GFP+ spread in interferon-treated Huh-7.5.1 cells (Fig. 6C). Taken together, these data suggest that insufficient SAMD9 could explain the permissiveness of Huh-7.5.1 cells in the absence of interferon.
FIG 6 .
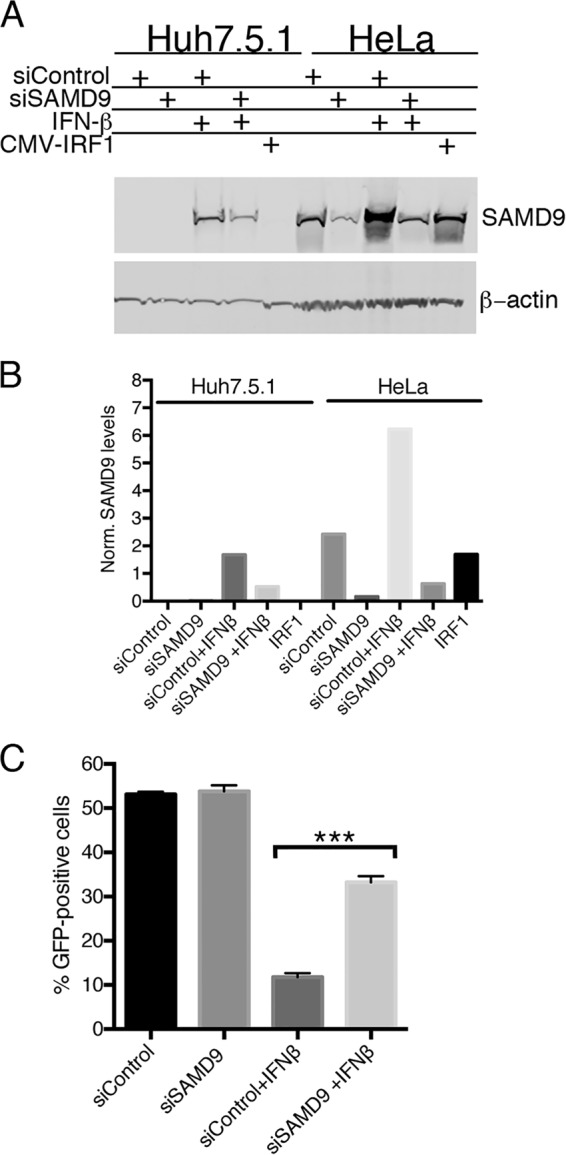
Interferon induces SAMD9 expression in Huh-7.5.1 cells and inhibits replication of vK1L−C7L−/GFP+. (A) Induction of SAMD9 in Huh-7.5.1 cells. Huh-7.5.1 and HeLa cells were transfected with control siRNA or SAMD9 siRNA, and 24 h later, the cells were treated with 200 U/ml of IFN-β or left untreated. After an additional 24 h, the cells were lysed and analyzed by Western blotting with antibodies to SAMD9 and β-actin as a loading control. (B) Quantification of SAMD9. The bands in panel A were quantified using Image Studio software from LI-COR. The intensities of SAMD9 bands were normalized to the intensities of the β-actin bands. (C) Inhibition of vK1L−C7L−/GFP+ replication in Huh-7.5.1 cells treated with IFN-β and partial reversal with SAMD9 siRNA. Huh-7.5.1 cells were transfected with control siRNA or siRNA to SAMD9 for 24 h and then were left untreated or treated with 200 U/ml of IFN-β for 24 h. After infection with 0.01 PFU of vK1L−C7L−/GFP+ per cell for 18 h, GFP was measured by flow cytometry. Data from two experiments each performed in triplicate were combined. Values are means plus standard deviation (error bars). Values that are significantly different (P ≤ 0.001) calculated as Bonferroni test after one-way ANOVA using PRISM GraphPad software are indicated (***).
Further analysis of the effect of WDR6 on vK1L−C7L− replication.
WDR6 was the second strongest hit in the genome-wide screen. In order to confirm the siRNA data, we used CRISPR/Cas9 technology to inactivate the WDR6 gene in HeLa cells. There was a partial reduction of WDR6 expression in cell line 1 and a more complete inactivation in cell line 2, suggesting knockout of one and two alleles, respectively (Fig. 7A). In neither cell line, however, was a reduction in SAMD9 noted (Fig. 7A). Moreover, SAMD9 retained the ability to interact with C7 and K1 proteins in the absence of WDR6. Replication of vK1L−C7L− corresponded inversely to the WDR6 level: WDR6#2 > WDR6#1 > HeLa cells (Fig. 7B).
FIG 7 .
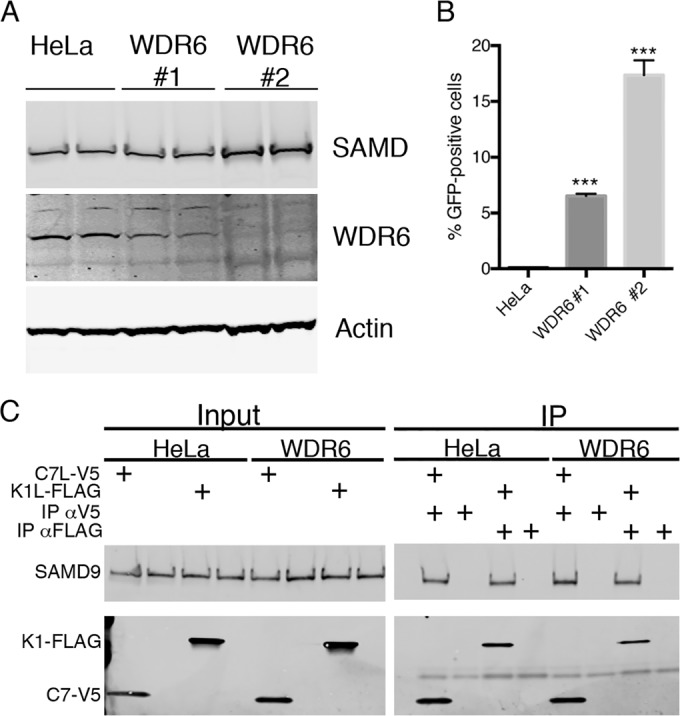
WDR6 is a restriction factor for vK1L−C7L−/GFP+. (A) Generation of WDR6-depleted cells by CRISPR/Cas9 technology. HeLa cells were transfected with the CRISPR/Cas9 components as described in Materials and Methods. Cells from individual colonies 1 and 2 were lysed, and their proteins were resolved by SDS-PAGE and analyzed by Western blotting to detect endogenous WDR6, SAMD9, and actin. (B) HeLa cells and two WDR6 depleted colonies were infected with vK1L−C7L−/GFP+ (0.01 PFU per cell) and incubated for 18 h. GFP-positive cells were quantified using flow cytometry. Data from three experiments each performed in triplicate were combined. Values are means plus standard deviations (error bars). The values that are significantly different (P ≤ 0.001) relative to the value for HeLa cells calculated by Bonferroni test after one-way ANOVA using PRISM GraphPad software are indicated (***). (C) HeLa cells and cells of two WDR6 depleted colonies were infected with vK1L−C7L−/GFP+ at 3 PFU per cell and mock transfected or transfected with C7L-V5 or K1L-FLAG regulated by the T7 promoter. Eighteen hours later, the cells were lysed and incubated with antibodies for the V5 or FLAG epitope tag. Input and proteins captured by magnetic beads conjugated to protein G were resolved by SDS-PAGE and Western blotting for endogenous SAMD9 and V5 or FLAG epitope tag.
DISCUSSION
The present study demonstrates the power of a genome-wide siRNA screen to unambiguously identify cellular restriction factors for a virus host range mutant. Of the more than 20,000 human genes probed, only siRNAs to SAMD9 fully rescued the VACV K1L−C7L− host range mutant as determined by the facile GFP spread assay. Moreover, SAMD9 was the strongest hit for each of three individual siRNAs in the primary screen as well as for a pool of four siRNAs in the secondary screen. Only two additional gene targets, namely, WDR6 and FTSJ1, scored highly positive for all three individual siRNAs in the primary screen as well as the pooled siRNAs in the secondary screen. Further analyses confirmed that the siRNAs targeting SAMD9, WDR6, and FTSJ1 significantly enhanced spread of the mutant in the order SAMD9 > WDR6 > FTSJ1. On the basis of these results, we focused primarily on SAMD9 and to a lesser extent on WDR6. The roles of SAMD9 and WDR6 in preventing replication of vK1L−C7L− were confirmed by demonstrating that both SAMD9 and WDR6 CRISPR/Cas9 knockout human cell lines were permissive for replication of the mutant virus.
Although SAMD9 had been shown to be an antagonist of the myxoma virus C7 homolog MO62 prior to this study (15), we did not anticipate that it would also be a major restriction factor for the VACV K1L−C7L− mutant for the following reason. Although proteomic studies had identified SAMD9 as a binding partner of MO62 (15), the C7 protein was reported not to interact with SAMD9 when expressed by transfection in cells infected with myxoma virus, leading to the suggestion that C7 might act at another step, possibly in the SAMD9 antiviral pathway (13, 15). Furthermore, replication of the C7L−K1L− mutant is restricted in mouse cells (14), which do not carry genes that encode SAMD9 (27). Nevertheless, we found that C7 and K1 independently interacted with SAMD9 both in VACV-infected and uninfected cells. Differences in the experimental protocols likely contributed to the different conclusions. Interestingly, SAMD9 was not diminished in WDR6 knockout cells, and thus far, we have not detected an interaction between WDR6 and SAMD9, C7, or K1. After our screen was completed, Liu and McFadden (22) also reported that knocking down SAMD9 rescued a VACV K1L−C7L− mutant in human cells.
Although the K1L−C7L− VACV mutant is defective in most human cells, hepatoma Huh-7 cells are an exception (7). The VACV K1L−C7L− mutant was able to grow in Huh-7.5.1 cells derived from Huh-7 cells (28) but was inhibited by pretreatment of the cells with beta interferon. SAMD9 was detected by Western blotting in Huh-7.5.1 cells only after interferon treatment. Knockdown of SAMD9 with siRNA in interferon-treated Huh-7.5.1 cells partially restored replication of the mutant virus, suggesting that the innate level of SAMD9 in untreated Huh-7.5.1 cells may be too low to inhibit the VACV K1L−C7L− mutant.
The K1L−C7L− VACV mutant, and the corresponding myxoma virus mutant, exhibit impaired viral protein synthesis (8–10, 15). However, relatively little is known about SAMD9 function (27). In some cells, SAMD9 expression is upregulated by tumor necrosis factor, type I and II interferons, and IRF1 (26, 28–30), and mutations have been associated with the severe rare disease normophosphatemic familial tumoral calcinosis (31). Liu and McFadden (22) showed that SAMD9 associates with stress granules induced by sodium arsenate and cytoplasmic granules formed after infection with myxoma virus MO62 and VACV K1L−C7L− and VACV E3L− host range mutants. Still less is known about WDR6 than SAMD9. WDR6 belongs to the WD repeat protein family, found in all eukaryotes with roles in a variety of functions, including signal transduction, transcription, and cellular proliferation (32–34). However, we are unaware of any known relationship between SAMD9 and WDR6. Poxvirus host range mutants may provide a handle to determine the cellular functions of SAMD9 and WDR6. In addition, the successful use of a poxvirus host range mutant for screening against a human genome-wide library suggests that this approach should be applicable to other virus families.
MATERIALS AND METHODS
Cells and viruses.
BS-C-1 (ATCC CCL-26) and HeLa (ATCC CCL-2) cells were grown in minimum essential medium with Earle’s salt and Dulbecco minimum essential medium, respectively, supplemented with 10% fetal bovine serum, 100 U of penicillin, and 100 µg of streptomycin per ml (Quality Biologicals, Gaithersburg, MD). Huh-7.5.1 cell were grown in Dulbecco minimum essential medium supplemented with 10% fetal bovine serum, 10 mM HEPES, and 1× nonessential amino acids. VACV with deletions of the C7L and K1L ORFs and expressing GFP and bacteriophage T7 RNA polymerase (7) was a generous gift of Yan Xiang.
Plasmids.
The following primers were used for PCR to construct plasmids in which K1L, C7L, and SAMD9 were regulated by bacteriophage T7 promoters: C7L forward, TAATACGACTCACTATAGGGAATTGTGAGCGCTCGCCACATGGGTATACAGCACGAATTCGAC; C7L reverse, TACGTAGAATCGAGACCGAGGAGAGGGTTAGGGATAGGCTTACCATCCATGGACTCATAATCTCTATACGG; K1L forward, TAATACGACTCACTATAGGGAATTGTGAGCGCTCGCCACATGGATCTGTCACGAATTAATACTTGG; K1L reverse, CTACTTGTCGTCATCGTCTTTGTAGTCTACGTTTTTCTTTACACAATTGACGTACATG; SAMD9 forward, TAATACGACTCACTATAGGGAATTGTGAGCGCTCGCCACATGGCAAAGCAACTTAACCTTCCAG; and SAMD9 reverse, (TTAAGCGTAATCTGGAACATCGTATGGGTAAACAATTTCAATGTCATAAGCAAGTGG.
PCR products were cloned into the Zero Blunt PCR cloning vector (Life Sciences Technologies) and sequenced. CMV-C7L-HA and CMV-K1L were codon optimized and synthetized by GeneArt, Life Sciences Technologies.
Antibodies and other reagents.
Antibodies specific for human SAMD9 and epitope tags for V5, myc, HA and FLAG were purchased from Sigma. IFN-β was purchased from Antigenix America Inc. Silencer Select predesigned siRNAs were purchased from Ambion (Life Sciences Technologies).
High-throughput screen.
Screening was conducted as previously described (24) using the Ambion Silencer Select Human Genome siRNA Library version 4, which targets ~21,500 genes with the vast majority consisting of three nonoverlapping and nonpooled siRNAs and the Dharmacon On-Target Plus SMARTpool siRNA consisting of four unique siRNA duplexes per gene in a single well. Image acquisition with a Molecular Devices ImageXpress Micro high-content platform integrated into an Agilent BioCel robotic system and image processing were previously described (24). A number of parameters were calculated using associated MetaXpress software. These included the percentage of cells positive for virus and total nuclei. Data were ranked by the percentage of cells positive for virus. The redundant siRNA analysis tool (RSA) was used to minimize the impact of off-target activities (23).
Western blot analysis.
Proteins of whole-cell lysates were separated in 4 to 12% Novex NuPAGE acrylamide gels with 2-(N-morpholino)ethanesulfonic acid buffer or 3 to 8% Tris-acetate gels and transferred to nitrocellulose membranes using the iBlot system (Invitrogen). The membrane was blocked with 5% nonfat milk in Tris-buffered saline and then incubated for 2 h at room temperature or overnight at 4°C in the same solution with 0.05% Tween 20 and primary antibodies at appropriate dilutions. Excess antibodies were removed by washing with Tris-buffered saline containing Tween 20 followed by phosphate-buffered saline without detergent. IRDye 800- or 700-conjugated secondary antibodies against mouse and rabbit antibodies were added, and the mixture was incubated for 1 h at room temperature, washed, and developed using an Odyssey infrared imager (LI-COR Biosciences, Lincoln, NE). The images were acquired with Image Studio software (LI-COR Biosciences, Lincoln, NE) and prepared with Adobe Photoshop.
Immunoprecipitation.
Cells from six-well plates were washed with cold phosphate-buffered saline, scraped off, and lysed in 1% NP-40, 150 mM NaCl, 50 mM Tris-HCl (pH 7), and Complete protease inhibitors (Roche) for 15 min on ice. Lysates were cleared by high-speed centrifugation for 5 min, and portions (10%) of the supernatants were kept as input controls. The lysates were incubated with 2 µg IgG for 2 h, followed by 1-h incubation with protein G conjugated to Dynabeads magnetic beads (Life Sciences Technologies). The beads were washed with lysis buffer, and the proteins were eluted by boiling in reducing sample buffer.
Generation of knockout cells using CRISPR/Cas9.
HeLa cells with an inactivated SAMD9 gene were generated by using the Edit-R CRISPR-Cas9 Gene Engineering kit (Dharmacon) according to the manufacturer’s instructions. Transfections were carried out with Dharmafect Duo transfection reagent (Dharmacon). Targeting sequences were designed using the web tool CRISPR Design at http://crispr.mit.edu/. After transfection, cells were treated with puromycin (1 µg/ml) for 48 h. The surviving cells were plated in the absence of puromycin in 96-well plates with ~1 to 5 cells/well. Colonies were selected according to their ability to support replication of vC7L−K1L−/GFP.
Statistical analysis.
Figures with graphs with error bars show the means of two or three independent experiments performed in triplicate, and P values were calculated using one-way analysis of variance (ANOVA) and multiple test correction using the Bonferroni method. The calculations were performed in GraphPad PRISM 6.
SUPPLEMENTAL MATERIAL
Data set from Ambion Silencer Select siRNA library part 1.
Data set from Ambion Silencer Select siRNA library part 2.
Data set from Ambion Silencer Select siRNA library part 3.
Data set from the Dharmacon OnTargetPlus siRNA library.
ACKNOWLEDGMENTS
We thank Yan Xiang for the K1L−C7L− deletion virus.
The research was supported by funds from the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Citation Sivan G, Ormanoglu P, Buehler EC, Martin SE, Moss B. 2015. Identification of restriction factors by human genome-wide RNA interference screening of viral host range mutants exemplified by discovery of SAMD9 and WDR6 as inhibitors of the vaccinia virus K1L−C7L− mutant. mBio 6(4):e01122-15. doi:10.1128/mBio.01122-15.
REFERENCES
- 1.Moss B. 2013. Poxviridae, p 2129–2159. In Knipe DM, Howley PM (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Upton C, Slack S, Hunter AL, Ehlers A, Roper RL. 2003. Poxvirus orthologous clusters: toward defining the minimum essential poxvirus genome. J Virol 77:7590–7600. doi: 10.1128/JVI.77.13.7590-7600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bratke KA, McLysaght A, Rothenburg S. 2013. A survey of host range genes in poxvirus genomes. Infect Genet Evol 14:406–425. doi: 10.1016/j.meegid.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haller SL, Peng C, McFadden G, Rothenburg S. 2014. Poxviruses and the evolution of host range and virulence. Infect Genet Evol 21:15–40. doi: 10.1016/j.meegid.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gillard S, Spehner D, Drillien R, Kirn A. 1986. Localization and sequence of a vaccinia virus gene required for multiplication in human cells. Proc Natl Acad Sci U S A 83:5573–5577. doi: 10.1073/pnas.83.15.5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perkus ME, Goebel SJ, Davis SW, Johnson GP, Limbach K, Norton EK, Paoletti E. 1990. Vaccinia virus host range genes. Virology 179:276–286. doi: 10.1016/0042-6822(90)90296-4. [DOI] [PubMed] [Google Scholar]
- 7.Meng X, Jiang C, Arsenio J, Dick K, Cao J, Xiang Y. 2009. Vaccinia virus K1L and C7L inhibit antiviral activities induced by type I interferons. J Virol 83:10627–10636. doi: 10.1128/JVI.01260-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drillien R, Koehren F, Kirn A. 1981. Host range deletion mutant of vaccinia virus defective in human cells. Virology 111:488–499. doi: 10.1016/0042-6822(81)90351-2. [DOI] [PubMed] [Google Scholar]
- 9.Sutter G, Ramsey-Ewing A, Rosales R, Moss B. 1994. Stable expression of the vaccinia virus K1L gene in rabbit cells complements the host range defect of a vaccinia virus mutant. J Virol 68:4109–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramsey-Ewing AL, Moss B. 1996. Complementation of a vaccinia virus host range K1L gene deletion by the non-homologous CP77 gene. Virology 222:75–86. doi: 10.1006/viro.1996.0399. [DOI] [PubMed] [Google Scholar]
- 11.Hsiao JC, Chung CS, Drillien R, Chang W. 2004. The cowpox virus host range gene, CP77, affects phosphorylation of eIF2 alpha and vaccinia viral translation in apoptotic HeLa cells. Virology 329:199–212. doi: 10.1016/j.virol.2004.07.032. [DOI] [PubMed] [Google Scholar]
- 12.Backes S, Sperling KM, Zwilling J, Gasteiger G, Ludwig H, Kremmer E, Schwantes A, Staib C, Sutter G. 2010. Viral host-range factor C7 or K1 is essential for modified vaccinia virus Ankara late gene expression in human and murine cells, irrespective of their capacity to inhibit protein kinase R-mediated phosphorylation of eukaryotic translation initiation factor 2 alpha. J Gen Virol 91:470–482. doi: 10.1099/vir.0.015347-0. [DOI] [PubMed] [Google Scholar]
- 13.Liu J, Rothenburg S, McFadden G. 2012. The poxvirus C7L host range factor superfamily. Curr Opin Virol 2:764–772. doi: 10.1016/j.coviro.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meng XZ, Chao J, Xiang Y. 2008. Identification from diverse mammalian poxviruses of host-range regulatory genes functioning equivalently to vaccinia virus C7L. Virology 372:372–383. doi: 10.1016/j.virol.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu J, Wennier S, Zhang LL, McFadden G. 2011. M062 is a host range factor essential for myxoma virus pathogenesis and functions as an antagonist of host SAMD9 in human cells. J Virol 85:3270–3282. doi: 10.1128/JVI.02243-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bradley RR, Terajima M. 2005. Vaccinia virus K1L protein mediates host-range function in RK-13 cells via ankyrin repeat and may interact with a cellular GTPase-activating protein. Virus Res 114:104–112. doi: 10.1016/j.virusres.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 17.Meng XZ, Xiang Y. 2006. Vaccinia virus K1L protein supports viral replication in human and rabbit cells through a cell-type-specific set of its ankyrin repeat residues that are distinct from its binding site for ACAP2. Virology 353:220–233. doi: 10.1016/j.virol.2006.05.032. [DOI] [PubMed] [Google Scholar]
- 18.Li YC, Meng XZ, Xiang Y, Deng JP. 2010. Structure function studies of vaccinia virus host range protein K1 reveal a novel functional surface for ankyrin repeat proteins. J Virol 84:3331–3338. doi: 10.1128/JVI.02332-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shisler JL, Jin XL. 2004. The vaccinia virus K1L gene product inhibits host NF-κB activation by preventing IκBα degradation. J Virol 78:3553–3560. doi: 10.1128/JVI.78.7.3553-3560.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Willis KL, Patel S, Xiang Y, Shisler JL. 2009. The effect of the vaccinia K1 protein on the PKR-eIF2 alpha pathway in RK13 and HeLa cells. Virology 394:73–81. doi: 10.1016/j.virol.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meng XZ, Schoggins J, Rose L, Cao JX, Ploss A, Rice CM, Xiang Y. 2012. C7L family of poxvirus host range genes inhibits antiviral activities induced by type I interferons and interferon regulatory factor 1. J Virol 86:4538–4547. doi: 10.1128/JVI.06140-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu J, McFadden G. 2015. SAMD9 is an innate antiviral host factor with stress response properties that can be antagonized by poxviruses. J Virol 89:1925–1931. doi: 10.1128/JVI.02262-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.König R, Chiang C, Tu BP, Yan SF, DeJesus PD, Romero A, Bergauer T, Orth A, Krueger U, Zhou Y, Chanda SK. 2007. A probability-based approach for the analysis of large-scale RNAi screens. Nat Methods 4:847–849. doi: 10.1038/nmeth1089. [DOI] [PubMed] [Google Scholar]
- 24.Sivan G, Martin SE, Myers TG, Buehler E, Szymczyk KH, Ormanoglu P, Moss B. 2013. Human genome-wide RNAi screen reveals a role for nuclear pore proteins in poxvirus morphogenesis. Proc Natl Acad Sci U S A 110:3519–3524. doi: 10.1073/pnas.1300708110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuerst TR, Niles EG, Studier FW, Moss B. 1986. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci U S A 83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lemos de Matos A, Liu J, McFadden G, Esteves PJ. 2013. Evolution and divergence of the mammalian SAMD9/SAMD9L gene family. BMC Evol Biol 13:121. doi: 10.1186/1471-2148-13-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chefetz I, Ben Amitai D, Browning S, Skorecki K, Adir N, Thomas MG, Kogleck L, Topaz O, Indelman M, Uitto J, Richard G, Bradman N, Sprecher E. 2008. Normophosphatemic familial tumoral calcinosis is caused by deleterious mutations in SAMD9, encoding a TNF-alpha responsive protein. J Invest Dermatol 128:1423–1429. doi: 10.1038/sj.jid.5701203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanaka M, Shimbo T, Kikuchi Y, Matsuda M, Kaneda Y. 2010. Sterile alpha motif containing domain 9 is involved in death signaling of malignant glioma treated with inactivated Sendai virus particle (HVJ-E) or type I interferon. Int J Cancer 126:1982–1991. doi: 10.1002/ijc.24965. [DOI] [PubMed] [Google Scholar]
- 30.Hershkovitz D, Gross Y, Nahum S, Yehezkel S, Sarig O, Uitto J, Sprecher E. 2011. Functional characterization of SAMD9, a protein deficient in normophosphatemic familial tumoral calcinosis. J Invest Dermatol 131:662–669. doi: 10.1038/jid.2010.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Topaz O, Indelman M, Chefetz I, Geiger D, Metzker A, Altschuler Y, Choder M, Bercovich D, Uitto J, Bergman R, Richard G, Sprecher E. 2006. A deleterious mutation in SAMD9 causes normophosphatemic familial tumoral calcinosis. Am J Hum Genet 79:759–764. doi: 10.1086/508069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li D, Burch P, Gonzalez O, Kashork CD, Shaffer LG, Bachinski LL, Roberts R. 2000. Molecular cloning, expression analysis, and chromosome mapping of WDR6, a novel human WD-repeat gene. Biochem Biophys Res Commun 274:117–123. doi: 10.1006/bbrc.2000.3012. [DOI] [PubMed] [Google Scholar]
- 33.Xie X, Wang Z, Chen Y. 2007. Association of LKB1 with a WD-repeat protein WDR6 is implicated in cell growth arrest and p27Kip1 induction. Mol Cell Biochem 301:115–122. doi: 10.1007/s11010-006-9402-5. [DOI] [PubMed] [Google Scholar]
- 34.Chiba T, Inoue D, Mizuno A, Komatsu T, Fujita S, Kubota H, Luisa Tagliaro M, Park S, Trindade LS, Hayashida T, Hayashi H, Yamaza H, Higami Y, Shimokawa I. 2009. Identification and characterization of an insulin receptor substrate 4-interacting protein in rat brain: implications for longevity. Neurobiol Aging 30:474–482. doi: 10.1016/j.neurobiolaging.2007.07.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data set from Ambion Silencer Select siRNA library part 1.
Data set from Ambion Silencer Select siRNA library part 2.
Data set from Ambion Silencer Select siRNA library part 3.
Data set from the Dharmacon OnTargetPlus siRNA library.