

In this review, Ornitz and Marie examine progress made on understanding the functions of the fibroblast growth factor (FGF) signaling pathway during critical stages of skeletogenesis and explore the mechanisms by which mutations in FGF signaling molecules cause skeletal malformations in humans.
Keywords: FGF, FGFR, cartilage, bone, chondrocyte, osteoblast
Abstract
Fibroblast growth factor (FGF) signaling pathways are essential regulators of vertebrate skeletal development. FGF signaling regulates development of the limb bud and formation of the mesenchymal condensation and has key roles in regulating chondrogenesis, osteogenesis, and bone and mineral homeostasis. This review updates our review on FGFs in skeletal development published in Genes & Development in 2002, examines progress made on understanding the functions of the FGF signaling pathway during critical stages of skeletogenesis, and explores the mechanisms by which mutations in FGF signaling molecules cause skeletal malformations in humans. Links between FGF signaling pathways and other interacting pathways that are critical for skeletal development and could be exploited to treat genetic diseases and repair bone are also explored.
Skeletal development: an overview
Development of the vertebrate skeleton occurs through the processes of endochondral and intramembranous bone formation. Endochondral-derived bones comprise the appendicular skeleton, facial bones, vertebrae, and medial clavicles, while intramembranous bones comprise the cranium and lateral clavicles. Endochondral ossification initiates with the condensation of mesenchyme, followed by the formation of a cartilaginous template that patterns the developing skeleton and the segmentation of this template into cartilaginous joints that are stabilized by a flexible fibrous capsule and mineralized cortical and trabecular (spongy) bone. Intramembranous bone forms from a mesenchymal condensation that directly gives rise to bone.
Chondrogenesis
Mesenchymal condensations are the first histologically visible features of the developing appendicular skeleton (Fig. 1A). Mesenchymal condensations form in the developing limb bud and are characterized by the aggregation of loose mesenchymal cells and the expression of extracellular matrix proteins, cell adhesion molecules, transcription factors, and receptors for signaling molecules. The transcription factor Sox9 is required for the differentiation of the chondrogenic lineage, and bone morphogenetic protein (BMP) receptors 1a and 1b are required for chondrogenesis and the maintenance of Sox9 expression (Kosher et al. 1986; Nah et al. 1988; Bi et al. 1999; Akiyama et al. 2002; Yoon et al. 2005; Yeung Tsang et al. 2014; Lim et al. 2015). Proliferating cells within the forming mesenchymal condensation express type II collagen (Col2a1) centrally in chondroprogenitors and type I collagen (Col1a1) peripherally in osteoprogenitors (Kosher et al. 1986).
Figure 1.
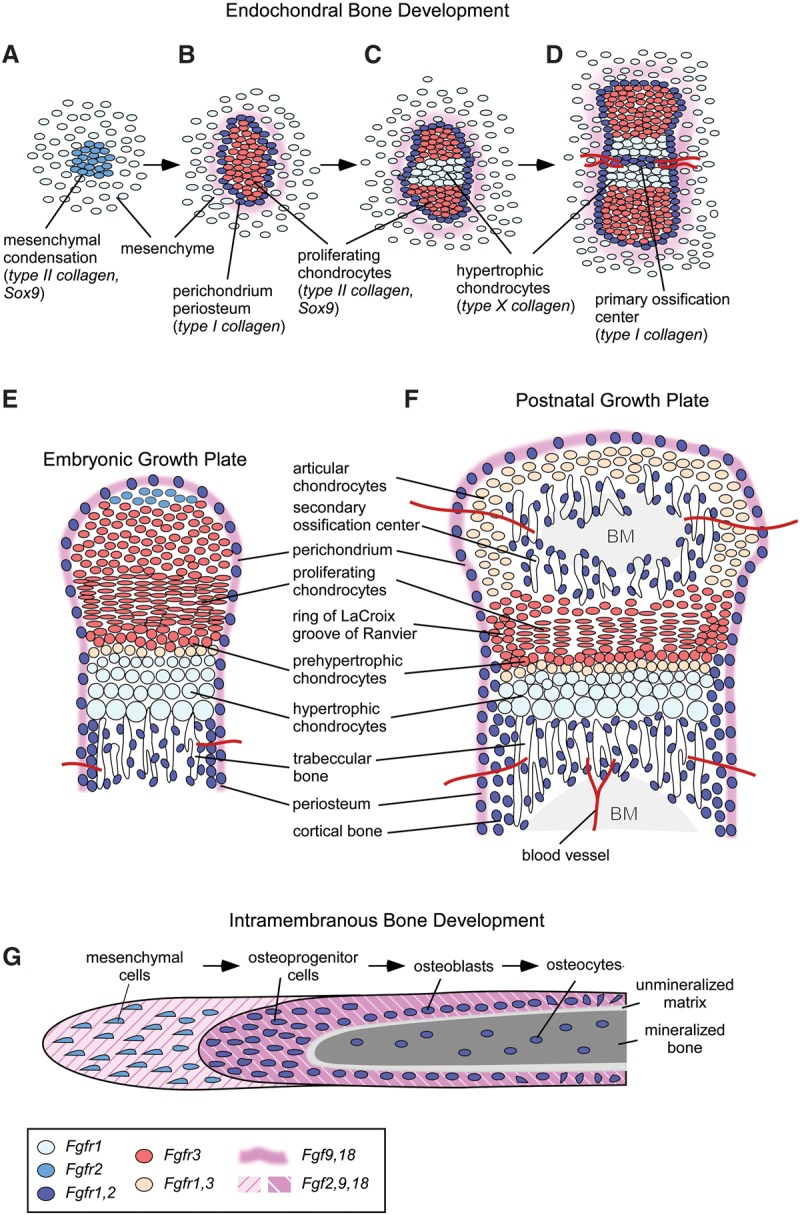
Fibroblast growth factor (FGF) and FGF receptor (FGFR) expression patterns during endochondral bone development. (A–D) Progression of endochondral bone development from the mesenchymal condensation to formation of the primary ossification center. (E) Embryonic growth plate. (F) Postnatal growth plate after formation of the secondary ossification center. (G) Developmental progression of intramembranous bone development. Cells and tissues are color-coded for expression domains of FGFs and FGFRs. (BM) Bone marrow.
Proliferating chondrocytes form columns oriented along the longitudinal axis of the developing bone. Midway along the longitudinal axis of the mesenchymal condensation, proliferating chondrocytes exit the cell cycle and differentiate into prehypertrophic chondrocytes (Fig. 1B,C) that are marked by expression of Indian Hedgehog (IHH), parathyroid hormone (PTH)-related peptide receptor (PTH1R), and type X collagen (Col10a1). Several transcription factors are required for the initiation and progression of chondrocyte hypertrophy. Sox9 is required to maintain chondrocyte identity and prepare chondrocytes for subsequent hypertrophy but must be down-regulated for progression to hypertrophy (Ikegami et al. 2011; Dy et al. 2012). Runx2 (Cbfa1) and Runx3 (Cbfa3) are required for chondrocyte maturation and indirectly function to enhance chondrocyte proliferation through promoting Ihh expression (Yoshida et al. 2004). The balance between chondrocyte proliferation and hypertrophy is regulated by interactions between several signaling pathways, including IHH, PTH-related peptide (PTHLH), BMP, Wnt, and fibroblast growth factor (FGF) (Long and Ornitz 2013; Kozhemyakina et al. 2015). The activity of Wnt and PTHLH signals regulates the level of calcium/calmodulin-dependent protein kinase II (CAMK2), which induces chondrocyte hypertrophy in part through increasing RUNX2 and β-catenin activity (Li et al. 2011; Long and Ornitz 2013). Chondrocyte hypertrophy leads to an expansive force that is required for bone elongation (Hunziker et al. 1987; Hunziker and Schenk 1989; Noonan et al. 1998). Hypertrophic chondrocytes can also begin to mineralize their extracellular matrix and have the capacity to differentiate into osteoblasts within the primary spongiosa (Yang et al. 2014; Zhou et al. 2014).
Endochondral bone formation
Mineralized bone originates with the establishment of a primary ossification center (Fig. 1D) that initiates with the differentiation of type I collagen-producing osteoblasts and the formation of a bone collar (the precursor of cortical and trabecular bone) surrounding the central hypertrophic chondrocyte zone. Lineage tracing studies show that osteoprogenitor cells (Osterix lineage) in the perichondrium/periosteum give rise to trabecular osteoblasts, while more mature osteoblasts (Collagen I lineage) remain in the perichondrium and populate cortical bone (Maes et al. 2010). Vascular endothelial growth factor (VEGF-A), expressed in hypertrophic chondrocytes, supports the invasion of endothelial cells, osteoprogenitor cells, and osteoclasts, which remodel the chondrogenic matrix and organize to form the primary ossification center (Liu and Olsen 2014). Bidirectional expansion of the primary ossification center along the longitudinal axis replaces growth plate chondrocytes with trabecular bone (primary spongiosa). After formation of a secondary ossification center within the distal resting chondrocyte zone, the definitive growth plate can be identified by well-demarcated zones of cells (Fig. 1E,F). Toward the epiphysis of a developing bone, slowly growing resting chondrocytes and cells within the adjacent groove of Ranvier and ring of LaCroix serve as a pool of progenitor cells that populate the rapidly dividing cells within the proliferating chondrocyte zone, which differentiate into prehypertrophic and then hypertrophic chondrocytes (Robinson et al. 1999; Abad et al. 2002; Karlsson et al. 2009). At the distal end of the epiphyseal growth plate (toward the midpoint of a long bone), hypertrophic chondrocytes either die by apoptosis or differentiate into trabecular osteoblasts (Yeung Tsang et al. 2014). Continued growth of the vascular network and influx of hematopoietic-derived cells, including osteoclasts, remodels the primary spongiosa to form the bone marrow cavity.
Intramembranous bone formation
During development, intramembranous ossification begins with the condensation of mesenchymal cells and their progressive differentiation into osteoblasts to form a mineralized matrix within ossification centers (Hall and Miyake 2000). In the skull, these centers expand during development but do not fuse at the junction with other cranial bones, allowing skull expansion during growth (Opperman 2000). The junction between cranial bones, called a suture, is responsible for the maintenance of separation between two membrane bones. Most cells surrounding the suture are mesenchymal cells, a minority of which differentiate into preosteoblasts and then into mature osteoblasts, which are found along the developing bone trabeculae. At the end of the bone formation period, osteoblasts die by apoptosis or become embedded in the matrix as osteocytes, which then eventually also undergo apoptosis (Fig. 1G; Rice et al. 1999; Fromigue et al. 2005). The early commitment of mesenchymal stem cells into osteoblasts requires expression of Runx2, a master transcription factor that regulates several genes in osteoblasts, such as type I collagen, bone sialoprotein (BSP), osteopontin (OP), transforming growth factor (TGFβ), and osteocalcin (OC) (Marie 2008). Transcription factors, such as MSX2 and DLX5, play important roles in osteoblast differentiation during cranial bone formation in part by interacting with RUNX2 (Dailey et al. 2005; Rice and Rice 2008). The regulation of intramembranous bone formation involves several regulatory factors, including TGFβs, BMPs, FGFs, and Wnt signaling, all of which were shown to regulate cell differentiation and survival in a spatiotemporal manner (Opperman 2000; Dailey et al. 2005; Lenton et al. 2005; Rice and Rice 2008).
FGF signaling
The canonical and endocrine branches of the mammalian FGF family comprise 18 secreted signaling proteins that bind to and activate four receptor tyrosine kinase molecules (FGF receptors [FGFRs]) (Ornitz and Itoh 2015). Fifteen canonical FGFs are differentially expressed in most tissues of the developing embryo, where they function as essential regulators of the earliest stages of development and organogenesis. Canonical FGFs are also expressed in postnatal and adult tissues, where they regulate growth and function as factors for tissue maintenance, repair, and regeneration. Three endocrine FGFs have critical postnatal roles in the regulation of phosphate, bile acid, carbohydrate, and lipid metabolism.
Of the four tyrosine kinase FGFRs, FGFR1, FGFR2, and FGFR3 undergo alternative splicing to produce variants that have distinct ligand-binding specificities and that are differentially expressed in epithelial and mesenchymal lineages. In general, FGFRb splice variants are found in epithelial tissues and bind FGFs such as FGF7 and FGF10 that are expressed in mesenchymal tissues. FGFRc splice variants are found in mesenchymal tissues and bind FGF ligands that are expressed in both epithelial and mesenchymal lineages. A fifth nontyrosine kinase FGFR, FGFR5 or FGFRL1, is homologous to FGFR1–4 in the extracellular ligand-binding domain but lacks an intracellular tyrosine kinase domain. Fgfrl1 was originally identified in human cartilage (Wiedemann and Trueb 2000).
The biological activity of canonical FGFs is enhanced by heparan sulfate (HS) and other sulfated polysaccharides (Shimokawa et al. 2011; Ornitz and Itoh 2015). A class of cell surface and extracellular matrix proteins primarily consisting of HS proteoglycans (HSPGs) contain HS covalently attached to a protein core. HSPGs include cell surface transmembrane proteins (syndecans), cell surface glycerophosphatidylinositide-anchored proteins (glypicans), and diffusible protein components of the extracellular matrix (perlecan and agrin) (Ornitz and Itoh 2015). Cell surface and extracellular matrix HSPGs affect diffusion of FGFs in tissues and interact with FGFs and FGFRs to increase the affinity of FGF–FGFR heterodimers to enhance receptor activation (Wu et al. 2003; Belov and Mohammadi 2013; Xu and Esko 2014). In contrast to canonical FGFs, endocrine FGFs require a protein cofactor to enhance receptor binding and activation. Members of the Klotho family were found to fulfill this role based on phenotypic similarities of αKlotho and Fgf23 knockout mice (Kurosu et al. 2006; Urakawa et al. 2006; Kuro-o 2008).
Binding of FGF ligands to FGFRs induces dimerization and juxtaposition of the tyrosine kinase domains to initiate the sequential transphosphorylation of at least six tyrosine residues (Furdui et al. 2006; Goetz and Mohammadi 2013; Ornitz and Itoh 2015). Activation of the FGFR tyrosine kinase domain allows the direct phosphorylation of the docked adaptor protein FGFR substrate 2α (FRS2α) and binding of other adaptor proteins, including phospholipase Cγ (PLCγ), signal transducer and activator of transcription 1 (STAT1), STAT3, and STAT5 (Ornitz and Itoh 2015). In chondrocytes, FGFR3 has been found to specifically activate STAT1 (Su et al. 1997; Sahni et al. 1999). Phosphorylation of FRS2α recruits the adaptor protein GRB2, which activates the MAPK pathway thorough SOS and the PI3K–AKT pathway through GAB1 (Kouhara et al. 1997; Lamothe et al. 2004). Downstream from RAS and PI3K, FGFs can activate several distinct MAPKs, including ERK1/2, JNK, and p38 (Tan et al. 1996; Tsang and Dawid 2004; House et al. 2005; Liao et al. 2007; Kanazawa et al. 2010). Binding of CRKL to the activated FGFR enhances phosphorylation of FRS2α, MAPK pathway signaling, and ERK1/2 activation (Moon et al. 2006).
The four known branches of the FGFR intracellular signaling cascades are also regulated by inhibitory molecules that include high-level expression of GRB2 (which interferes with PLCγ binding to the FGFR) and Sprouty (Spry) proteins (which interact with GRB2 to block MAPK and PI3K signaling) (Hanafusa et al. 2002; Timsah et al. 2014). Further downstream, SEF (similar expression to FGF) antagonizes the MAPK pathway through interaction with MEK (Torii et al. 2004), and DUSP6 (dual-specificity phosphatase 6) suppresses MAPK signaling through dephosphorylation of ERK1/2 (Camps et al. 1998). The E3 ubiquitin ligase CBL inhibits FGFR signaling by forming a ternary complex with phospho-FRS2 and GRB2, where it promotes the ubiquitination and degradation of the FGFR and FRS2 (Wong et al. 2002). CBL also interacts with PI3K, leading to its ubiquitination and degradation (Dufour et al. 2008). The adaptor protein Grb14 binds to FGFR phospho-Tyr766 and interferes with phosphorylation and activation of PLCγ (Browaeys-Poly et al. 2010).
FGF signaling during skeletal development
The endochondral mesenchymal condensation
Fgfs, Fgfrs, and their co-receptor, HS, are expressed in a time- and space-dependent manner during all stages of skeletal development (Fig. 1). At precondensation stages, distal limb bud mesenchyme expresses both Fgfr1 and Fgfr2 (Orr-Urtreger et al. 1991; Sheeba et al. 2010), whereas Fgfr3 and Fgfr4 are not detected in undifferentiated distal limb bud mesenchyme (Sheeba et al. 2010). Distal limb bud FGFRs respond to FGFs produced by the apical ectodermal ridge (AER) of the limb bud (Zeller et al. 2009). AER FGFs (primarily FGF4 and FGF8) signal to limb mesenchyme and are required for proximo–distal outgrowth of the limb bud. The function of mesenchymal FGFRs in the limb bud has been investigated through conditional gene inactivation using the Cre/LoxP system and by expressing inhibitory RNAs. Conditional inactivation of Fgfr1 and Fgfr2 in limb bud mesenchyme (using Prx1-Cre) or inactivation of Fgfr1 in all mesenchyme [using T(brachyury)-Cre] results in severe skeletal hypoplasia (Verheyden et al. 2005; Yu and Ornitz 2008). In contrast, inactivation of Fgfr1 or Fgfr2 in distal limb bud mesenchyme (using AP2-Cre) results in considerably milder skeletal patterning phenotypes (Coumoul et al. 2005; Li et al. 2005). These experiments collectively demonstrate redundancy between FGFR1 and FGFR2 in distal limb mesenchyme.
In condensing mesenchyme (Fig. 1A) Fgfr2 expression is increased (compared with the surrounding mesenchyme) and initially overlaps with domains of Sox9 expression (Orr-Urtreger et al. 1991; Peters et al. 1992; Szebenyi et al. 1995; Delezoide et al. 1998; Eswarakumar et al. 2002; Sheeba et al. 2010). Fgfr1 is more uniformly expressed throughout limb bud mesenchyme. Although Fgfr3 and Fgfr4 are excluded from distal limb bud mesenchyme, these Fgfrs are expressed in more proximal locations in tissues corresponding to developing muscle (Orr-Urtreger et al. 1991; Peters et al. 1992; Szebenyi et al. 1995; Delezoide et al. 1998; Sheeba et al. 2010). Cells in the periphery of the condensation that will form perichondrium and periosteum express both Fgfr1 and Fgfr2 (Fig. 1B–D; Delezoide et al. 1998; Eswarakumar et al. 2002). As soon as condensed mesenchyme begins to differentiate into chondrocytes, Fgfr3 expression is activated (Fig. 1B) along with Sox9 and type II collagen, and Fgfr2 expression is decreased (Peters et al. 1992, 1993; Purcell et al. 2009). As central chondrocytes begin to hypertrophy, Fgfr3 expression is decreased, and Fgfr1 expression is increased (Fig. 1C; Peters et al. 1992, 1993; Deng et al. 1996; Naski et al. 1998; Jacob et al. 2006; Karolak et al. 2015).
The initiation of the mesenchymal condensation and its differentiation to the chondrogenic lineage are at least partially dependent on FGFR signaling (Murakami et al. 2000; Mariani et al. 2008; Yu and Ornitz 2008; Kumar and Lassar 2014). In primary chondrocytes and undifferentiated mesenchymal cell lines, FGF signaling increases Sox9 expression (Murakami et al. 2000; Shung et al. 2012). FGF activation of ERK1/2 also maintains competence of limb bud mesenchyme to differentiate into chondrocytes by blocking Wnt-induced methylation and silencing of the Sox9 promoter (Ten Berge et al. 2008; Kumar and Lassar 2014). Fgfr3 expression in proliferating chondrocytes requires Sox9, and Sox9-binding sites are found in the Fgfr3 gene (Oh et al. 2014). FGFR3 signaling is mitogenic for immature proliferating chondrocytes and is likely activated by FGF9 and FGF18, which are expressed in adjacent mesenchyme (Fig. 1; Iwata et al. 2000, 2001; Liu et al. 2002, 2007; Ohbayashi et al. 2002; Hung et al. 2007; Havens et al. 2008).
The forming perichondrium, bone collar, and trabecular bone express FGFR1 in mesenchymal progenitors and FGFR2 in differentiating osteoblasts (Fig. 1E,F; Molteni et al. 1999b; Britto et al. 2001; Ohbayashi et al. 2002; Jacob et al. 2006; Coutu et al. 2011). FGFR3 is expressed more intensely in chondroprogenitor cells located in the groove of Ranvier and ring of LaCroix (Robinson et al. 1999), and FGFR1 and FGFR3 are expressed in mouse and human articular chondrocytes (Fig. 1F; Yan et al. 2011; Weng et al. 2012). Immature cultured osteoblasts in vitro expressed relatively higher levels of Fgfr1, whereas mature osteoblasts express relatively higher levels of Fgfr2 (Rice et al. 2003). Differential expression of these Fgfrs may reflect a distinct response to exogenous FGFs such as FGF2 (Cowan et al. 2003).
Intramembranous mesenchymal condensations
The spatiotemporal pattern of expression of Fgfs and Fgfrs that control FGF signaling during membranous bone formation is reviewed in Ornitz and Marie (2002). Fgf2, Fgf4, Fgf9, Fgf18, Fgfr1, Fgfr2, and Fgfr3 are expressed throughout the early stages of intramembranous bone development (Fig. 1G; Kim et al. 1998; Rice et al. 2000; Britto et al. 2001; Liu et al. 2002; Ohbayashi et al. 2002; Quarto et al. 2009). The FGFR2c splice variant is expressed in early mesenchymal condensations and then in sites of intramembranous ossification, where it interacts with FGF18 (Eswarakumar et al. 2002). At later stages during cranial bone development, Fgf18 is expressed in mesenchymal cells and differentiating osteoblasts (Ohbayashi et al. 2002; Reinhold and Naski 2007). Fgf9 is expressed throughout calvarial mesenchyme during mid to late stages of embryonic development (Kim et al. 1998). Mice lacking both Fgf9 and Fgf18 have severely deficient cranial bone formation (IH Hung, CG Schoenwolf, M Lewandoski, and DM Ornitz, in prep.). Fgfr1 and Fgfr2, which are expressed in preosteoblasts and osteoblasts (Rice et al. 2003), are likely receptors for FGF9 and FGF18 in developing membranous bone. (Ornitz and Marie 2002)
The growth plate
Growth plate chondrocytes express very low levels of Fgfr2 in the resting zone, high levels of Fgfr3 in the proliferating and prehypertrophic zone, and high levels of Fgfr1 in hypertrophic chondrocytes (Fig. 1E,F; Peters et al. 1993; Delezoide et al. 1998; Hamada et al. 1999; Eswarakumar et al. 2002; Ornitz and Marie 2002; Yu et al. 2003; Jacob et al. 2006; Lazarus et al. 2007; Karolak et al. 2015). To investigate the potential functions of FGFR2 in skeletal development, the Fgfr2 gene was conditionally inactivated in limb bud mesenchyme with Dermo1(Twist2)-Cre (Yu et al. 2003). Although these mice showed decreased postnatal growth, chondrocyte proliferation and the length of the proliferating chronfrocyte zone were not affected, suggesting that FGFR2 does not have a nonredundant function in resting or proliferating chondrocytes. The reduced stature of these mice was attributed to decreased osteogenesis and increased osteoclast activity at the chondro–osseous junction (Yu et al. 2003).
In early embryonic stages of skeletal development, during establishment of the growth plate, FGFR3 has promitogenic activity in chondrocytes (Iwata et al. 2000, 2001; Havens et al. 2008). However, following formation of the secondary ossification center and throughout postnatal skeletal growth, FGFR3 signaling functions to inhibit chondrogenesis, primarily acting on proliferating chondrocytes and their differentiation to prehypertrophic and hypertrophic chondrocytes (Colvin et al. 1996; Deng et al. 1996). This paradoxical activity of FGFR3 underlies the etiology of chondrodysplastic disorders in which gain-of-function mutations in FGFR3 cause decreased proliferation and differentiation of proliferating chondrocytes during prepubertal skeletal growth, resulting in skeletal dwarfism (Naski et al. 1996, 1998). The mechanisms that regulate FGFR3 expression and signaling during prepubertal skeletal growth are an ongoing and important topic of research. In addition to providing an understanding of the mechanisms regulating bone growth, these studies are providing potential therapeutic avenues for the treatment of achondroplasia and other forms of skeletal dwarfism.
FGFR3 signaling in growth plate chondrocytes
Signaling through FGFR3 in growth plate chondrocytes activates STAT1, ERK1/2, and p38 intracellular signals; increases expression of Snail1 (Snai1); down-regulates AKT; activates protein phosphatase 2a (PP2a); and leads to dephosphorylation (activation) of the retinoblastoma family members p107 and p130 (Fig. 2; Su et al. 1997; Laplantine et al. 2002; Raucci et al. 2004; Priore et al. 2006; de Frutos et al. 2007; Kolupaeva et al. 2008; Kurimchak et al. 2013). Ultimately, suppression of chondrocyte proliferation resulting from activation of FGFR3 is effected through increased expression of the cell cycle inhibitor p21Waf1/Cip1 and activation of p107 (and p130) (Cobrinik et al. 1996; Su et al. 1997; Aikawa et al. 2001; Laplantine et al. 2002; Dailey et al. 2003; Legeai-Mallet et al. 2004; Kolupaeva et al. 2008, 2013).
Figure 2.
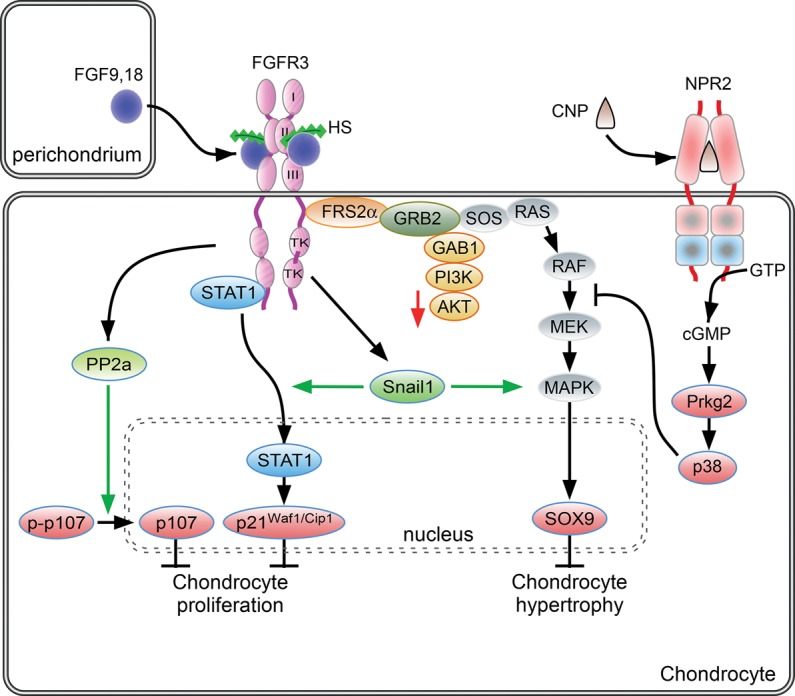
FGF signaling pathways in proliferating chondrocytes. During endochondral bone development, FGF9 and FGF18, derived from the perichondrium and surrounding tissue, signal to chondrocytes. Activity is mediated in part by regulated diffusion through the extracellular matrix through affinity for HS and potentially other sulfated glycosaminoglycans. Activation of FGFR3 in proliferating chondrocytes activates the STAT1 and MAPK signaling pathways. FGFR3 signaling results in increased expression of Snail1, which in turn is required for STAT1 and MAPK signaling. FGFR3 signaling can also activate PP2a. Activation of downstream signals, p107, p21Waf1/Cip1, and Sox9 regulates chondrocyte proliferation and differentiation to hypertrophic chondrocytes. C-type natriuretic peptide (CNP) signals through the natriuretic peptide receptor 2 (NPR2), a guanylyl cyclase. cGMP activates cyclic GMP-dependent protein kinase II (cGKII), which, through p38 MAPK activation, functionally antagonizes RAF1 activation of MEK. See the text for more details.
SNAIL1 is integral to the FGFR3 signaling pathway in chondrocytes (Fig. 2). Snail1 expression is induced by FGFR3, and Snail1 is expressed at high levels in human thantophoric dysplasia bone tissue. Experimentally, ectopic activation of SNAIL1 in mice resulted in an achondroplasia phenotype with decreased chondrocyte proliferation and shortened bones at late embryonic stages (de Frutos et al. 2007). Functionally, SNAIL1 appears to activate both the STAT1 and MAPK branches of the FGFR3 signaling pathway, as ectopic activation of SNAIL1 resulted in nuclear translocation of STAT1 and increased phosphorylation of ERK1/2 (de Frutos et al. 2007). Although the mechanism by which SNAIL1 regulates STAT1 and MAPK is not known, a recent report showed that SNAIL1 may regulate the nuclear localization of p-ERK (Smith et al. 2014). Conversely, activated ERK2 directly phosphorylates SNAIL1, leading to nuclear accumulation and increased protein stability (Zhang et al. 2013). In support of a requirement for SNAIL proteins in regulating chondrogenesis, conditional inactivation of both Snail1 and Snail2 resulted in increased p21Waf1/Cip1 and decreased chondrocyte proliferation (Chen and Gridley 2013).
Decreased bone growth, caused by activating mutations in FGFR3, results from decreased chondrocyte proliferation and decreased hypertrophy. The underlying mechanisms suggest that these phenotypes are distinct and are regulated by different branches of the FGFR3 signaling pathway. Evidence that the STAT1 branch of the pathway regulates chondrocyte proliferation is derived from in vivo rescue experiments in which chondrocyte proliferation defects in mice with activating mutations in FGFR3 can be rescued by biallelic inactivation of Stat1 (Murakami et al. 2004). However, in this model, inactivation of Stat1 still does not rescue the overall achondroplasia phenotype in which impaired chondrocyte hypertrophy leads to decreased bone growth. Further studies showed that, downstream from FGFR3, activation of MAPK signaling suppressed chondrocyte hypertrophy, resulting in skeletal dwarfism (Murakami et al. 2004). The mechanism may involve suppression of SOX9 down-regulation in prehypertrophic chondrocytes by activated FGFR3 signaling (Fig. 2; Murakami et al. 2000; Shung et al. 2012; Zhou et al. 2015). Additionally, activation of the PP2A–B55α holoenzyme by FGFR3, which dephosphorylates and activates p107, may independently contribute to decreased chondrocyte hypertrophy (Fig. 2; Kolupaeva et al. 2013; Kurimchak et al. 2013).
Further support of the model in which STAT1 activation suppresses chondrocyte proliferation and MAPK signaling regulates matrix production and chondrocyte differentiation is derived from studies that examined interactions between FGFR3 and the C-type natriuretic peptide (CNP) signaling pathways (Fig. 2; Yasoda et al. 2004). CNP expression was found to attenuate the phenotype of achondroplasia mice through inhibition of MAPK signaling, which restored matrix production and hypertrophic differentiation (see below). The activity of CNP was notably independent of STAT1 activity and did not affect chondrocyte proliferation.
The function of the STAT1 and MAPK branches of the FGFR3 signal pathway may not be completely distinct. In cultured chondrocytes, FGFR3 suppression of chondrocyte proliferation required MAPK signaling and was independent of STAT signaling (Raucci et al. 2004; Krejci et al. 2008a). In support of this observation, mice lacking ERK1 and conditionally lacking ERK2 (Col2a1-Cre) in chondrocytes showed increased chondrocyte proliferation at late embryonic stages (Matsushita et al. 2009a; Sebastian et al. 2011). Some of the variability in experimental results may be due to in vitro culture conditions, the developmental stage being examined (embryonic vs. postnatal), and potential indirect effects on proliferating chondrocytes resulting from interactions with other signaling pathways in nearby cells within the groove of Ranvier, ring of LaCroix, and perichondrium.
Indirect mechanisms by which FGFR3 signaling may regulate growth plate development include the regulation of BMP, Wnt, IHH, and PTHLH/PTH1R expression and activity. Comparisons of activating mutations in Fgfr3 and mice lacking Fgfr3 (Fgfr3−/−) show that FGFR3 signaling suppresses expression of Bmp4, BMPR1a, Ihh, and Pth1r in postnatal growth plate chondrocytes (Naski et al. 1998; Chen et al. 2001; Qi et al. 2014). Additionally, in a chondrocyte cell line, overexpression of FGFR3 suppresses expression of both Pthlh and its receptor, Pth1r (Li et al. 2010), and, in chondrocyte cell lines and micromass cultures, FGF signaling activates Wnt/β-catenin signaling through phosphorylation of LRP6 and functions to suppress hypertrophic differentiation (Krejci et al. 2012; Buchtova et al. 2015).
Regulation of Fgfr3 expression
Regulation of FGFR3 expression and activation is essential for chondrogenesis. In cultured cells, thyroid hormone has been shown to inhibit chondrocyte proliferation and promote chondrocyte hypertrophy, activities that are similar to that of signaling through FGFR3. Consistent with this observation, treatment of cultured chondrocytes with thyroid hormone increased Fgfr3 expression, and mice lacking thyroid hormone receptor had decreased levels of Fgfr3 in growth plate chondrocytes (Barnard et al. 2005). PTHLH may also regulate FGFR3 expression. Treatment of thantophoric dysplasia mice with intermittent PTH injection partially rescued the lethality and skeletal growth phenotype. In cultured cells, PTH treatment inhibited phosphorylation of FGFR3, suggesting that PTH may function in part through regulation of FGFR3 signaling (Xie et al. 2012). Additionally, analysis of the Fgfr3 promoter identified a transcriptional regulatory element, CSRh, which was repressed by PTH through a cAMP and protein kinase A (PKA)-dependent mechanism (McEwen et al. 1999). A third mechanism that could regulate FGFR3 is hypoxia, a characteristic feature of growth plate chondrocytes. Although this has not been investigated in chondrocytes, in bladder cancer cells, Fgfr3 expression was induced by hypoxia in a transcriptional and HIF-1α-dependent manner (Blick et al. 2013).
FGFR1 signaling in hypertrophic chondrocytes
Fgfr1 is prominently expressed in prehypertrophic and hypertrophic chondrocytes (Fig. 1) and overlaps with Fgfr3 in prehypertrophic chondrocytes (Fig. 1). The functions of FGFR1 signaling in hypertrophic chondrocytes are not entirely understood due to imprecise genetic tools and difficulties in distinguishing cell-autonomous functions in hypertrophic chondrocytes from activities in the adjacent perichondrium and other tissues. Activating mutations in FGFR1 that cause osteoglophonic dysplasia result in severe dwarfism in humans (White et al. 2005); however, an activating mutation in Fgfr1 in mice did not affect endochondral ossification (Zhou et al. 2000). Possible functions of FGFR1 signaling have been investigated in mice through targeted inactivation of Fgfr1 (Col2a1-Cre or Dermo1-Cre). At early stages of development, Fgfr1 conditional knockout mice (Dermo1-Cre) show impaired chondrocyte hypertrophy (Hung et al. 2007). In contrast, at later stages of development, Fgfr1 conditional knockout mice (Col2a1-Cre) have an expanded hypertrophic chondrocyte zone (Jacob et al. 2006). Both Dermo1-Cre and Col2a1-Cre efficiently target chondrocytes but also target the osteoblast lineage (Yu et al. 2003; Jacob et al. 2006; Ford-Hutchinson et al. 2007; Wang et al. 2011; Ono et al. 2014). Therefore, defects in osteoblast development could indirectly affect chondrogenesis (Jacob et al. 2006; Karolak et al. 2015), and a definitive assessment of FGFR1 function in hypertrophic chondrocytes will thus require more precise targeting of the chondrocyte lineage.
FGF ligands in endochondral bone growth
Fgf9 and Fgf18 are expressed in the perichondrium and periosteum, and Fgf2 is expressed in chondrocytes (Gonzalez et al. 1996; Liu et al. 2002; Ohbayashi et al. 2002; Hung et al. 2007; Reinhold and Naski 2007). Mice lacking Fgf2 have decreased bone mass but no apparent change in growth plate structure or function (Montero et al. 2000). In contrast, mice lacking Fgf9 exhibit rhizomelic (affecting proximal skeletal elements) shortening of the appendicular skeleton. FGF9 and FGF18 also have stage-specific effects on chondrogenesis. At early stages of development of the humerus and femur, chondrocyte proliferation is decreased, and chondrocyte hypertrophy is impaired in Fgf9−/− mice (Hung et al. 2007). Mice lacking Fgf18 show decreased chondrocyte proliferation in more distal skeletal elements (Liu et al. 2007). This phenotype is consistent with FGF9 and FGF18 signaling to FGFR3 and the promitogenic properties of FGFR3 on immature chondrocytes (Iwata et al. 2000, 2001; Hung et al. 2007). At later stages of development, Fgf9−/− and Fgf18−/− mice show an expansion of the femoral hypertrophic chondrocyte zone, a phenotype that is consistent with FGFR3 functioning to suppress chondrocyte proliferation and differentiation in the mature growth plate (Liu et al. 2002; Ohbayashi et al. 2002; Hung et al. 2007). In contrast to mice lacking Fgf9, mice lacking Fgf18 (Fgf18−/−) have defects in chondrogenesis that affect both proximal and distal skeletal elements. Fgf18−/− mice also have defects in osteoblast proliferation and differentiation that suggest signaling to FGFR1 and FGFR2 in osteoprogenitor cells (discussed below). Mice lacking both Fgf9 and Fgf18 have a severe osteochondrodysplasia that affects proximal and distal skeletal elements (IH Hung, CG Schoenwolf, M Lewandoski, and DM Ornitz, in prep.).
Autosomal dominant mutations in FGF9 in both mice and humans affect chondrogenesis and formation of joints. The elbow knee synostosis (EKS) mutation in mice and the multiple synostosis syndrome in humans reduce the affinity of FGF9 for its coreceptor, HS. Although reduced affinity for HS impairs receptor binding, the resulting increased diffusion of FGF9 through the extracellular matrix effectively increases its domain of activity in developing chondrocytes and surrounding tissue (Harada et al. 2009; Kalinina et al. 2009; Wu et al. 2009). The joint fusion phenotype is very similar to mice that cell-autonomously overexpress an activated FGFR in developing chondrocytes (Wang et al. 2001).
Cortical, trabecular, and intramembranous bone
FGFR signaling in osteoblasts
In cells of the osteoblast lineage, FGFR-mediated activation of ERK1/2, PLCγ/PKCα, and PI3K/Akt signaling results in the modulation of cell proliferation, differentiation, and apoptosis, depending on the stage of cell differentiation (Fig. 3; Dailey et al. 2005; Marie et al. 2005). In early osteoblast precursor cells, activation of ERK1/2 by FGFR signaling primarily leads to increased cell proliferation (Choi et al. 2008; Miraoui et al. 2009). In more mature cells, ERK1/2 activation by FGF2 enhances acetylation and stabilization of RUNX2, a key transcription factor involved in osteoblastogenesis and bone formation (Xiao et al. 2002; Park et al. 2010; Yoon et al. 2014). ERK1/2 signaling is also involved in Runx2 expression and osteoblast differentiation induced by FGF18-mediated activation of FGFR1/FGFR2 in murine osteoblast precursor cells (Hamidouche et al. 2010). PKCδ activation also plays a central role in FGF/FGFR-stimulated expression and transactivation activity of RUNX2 (Kim et al. 2003; Niger et al. 2013). In turn, RUNX2 enhances the expression of Fgfr2, Fgf18, and proteoglycans that are involved in FGF signaling in the perichondrium (Hinoi et al. 2006; Reinhold and Naski 2007; Teplyuk et al. 2009). Finally, in an osteocyte cell line, FGF2-stimulated ERK1/2 activation was found to increase the expression of Dmp1, a marker of osteocytes. In vivo, Dmp1 expression is reduced in limbs of mice lacking ERK1 and conditionally lacking ERK2 (Prx1-Cre), which show no osteocytes, indicating that FGF signaling coordinately regulates Dmp1 expression and osteocyte differentiation (Kyono et al. 2012). Overall, these studies establish that multiple pathways activated by FGF/FGFR signaling control all steps of osteoblastogenesis (Fig. 3).
Figure 3.
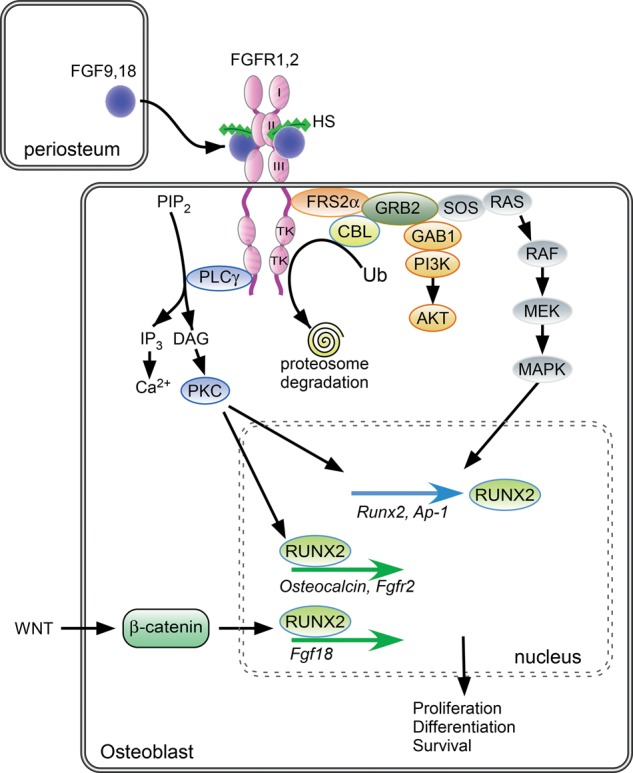
FGF/FGFR signaling in cells of the osteoblast lineage. FGF9 and FGF18, expressed in periosteal and surrounding tissue, interact with HS, FGFR1, and FGFR2. Activation of the FGFR tyrosine kinase domain leads to the recruitment of various substrates and activation of phospholipase Cγ/PKCα, ERK MAPKs, and PI3K/AKT, resulting in the modulation of transcription factors that control cell proliferation, differentiation, and survival in cells of the osteoblast lineage. Following FGFR activation, CBL is recruited and mediates FGFR ubiquitination and proteasome degradation, leading to FGFR down-regulation. Activated FGFRs may also be retained in intracellular compartments and degraded following c-Cbl-dependent ubiquitination. FGFR signaling activates expression of the transcription factor RUNX2, which regulates the expression of osteoblast-specific genes. In cooperation with Wnt/β-catenin signaling, RUNX2 activates Fgf18 gene expression. See the text for more details.
FGFR signaling is down-regulated by negative feedback mechanisms involving receptor internalization and degradation (Schlessinger 2000; Lemmon and Schlessinger 2010). This down-regulation process involves FGFR interaction with multiple proteins, including the docking protein FRS2α and the ubiquitin ligase c-CBL, an adaptor protein that mediates FGFR ubiquitination after ligand binding (Schmidt and Dikic 2005). In response to FGF stimulation, tyrosine phosphorylation of FRS2α forms a ternary complex with CBL and GRB2 (Schlessinger 2003). Increased binding of activated FGFR2 to FRS2α and c-CBL results in ubiquitination of the FGFR and its subsequent degradation by the proteasome (Schlessinger 2003). Depending on the cell type, a proportion of the constitutively activated FGFRs may be retained in intracellular compartments and degraded following c-Cbl-dependent ubiquitination (Hatch et al. 2006). In osteoblasts, CBL plays an important role in down-regulation of activated FGFR signaling and subsequently in the control of osteoblastogenesis (Severe et al. 2013). FGFR2 ubiquitination by c-CBL leads to FGFR2 down-regulation and subsequent reduction in osteogenic differentiation capacity, whereas inhibition of c-CBL interaction with FGFR2 promotes osteoblast differentiation and osteogenic differentiation through increased ERK1/2 and PI3K signaling (Dufour et al. 2008). This highlights the essential role of c-CBL-mediated regulation of FGFR signaling in osteoblastogenesis (Severe et al. 2013).
Interactions with other pathways
In addition to cell signaling directly generated by activated FGF/FGFRs, FGF interacts with other pathways, such as BMP signaling, to regulate osteoblastogenesis. FGF2 treatment stimulates Bmp2 gene expression in osteoblasts during cranial bone ossification (Choi et al. 2005; Fakhry et al. 2005). Consistently, BMP2 expression is reduced in bones of Fgf2-null mice (Naganawa et al. 2008). In line with this concept, BMP2 and FGF2 have a synergistic stimulatory effect on osteogenic function in human bone cells and in mice (Nakamura et al. 2005; Kuhn et al. 2013). Mechanistically, FGF2 enhances canonical BMP2 signaling in osteoblasts through promoting nuclear localization of RUNX2 and phospho-SMAD1/5/8 (Agas et al. 2013). Moreover, FGF2 isoforms specifically modulate BMP2 function and osteoblast differentiation (Sabbieti et al. 2013), and FGF2, FGF9, and FGF18 inhibit the expression of the BMP antagonist Noggin in vitro and in vivo (Warren et al. 2003; Reinhold et al. 2004; Fakhry et al. 2005). Thus, multiple mechanisms mediate FGF and BMP signaling interactions, leading to regulation of osteoblast differentiation.
Genetic and functional evidence indicates that FGF signaling functionally interacts with Wnt/β-catenin signaling to control mesenchymal stem cell fate and differentiation (Dailey et al. 2005; Miraoui and Marie 2010). During cranial suture development, WNT/β-catenin signaling controls stem cell renewal, proliferation, and lineage specification by balancing the activity of the FGF and BMP signaling pathways (Maruyama et al. 2010). Mechanistically, WNT/β-catenin signaling induces Fgf18 expression through induction of a complex with RUNX2 and TCF/LEF transcription factors on the Fgf18 promoter (Fig. 3; Reinhold and Naski 2007). In osteoprogenitor cells, FGF2 was found to antagonize WNT/β-catenin signaling (Mansukhani et al. 2005; Ambrosetti et al. 2008). In vivo, however, Wnt genes are decreased significantly in osteoblasts from Fgf2−/− mice, which display reduced osteoblast differentiation (Fei et al. 2011). Consistent with this, exogenous FGF2 can promote β-catenin nuclear accumulation and rescue osteoblast differentiation in Fgf2−/− mice, indicating that endogenous FGF2 stimulates cell differentiation in more mature osteoblasts in part by activating WNT/β-catenin signaling (Fei et al. 2011).
FGF signaling also interacts with the PTH signaling pathway as PTH stimulates Fgf2, Fgfr1, and Fgfr2 in osteoblast cells (Hurley et al. 1999). Consistently, the anabolic response to intermittent PTH is impaired in Fgf2-null or heterozygous mice (Hurley et al. 1999). Functional studies confirmed that endogenous FGF2 is required for the positive PTH effects on osteoblast proliferation, differentiation, and survival, suggesting that the anabolic PTH effect is dependent in part on FGF2 expression (Sabbieti et al. 2009). These genetic and functional analyses support the concept that FGF/FGFR signaling by itself and its interactions with other major signaling pathways play essential roles in controlling osteoblastogenesis and bone formation in adult life.
FGF and bone homeostasis
FGF/FGFR signaling in osteoblastogenesis
Bone formation is a complex process that involves the commitment of mesenchymal cells that progressively differentiate into osteoblast precursor cells and then into mature bone-forming cells under the control of transcription factors, systemic hormones, and local growth factors as well as cell–cell and cell–matrix interactions (Lian et al. 2006; Marie 2008; Long 2012). While it is well accepted that FGF/FGFR signaling plays an important role in osteoblastogenesis, it is now clear that the effects in osteogenic cells are complex, as they depend on the type of FGFs and FGFRs expressed, the stage of cell maturation, and the microenvironment (proteoglycans and interacting proteins) that may either enhance or attenuate FGF/FGFR signaling in bone cells. The collective data suggest that FGF2 signaling increases cell proliferation in immature osteoblasts and thereby expands the pool of osteoblast precursor cells that then fully differentiate in the bone environment (Debiais et al. 1998; Shimoaka et al. 2002; Ignelzi et al. 2003; Fakhry et al. 2005). Accordingly, blocking FGF2 biological activity reduces osteogenesis in vivo (Moore et al. 2002). Recent data indicate that FGF18 is also an essential autocrine-positive regulator of the osteogenic differentiation program (Jeon et al. 2012), mediated by FGFR1/FGFR2 activation of ERK1/2 and PI3K signaling (Hamidouche et al. 2010). Genetic studies in mice also demonstrated that both FGF2 and FGF18 are important regulators of skeletal cell proliferation and differentiation during osteogenesis. Mice lacking Fgf2 display reduced bone formation due to decreased osteoblast differentiation (Montero et al. 2000; Xiao et al. 2010). Fgf18-deficient mice show decreased osteogenic mesenchymal cell proliferation, reduced osteoblast differentiation, and delayed ossification (Liu et al. 2002; Ohbayashi et al. 2002). In addition to controlling osteoblast proliferation and differentiation, FGF signaling regulates osteoblast apoptosis. In osteoblast precursor cells, FGF2 induces osteoblast survival through activation of PI3K/AKT signaling (Debiais et al. 1998) and FGFR1-mediated increased Bcl2/Bax ratio (Agas et al. 2008). In more differentiated osteoblasts, FGF treatment or overexpression of FGF2 in transgenic mice induces apoptosis in mouse calvaria (Mansukhani et al. 2000; Ignelzi et al. 2003), which limits the increase in the osteoblast population.
FGF regulation of bone metabolism
Recent data indicate that some isoforms of FGF2 have distinct effects on bone metabolism. Overexpression of the low-molecular-weight (18-kDa) isoform of FGF2 in osteoblasts causes increased trabecular and cortical bone mass resulting from increased bone formation, whereas its inactivation results in opposite effects, indicating that low-molecular-weight FGF2 is a critical determinant of bone mass in mice (Xiao et al. 2009). Mechanistic analysis showed that these effects are related to modulation of the WNT/β-catenin signaling pathway (Xiao et al. 2009). In contrast, overexpression of nuclear-localized high-molecular-weight FGF2 isoforms in osteoblasts causes a phenotype similar to X-linked hypophosphatemia (XLH), a disease characterized by increased expression of FGF23 by osteocytes.
FGF23 is a phosphaturic agent, and its overexpression results in phosphate wasting, hypophosphatemia, and rickets/osteomalacia. High-molecular-weight FGF2 overexpression in osteoblast precursor cells results in the inhibition of osteoblast differentiation and matrix mineralization through FGF23/FGFR/MAPK signaling, independently of phosphate wasting (Xiao et al. 2013). Conversely, genetic inactivation of the FGF2 high-molecular-weight isoforms caused increased bone mass due to increased osteoblast differentiation and decreased oscteoclast activity associated with decreased FGF23 expression, indicating a negative impact of high-molecular-weight FGF2 isoforms on bone cell metabolism and phosphate homeostasis (Homer-Bouthiette et al. 2014). These findings highlight the functional implications of distinct FGF2 isoforms on FGF23 secretion, bone cell activity, and bone matrix mineralization.
Functionally, the expression of FGF23 in osteoblasts and osteocytes is controlled by FGFR1 signaling (Martin et al. 2011), which in turn controls phosphate homeostasis and bone mineralization (Quarles 2012; Feng et al. 2013). Low-molecular-weight FGF2 stimulates FGF23 promoter activity through PLCγ/NFAT and MAPK signaling, whereas high-molecular-weight FGF2 promotes FGF23 promoter activity through cAMP-dependent binding of FGFR1 and CREB to a conserved cAMP response element in the FGF23 promoter (Han et al. 2015). Consistently, conditional deletion of Fgfr1 in osteocytes of Hyp mice, a mouse model of XLH, reduced the excessive FGF23 levels produced by osteocytes and partially rescued the bone phenotype in these mice (Xiao et al. 2014). Moreover, pharmacological activation of FGFR1 in osteoblasts leads to increased FGF23 secretion and hypophosphatemia in adult mice (Wu et al. 2013), whereas pharmacological FGFR inhibition ameliorates FGF23-mediated hypophosphatemic rickets (Wohrle et al. 2013). These data support an important role for FGF23/FGFR1 signaling in the control of bone mass and mineralization in vivo. The role of FGF23 signaling in nonskeletal tissues and on phosphate metabolism has been extensively reviewed (Quarles 2012; Liao 2013; White et al. 2014).
Stage-specific activity of FGFs in osteoblastogenesis
FGFR expression and function control cells of the osteoblast lineage at different stages of differentiation. In mesenchymal osteoprogenitor cells, FGFR1/2 are important for maintaining mesenchymal stem cell stemness by inhibiting cell senescence (Coutu et al. 2011; Di Maggio et al. 2012). During the osteogenic differentiation process, initially high Fgfr1/Fgfr2 expression is associated with FGF2 responsiveness and a robust proliferative response (Cowan et al. 2003; Haupt et al. 2009). In more mature osteoblasts, decreased expression of Fgfr1 and increased expression of Fgfr2, Fgfr3, and Fgfr4 are associated with an inability to sustain an FGF2 response (Haupt et al. 2009). In vivo studies confirmed that FGFR1 controls osteoblasts at different stages of maturation (Jacob et al. 2006). Conditional inactivation of Fgfr1 in osteoprogenitor cells results in increased proliferation and delayed differentiation and matrix mineralization, whereas Fgfr1 deficiency in differentiated osteoblasts caused enhanced mineralization and increased expression of Fgfr3 (Jacob et al. 2006). FGFR2 also plays an important role in osteoblastogenesis. In mesenchymal cells, constitutive Fgfr2 activation results in increased osteoblast differentiation and decreased adipocyte differentiation through ERK1/2 and PKCα signaling, resulting in increased Runx2 expression (Miraoui et al. 2009, 2010b). Consistently, mice conditionally lacking Fgfr2 or harboring a mutation in Fgfr2c, the mesenchymal splice variant of Fgfr2, show decreased Runx2 expression, retarded ossification, and decreased bone mineral density, suggesting that Fgfr2 is a positive regulator of osteoblast maturation (Eswarakumar et al. 2002; Yu et al. 2003). Although FGFR3 is mainly expressed in chondrocytes, young adult Fgfr3-null mice are osteopenic due to reduced osteoblast maturation and defective trabecular bone mineralization, suggesting a role for FGFR3 in postnatal bone metabolism (Valverde-Franco et al. 2004). Osteoblasts derived from Stat1−/− mice have decreased expression of Fgfr3 and increased expression of Fgf18, which acts through FGFR1 or FGFR2, suggesting that changes in the repertoire of Fgfr expression affects feedback loops that control FGF-dependent osteoblastogenesis (Xiao et al. 2004).
FGFs bind cell surface HSPGs, which serve as low-affinity coreceptors that enhance FGF–FGFR binding and signaling (Eswarakumar et al. 2005; Ornitz and Itoh 2015). Early studies showed that some HSPGs, such as syndecans, expressed in the bone environment affect the osteoblast response to FGF2 during osteogenesis (Molteni et al. 1999a). In vitro data indicate that both cell surface and secreted HSPGs can further amplify FGF2 signaling and osteoblast differentiation (Jackson et al. 2007). Notably, the HSPG glypican-3 was found to be essential for Runx2 expression and subsequent osteoblast differentiation in vitro (Haupt et al. 2009). Since RUNX2 was found to enhance the expression of FGFRs and proteoglycans such as syndecans in osteoblasts, RUNX2 and the FGF/proteoglycan axis likely cooperate to control osteoblast proliferation and differentiation (Fromigue et al. 2004; Teplyuk et al. 2009). Thus, the regulation of osteogenesis by FGF/FGFR signaling is dependent on not only the expression of a repertoire of FGFs and FGFRs but also the expression of accessory extracellular molecules that regulate cell signaling and FGF/FGFR biological activity.
FGF signaling in osteoclasts
Early studies suggested that FGF2 regulates bone resorption directly through its effect on osteoclast precursor proliferation and indirectly by promoting prostaglandin G/H synthase-2 (Ptgs2) expression and prostaglandin formation (Kawaguchi et al. 1995, 2000; Chikazu et al. 2001). Additionally, FGF2 can act directly on mature osteoclasts to stimulate bone resorption through the activation of FGFR1 and MAPK (Chikazu et al. 2000; Kawaguchi et al. 2000). A role for FGF2 in bone resorption is supported by the finding that ostoclastogenesis in response to PTH is markedly impaired in Fgf2-null mice (Okada et al. 2003). FGF18 can also induce osteoclast formation and function indirectly through stimulating receptor activator of the nuclear factor-κB ligand (RANKL) and cyclooxygenase-2 expression in osteoblasts (Shimoaka et al. 2002). Another study showed that FGF6 increased the formation and resorbing activity of human primary mature osteoclasts in vitro (Bosetti et al. 2010), whereas FGF8a inhibited osteoclastogenesis, but not osteoclast function, in mouse bone marrow cultures.
FGFR1 was detected on isolated mouse osteoclasts (Chikazu et al. 2000). Osteoclasts with FGFR1 deficiency (LysM-Cre mice) displayed decreased expression of genes related to osteoclast activity, including TRAP and MMP9, and exhibited abnormal bone remodeling with reduced osteoclast number and impaired osteoclast function (Lu et al. 2009). On the other hand, osteoclast numbers and bone resorption area were increased in cultured bone marrow cells derived from Fgfr3G369C/+ mice, suggesting that gain-of-function mutations in FGFR3 increase osteoclast activity, which contributes to decreased bone mass in these mice (Su et al. 2010).
Mutations in FGFRs in human skeletal disease
Mutations in FGFRs, first revealed with the discovery of the genetic etiology of achondroplasia as a missense mutation in FGFR3 (Shiang et al. 1994), highlight the importance of FGF signaling in skeletal development. Following this initial discovery, many mutations have been identified in FGFR1–3 in related skeletal dwarfism syndromes, craniosynostosis syndromes, and skeletal overgrowth syndromes (Ornitz and Itoh 2015). The clinical and genetic features of these diseases have been extensively reviewed (Vajo et al. 2000; Ornitz and Marie 2002; Chen and Deng 2005; Marie et al. 2005; Ornitz 2005; Melville et al. 2010; Johnson and Wilkie 2011; Foldynova-Trantirkova et al. 2012; Krejci 2014).
Chondrodysplasia syndromes
Achondroplasia is the most common form of skeletal dwarfism in humans, and >97% of cases result from an autosomal dominant missense mutation (G380R) in FGFR3 (Shiang et al. 1994; Wilkin et al. 1998; Vajo et al. 2000). Related autosomal dominant chondrodysplasia syndromes include the more severe and usually lethal thanatophoric dysplasia types I and II, which are attributed to two mutations in FGFR3 (K650E and R248C, respectively), and the milder form of dwarfism, hypochondroplasia, which is caused by an N540K or K650N mutation in FGFR3 (Bellus et al. 1995, 2000; Tavormina et al. 1995; Bonaventure et al. 1996). A closely related disease, referred to as severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN) (Bellus et al. 1999; Tavormina et al. 1999), and a very rare lethal chondrodysplasia, platyspondylic lethal skeletal dysplasia, San Diego type (Brodie et al. 1999) are both caused by a K650M mutation in FGFR3. A dominant mutation (M528I) that causes proportionate short stature was recently identified in FGFR3 (Kant et al. 2015). Functional studies suggest that this mutation is activating, similar to that of the G380R mutation that causes achondroplasia; however, the mechanisms that determine proportionate versus rhizomelic limb shortening are not known.
The mutations in FGFR3 associated with dwarfism activate the receptor to varying degrees (Naski et al. 1996; Krejci et al. 2008b). The N540K mutation causing hypochondroplasia is a weak gain-of-function mutation, and mutations causing achondroplasia and thantophoric dysplasia result in progressively stronger activation and, in the case of the R248C mutation, ligand-independent constitutive activation. FGFR3 is expressed prominently in proliferating and prehypertrophic chondrocytes in the growth plate, and activation of FGFR3 results in phenotypes that include decreased chondrocyte proliferation, impaired hypertrophic differentiation, and increased apoptosis (discussed below; Legeai-Mallet et al. 1998, 2004; Naski et al. 1998; Wang et al. 1999, 2001; Krejci et al. 2008b; Pannier et al. 2009).
The genetic homogeneity and relatively high incidence of the achondroplasia G380R mutation suggested that the associated G1138A transition in FGFR3 might be one of the most highly mutable nucleotides in the human genome (Wilkin et al. 1998; Vajo et al. 2000). However, the paternal origin of the mutation and advanced paternal age associated with achondroplasia suggested that a germline selection model in which mutant spermatogonial stem cells have a proliferative or survival advantage over unmutated cells could better account for the observed paternal age effect (Dakouane Giudicelli et al. 2008; Shinde et al. 2013). This paternal age effect also occurs with gain-of-function mutations in FGFR2 that cause craniosynostosis syndromes (Goriely et al. 2003, 2005; Goriely and Wilkie 2012).
Skeletal overgrowth and camptodactyly, tall stature, and hearing loss (CATSHL) syndrome
Inactivation of FGFR3 in mice results in skeletal overgrowth (Colvin et al. 1996; Deng et al. 1996; Eswarakumar and Schlessinger 2007). Growth plate histology in mice lacking Fgfr3 (Fgfr3−/−) showed an expanded proliferating chondrocyte zone and hypertrophic chondrocyte zone beginning at late stages of embryonic development and persisting to adulthood. Bones of the appendicular skeleton were disproportionally long compared with body weight, and the mice developed elongation of vertebral bodies and scoliosis. Fgfr3−/− mice also have primary defects in inner ear development and are consequently deaf (Colvin et al. 1996). The viability of Fgfr3−/− mice and the unique combination of skeletal overgrowth and hearing loss phenotypes suggested that a similar condition resulting from loss of function of FGFR3 could occur in humans. Analysis of a four-generation family with dominantly inherited CATSHL identified a missense mutation (R621H) in the catalytic loop of the FGFR3 tyrosine kinase domain (Toydemir et al. 2006). This mutation inactivates the kinase domain and likely confers dominant-negative activity to the receptor. A homozygous mutation in the FGFR3 tyrosine kinase domain (T546K) has also been identified in two siblings with CATSHL from consanguineous parents (Makrythanasis et al. 2014). Interestingly, a similar recessive mutation in FGFR3(V700E) in sheep results in spider lamb syndrome, characterized by long limbs, kyphoscoliosis, malformed ribs and sternebrae, Roman noses, lack of body fat, and muscular atrophy (Beever et al. 2006). Sheep that are heterozygous for this mutation appear normal but show a mild increase in skeletal growth (Smith et al. 2006).
Craniosynostosis syndromes
Mutations in FGFR1, FGFR2, and FGFR3 cause skeletal dysplasias (craniosynostosis) in Apert, Crouzon, and other syndromes characterized by premature fusion of one or more cranial sutures (Ornitz and Marie 2002; Johnson and Wilkie 2011). Mutations in FGFRs also result in dysmorphology of the appendicular skeleton, such as in bent bone dysplasia, which is caused by a mutation in FGFR2 (Merrill et al. 2012). The molecular mechanisms that result from FGFR mutations are complex and include constitutive (ligand-independent) or ligand-dependent FGFR activation, loss of function, and altered cellular trafficking of receptors (Yu et al. 2000; Ibrahimi et al. 2001; Yu and Ornitz 2001; Neben et al. 2014). Genetic, cellular, and molecular studies in mice and humans provided clues to the abnormalities induced by gain-of-function FGFR mutations (Hajihosseini 2008; Marie et al. 2008; Senarath-Yapa et al. 2012). Specifically, Apert FGFR2(S252W) mutations were shown to induce abnormal mesodermal progenitor cell proliferation, differentiation, and cell fate in cranial sutures in various mouse models, although the cellular abnormalities vary among specific sutures and between cells at distinct stages of differentiation in a particular suture (Wang et al. 2005; Holmes et al. 2009; Heuze et al. 2014; Motch Perrine et al. 2014).
At an early stage of development, osteoprogenitor cell proliferation in cranial sutures was unchanged (Chen et al. 2003) or transiently and moderately increased by Apert- and Crouzon-activating FGFR2 mutations (Mansukhani et al. 2000, 2005). At a later stage, activating FGFR2 mutations were widely found to cause increased osteoblast marker gene expression and accelerated osteoblast maturation in cranial sutures (Eswarakumar et al. 2004; Yang et al. 2008; Yin et al. 2008; Holmes et al. 2009; Yeh et al. 2012; Liu et al. 2013a,b; Morita et al. 2014). This finding in mice supports the original observation in humans that Apert and Crouzon FGFR2 mutations cause increased osteoblast maturation and function in postnatal human suture development (Lomri et al. 1998; Lemonnier et al. 2001b; Tanimoto et al. 2004; Baroni et al. 2005; Marie 2015). FGFR2 mutations are also associated with increased osteoblast apoptosis in fused sutures in mice (Mansukhani et al. 2005; Chen et al. 2014) and humans (Lemonnier et al. 2001a; Lomri et al. 2001; Kaabeche et al. 2005); however, this appears to be a secondary event that occurs subsequent to osteoblast maturation and not the primary cause of the synostosis (Holmes et al. 2009). Overall, the current literature indicates that the premature cranial ossification induced by FGFR2 mutations relates to an accelerated maturation of osteoblasts, whereas the moderate and transient increase in osteoprogenitor cell recruitment and the late osteoblast apoptosis do not appear to contribute primarily to the pathogenesis of the synostosis. The mutations in FGFR2 that causes bent bone dysplasia (M391R or Y381D) reduce ligand-dependent receptor activation and lead to altered trafficking of the mutant receptor to the nucleolus. Increased nucleolar activity of FGFR2 increases ribosomal RNA expression to promote proliferation and suppresses differentiation of osteoprogenitor cells (Merrill et al. 2012; Neben et al. 2014).
Besides FGFR1 and FGFR2 mutations, activating FGFR3 mutations in achondroplasia may also cause partial premature fusion of the coronal sutures, suggesting a role for FGFR3 activation in membranous ossification (Twigg et al. 2009; Di Rocco et al. 2014). Indeed, chondrocyte-specific activation of Fgfr3 in mice was found to induce premature suture closure and indirectly enhance osteoblast differentiation through MAPK activation and BMP signaling (Matsushita et al. 2009b). In long bones, gain-of-function FGFR3 mutations in chondrocytes, but not in osteoblasts, caused increased osteoclast recruitment, defective mineralization, and decreased trabecular bone mass, indicating that overactive FGFR3 signaling indirectly affects trabecular bone development (Su et al. 2010; Mugniery et al. 2012).
Intracellular pathways involved in craniosynostosis
Genetic and functional studies indicate that the premature suture fusion induced by FGFR mutations occurs as a consequence of activation of signaling pathways and downstream target genes. In several mouse models, Apert FGFR2(S252W) mutations activate ERK1/2, p38 MAPK, AKT, β-catenin, and PLCγ in osteogenic cells, which may contribute to premature cranial ossification (Chen et al. 2003; Kim et al. 2003; Shukla et al. 2007; Yin et al. 2008; Wang et al. 2010; Suzuki et al. 2012). Interestingly, reduced dosage of ERF, an inhibitory ETS transcription factor directly bound by ERK1/2 signaling, was found to cause craniosynostosis in humans and mice, implying a role of ERF downstream from ERK signaling in cranial suture ossification (Twigg et al. 2013). In human craniosynostosis, increased osteoblast gene expression and cranial osteogenesis induced by Apert FGFR2(S252W) mutation are related to PLCγ and PKCα activation (Lemonnier et al. 2001b). The consequence is increased expression of Runx2, which is associated with premature fusion of cranial sutures in mice and humans (Zhou et al. 2000; Eswarakumar et al. 2002; Tanimoto et al. 2004; Baroni et al. 2005; Guenou et al. 2005). In both mice and humans, FGFR2 levels are decreased in response to the Apert FGFR2(S252W) mutation as a result of c-CBL-mediated FGFR2 ubiquitination and degradation (Kaabeche et al. 2004; Holmes et al. 2009). In addition, c-CBL recruitment by Apert FGFR2(S252W) in human osteoblasts causes ubiquitination and down-regulation of the Src family members LYN and FYN (Kaabeche et al. 2004) and α5β1 integrin (Kaabeche et al. 2005), resulting in increased osteoblast differentiation and apoptosis, respectively. Increased c-CBL recruitment in Apert osteoblasts also leads to PI3K ubiquitination and degradation, resulting in attenuation of PI3K signaling and reduced osteoblast survival (Dufour et al. 2008). Thus, both FGFR-mediated activation of signaling pathways and down-regulation of regulatory molecules contribute to the cellular events in craniosynostosis.
Recent studies indicate that FGFR signaling interacts with other pathways to induce craniosynostosis. In human osteoblasts, Apert FGFR2(S252W) mutations cause increased expression of platelet-derived growth factor receptor α (PDGFRα) and epidermal growth factor receptor (EGFR) in osteoblasts via activation of PKCα-mediated AP-1 transcription, which contributes to premature suture ossification (Miraoui et al. 2010a). Consistent with these data, transgenic mice conditionally expressing an constitutively activated PDGFRα in neural crest cells display activation of PLCγ, resulting in premature fusion at early postnatal stages (Moenning et al. 2009). FGFR2 activation caused by Apert FGFR2(S252W) mutations also leads to increased c-CBL–Sprouty2 interaction and c-CBL sequestration in human osteoblasts, causing increased EGFR levels and signaling and increased osteoblast gene transcription (Miraoui et al. 2010a). Thus, both transcriptional and post-transcriptional mechanisms mediate the aberrant signaling pathways induced by FGFR2 mutations in cells of the osteoblast lineage.
Therapeutic strategies for FGF-related skeletal disease
Therapeutic strategies in achondroplasia
Skeletal drawfism in hypochondroplasia, achondroplasia, and thanatophoric dysplasia results from increased FGFR3 signaling in proliferating and hypertrophic chondrocytes (Foldynova-Trantirkova et al. 2012). For achondroplasia, in which affected individuals attain adult heights that are well below the fifth percentile, pharmacological therapy promises to offer an alternative to painful and costly surgical therapies that are currently used to increase height and correct disproportional bone growth.
Several approaches have been proposed to suppress FGFR3 signaling with the primary goal of enhancing skeletal growth (Laederich and Horton 2012). Therapeutic strategies can target FGF ligands that activate FGFR3, the activated receptor, signaling pathways that are downstream from FGFR3, signaling pathways that modulate FGFR3 intracellular pathways, or signaling pathways that are independent of FGFR3 signaling. Tyrosine kinase inhibitors that are selective for FGFRs have thus far not been effective in vivo because of difficulties in achieving therapeutic levels of inhibitors in the avascular growth plate and potential side effects of systemic inhibition of FGFRs over the long periods for time that will be required for treatment (Laederich and Horton 2012).
Human growth hormone was evaluated as a therapy; however, no long-term benefits were observed (Kanazawa et al. 2003; Horton et al. 2007). An antihistamine drug, meclozine dihydrochloride, has been shown to inhibit FGFR3 signaling in chondrocytes by suppressing ERK1/2 phosphorylation. Meclozine functioned to enhance bone growth in explant culture and increase bone growth in wild-type and achondroplasia mice (Matsushita et al. 2013, 2015). Inhibition of ligands that activate FGFR3 can be achieved through expression of a soluble extracellular domain of FGFR3 (sFGFR3) that can compete with endogenous FGFR3 by binding FGF ligands such as FGF9 and FGF18 that functionally regulate the growth plate (Liu et al. 2002, 2007; Ohbayashi et al. 2002; Hung et al. 2007; Garcia et al. 2013). Subcutaneous injections of recombinant sFGFR3 in a mouse model for achondroplasia was found to decrease mortality and improve skeletal growth (Garcia et al. 2013). Inhibitory antibodies that specifically target the FGFR3 extracellular domain have been developed as potential cancer therapeutics but have not been evaluated for treatment of achondroplasia (Martinez-Torrecuadrada et al. 2005; Hadari and Schlessinger 2009; Qing et al. 2009). A recent drug screen of induced pluripotent stem cell-derived chondrocytes from patients with achondroplasia and thanatophoric dysplasia identified statin drugs as effective in improving chondrogenic differentiation in vitro and improving the phenotype (limb and body length) of achondroplasia mice in vivo (Yamashita et al. 2014).
The most promising therapy thus far for treatment of achondroplasia is the use of a stabilized form of CNP called BMN-111 (Lorget et al. 2012; Wendt et al. 2015). BMN-111 is currently undergoing clinical trials for the treatment of achondroplasia (https://clinicaltrials.gov/ct2/show/NCT02055157). Mice or humans that overexpress CNP or activate natriuretic peptide receptor 2 (Npr2; guanylyl cyclase B) have a skeletal overgrowth phenotype (Fig. 2; Yasoda et al. 2004; Bocciardi et al. 2007; Hannema et al. 2013; Miura et al. 2014), while mice that lack CNP (Nppc−/−) or humans with heterozygous mutations in NPR2 exhibit skeletal dwarfism (Tsuji and Kunieda 2005; Nakao et al. 2015).
CNP signals through NPR2 in chondrocytes and inhibits the MAPK signaling pathway at the level of RAF1 (Fig. 2). Several studies suggest a mechanism in which CNP-induced cGMP activates cyclic GMP-dependent protein kinase II (cGKII; encoded by PRKG2) and p38 (MAPK14), which functionally antagonizes RAF1 activation of MEK (MAP2K1), which is a critical pathway regulating chondrocyte hypertrophy (Murakami et al. 2004; Ozasa et al. 2005; Agoston et al. 2007; Hutchison 2012; Peake et al. 2014). Interestingly, FGFR3–MAPK signaling may also inhibit cGMP production in chondrocytes (Ozasa et al. 2005). Thus, signaling by FGF ligands (FGF9 and FGF18) through FGFR3 functionally antagonizes CNP signaling through NPR2 to regulate the rate of chondrocyte hypertrophy. Consistent with feedback regulation of the FGF and CNP pathways in chondrocytes, patients with activating mutations in FGFR3 have increased circulating levels of CNP (Olney et al. 2015).
Therapeutic strategies in craniosynostosis
The identification of the aberrant signals induced by FGFR mutations provided clues for potential therapeutic nonsurgical interventions in craniosynostosis (Wilkie 2007; Melville et al. 2010). In vitro, overactivated FGFR2 signaling by Apert and Crouzon mutations can be attenuated by specific glycosaminoglycans (McDowell et al. 2006), FGFR or tyrosine kinase inhibitors (Perlyn et al. 2006), or a soluble mutant form of FGFR2 that exerts a dominant-negative effect on FGFR2 (Tanimoto et al. 2004). In vivo, selective uncoupling of the docking protein FRS2α and the Crouzon-like activated FGFR2c mutant was found to attenuate FGFR signaling and prevent premature suture fusion in mice (Eswarakumar et al. 2006). In mice, treatments with a shRNA targeting the Apert FGFR2(S252W) mutation or the use of a ERK1/2 MAPK inhibitor (Shukla et al. 2007) or a soluble mutant form of FGFR2 (Morita et al. 2014; Yokota et al. 2014) were also found to prevent craniosynostosis. The recent finding that FGFR signaling interacts with PDGFRα signaling to induce premature suture fusion in mice and humans may offer alternative therapeutic strategies to indirectly attenuate signals induced by activating FGFR mutations (Moenning et al. 2009; Miraoui et al. 2010a). These recent studies may thus provide promising therapeutic strategies to efficiently reduce the aberrant FGFR signaling induced by Fgfr2 mutations in craniosynostosis.
FGF and bone regeneration
Fgf and Fgfr expression are regulated during fracture healing (Schmid et al. 2009), and there are accumulating data indicating that FGF/FGFR signaling plays active roles during the fracture repair process (Du et al. 2012; Fei et al. 2013). These studies led to the wide use of FGF2 as a therapeutic agent for promoting bone regeneration in animal models (Fei et al. 2013). Typically, the local implantation of FGF2 impregnated in a gelatin hydrogel or a molecular scaffold or the injection of chemically controlled FGF2-secreting cells was found to promote bone healing in cranial or long bone defects (Chen et al. 2004; Komaki et al. 2006; Kodama et al. 2009; Kawaguchi et al. 2010; Kwan et al. 2011). However, the effects on bone healing are dependent on the dose used, the time of application, and the duration of FGF treatment (Du et al. 2012; Fei et al. 2013). The anabolic effect of locally injected FGF2 results mainly from increased expansion of the pool of osteoblast precursor cells, resulting in their differentiation into osteoblasts (Nakamura et al. 1998; Nakajima et al. 2007). In addition, FGF2 may also increase angiogenesis at the site of injury. A close spatial and temporal relationship exists between angiogenesis and osteogenesis during bone repair and endochondral bone formation, and the connection between the two processes is essential for bone formation (Dirckx et al. 2013; Saran et al. 2014). Recent genetic studies indicate that FGF/FGFR signaling plays an essential role in both processes. It was shown that FGF9−/+ mice display reduced repair of cortical bone defects resulting from decreased osteoblast proliferation and differentiation associated with impaired neovascularization and reduced Vegf expression. This phenotype was rescued by exogenous FGF9, indicating that endogenous FGF9 is important for cortical bone repair in part through regulating Vegf expression (Behr et al. 2010). In addition to promoting angiogenesis, VEGF controls bone formation by stimulating the differentiation of mesenchymal stem cells to osteoblasts (Liu and Olsen 2014). Accordingly, VEGF alone has a potent bone repair potential by promoting angiogenesis and osteogenesis in vivo (Behr et al. 2012). Since FGF2 promotes Vegf expression in osteoblasts (Takai et al. 2007), these two factors may promote bone repair through activation of both angiogenesis and osteogenesis. Fgf18−/+ mice also showed reduced cortical bone defect healing capacity but due to decreased osteoblast differentiation with normal angiogenesis, showing that FGF18 is required for osteogenesis but not angiogenesis during bone repair (Behr et al. 2011). In a mouse model of skeletal prosthesis insertion, a local injection of FGF18 resulted in increased peri-implant bone formation in both Fgfr3+/+ and Fgfr3−/− mice, indicating that FGF18 can promote bone repair independently of FGFR3 (Carli et al. 2012), presumably through activation of FGFR1 and/or FGFR2. In line with this concept, it was recently found that DJ-1 (PARK7), an angiogenic protein, stimulates both osteoblast differentiation and angiogenesis via FGFR1 activation, resulting in enhanced long bone fracture repair in rats, further highlighting the importance of FGFR1 signaling in fracture repair (Kim et al. 2012). Overall, these studies uncover novel roles for FGF/FGFR signaling in bone repair through activation of both angiogenesis and osteogenesis, supporting the concept that FGFs may have therapeutic potential to stimulate bone regeneration.
Conclusion
During the past decade, significant advances have been made in our understanding of the role of FGF/FGFR signaling in skeletal development, bone formation, and bone diseases. Specifically, the analysis of genetic models in mice led to a better understanding of the complex role of FGF/FGFR signaling in endochondral and intramembranous ossification and bone repair. Also, the analysis of the skeletal phenotype induced by FGFR mutations in various mouse models and human skeletal dysplasias has improved our knowledge of the pathogenesis of chondrodysplasias and craniosynostosis. Finally, genetic and functional studies led to the identification of aberrant signaling mechanisms involved in skeletal diseases associated with FGFR mutations. This knowledge validated the concept that modulating FGFR signaling and downstream signaling pathways can prevent skeletal dysplasias induced by Fgfr mutations in mice, and this important concept may lead to novel therapeutic approaches in human skeletal dysplasias. In the future, further studies are needed to delineate the stage-specific role of FGF/FGFR signaling in cells of the chondrocyte and osteoblast lineage in vivo and characterize FGF interactions with other effectors during normal and aberrant chondrogenesis and osteogenesis. It is expected that a more complete understanding of the role FGF/FGFR signaling in prenatal and postnatal skeletal development will translate into novel therapeutic approaches for human skeletal dysplasias and improved skeletal repair.
Acknowledgments
We thank all team members who contributed to the work reviewed here. We apologize to the investigators whose work could not be cited due to space limitations. This work was supported by National Institutes of Health grant HD049808 (to D.M.O.), the Institut National de la Recherche Médicale, University Paris Diderot Sorbonne Paris Cité, the DIM Stem Pôle Ile de France, the French Minister of Research, and the Asssociation Prévention et Traitement des Décalcifications (Paris, France) (to P.J.M.).
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.266551.115.
References
- Abad V, Meyers JL, Weise M, Gafni RI, Barnes KM, Nilsson O, Bacher JD, Baron J. 2002. The role of the resting zone in growth plate chondrogenesis. Endocrinology 143: 1851–1857. [DOI] [PubMed] [Google Scholar]
- Agas D, Marchetti L, Menghi G, Materazzi S, Materazzi G, Capacchietti M, Hurley MM, Sabbieti MG. 2008. Anti-apoptotic Bcl-2 enhancing requires FGF-2/FGF receptor 1 binding in mouse osteoblasts. J Cell Physiol 214: 145–152. [DOI] [PubMed] [Google Scholar]
- Agas D, Sabbieti MG, Marchetti L, Xiao L, Hurley MM. 2013. FGF-2 enhances Runx-2/Smads nuclear localization in BMP-2 canonical signaling in osteoblasts. J Cell Physiol 228: 2149–2158. [DOI] [PubMed] [Google Scholar]
- Agoston H, Khan S, James CG, Gillespie JR, Serra R, Stanton LA, Beier F. 2007. C-type natriuretic peptide regulates endochondral bone growth through p38 MAP kinase-dependent and -independent pathways. BMC Dev Biol 7: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aikawa T, Segre GV, Lee K. 2001. Fibroblast growth factor inhibits chondrocytic growth through induction of p21 and subsequent inactivation of cyclin E–Cdk2. J Biol Chem 276: 29347–29352. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Chaboissier MC, Martin JF, Schedl A, de Crombrugghe B. 2002. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev 16: 2813–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosetti D, Holmes G, Mansukhani A, Basilico C. 2008. Fibroblast growth factor signaling uses multiple mechanisms to inhibit Wnt-induced transcription in osteoblasts. Mol Cell Biol 28: 4759–4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard JC, Williams AJ, Rabier B, Chassande O, Samarut J, Cheng SY, Bassett JH, Williams GR. 2005. Thyroid hormones regulate fibroblast growth factor receptor signaling during chondrogenesis. Endocrinology 146: 5568–5580. [DOI] [PubMed] [Google Scholar]
- Baroni T, Carinci P, Lilli C, Bellucci C, Aisa MC, Scapoli L, Volinia S, Carinci F, Pezzetti F, Calvitti M, et al. 2005. P253R fibroblast growth factor receptor-2 mutation induces RUNX2 transcript variants and calvarial osteoblast differentiation. J Cell Physiol 202: 524–535. [DOI] [PubMed] [Google Scholar]
- Beever JE, Smit MA, Meyers SN, Hadfield TS, Bottema C, Albretsen J, Cockett NE. 2006. A single-base change in the tyrosine kinase II domain of ovine FGFR3 causes hereditary chondrodysplasia in sheep. Anim Genet 37: 66–71. [DOI] [PubMed] [Google Scholar]
- Behr B, Leucht P, Longaker MT, Quarto N. 2010. Fgf-9 is required for angiogenesis and osteogenesis in long bone repair. Proc Natl Acad Sci 107: 11853–11858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr B, Sorkin M, Manu A, Lehnhardt M, Longaker MT, Quarto N. 2011. Fgf-18 is required for osteogenesis but not angiogenesis during long bone repair. Tissue Eng Part A 17: 2061–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr B, Sorkin M, Lehnhardt M, Renda A, Longaker MT, Quarto N. 2012. A comparative analysis of the osteogenic effects of BMP-2, FGF-2, and VEGFA in a calvarial defect model. Tissue Eng Part A 18: 1079–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellus GA, McIntosh I, Smith EA, Aylsworth AS, Kaitila I, Horton WA, Greenhaw GA, Hecht JT, Francomano CA. 1995. A recurrent mutation in the tyrosine kinase domain of fibroblast growth factor receptor 3 causes hypochondroplasia. Nat Genet 10: 357–359. [DOI] [PubMed] [Google Scholar]
- Bellus GA, Bamshad MJ, Przylepa KA, Dorst J, Lee RR, Hurko O, Jabs EW, Curry CJ, Wilcox WR, Lachman RS, et al. 1999. Severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN): phenotypic analysis of a new skeletal dysplasia caused by a Lys650Met mutation in fibroblast growth factor receptor 3. Am J Med Genet 85: 53–65. [PubMed] [Google Scholar]
- Bellus GA, Spector EB, Speiser PW, Weaver CA, Garber AT, Bryke CR, Israel J, Rosengren SS, Webster MK, Donoghue DJ, et al. 2000. Distinct missense mutations of the FGFR3 Lys650 codon modulate receptor kinase activation and the severity of the skeletal dysplasia phenotype. Am J Hum Genet 67: 1411–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov AA, Mohammadi M. 2013. Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology. Cold Spring Harb Perspect Biol 5: 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. 1999. Sox9 is required for cartilage formation. Nat Genet 22: 85–89. [DOI] [PubMed] [Google Scholar]
- Blick C, Ramachandran A, Wigfield S, McCormick R, Jubb A, Buffa FM, Turley H, Knowles MA, Cranston D, Catto J, et al. 2013. Hypoxia regulates FGFR3 expression via HIF-1α and miR-100 and contributes to cell survival in non-muscle invasive bladder cancer. Br J Cancer 109: 50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocciardi R, Giorda R, Buttgereit J, Gimelli S, Divizia MT, Beri S, Garofalo S, Tavella S, Lerone M, Zuffardi O, et al. 2007. Overexpression of the C-type natriuretic peptide (CNP) is associated with overgrowth and bone anomalies in an individual with balanced t(2;7) translocation. Hum Mutat 28: 724–731. [DOI] [PubMed] [Google Scholar]
- Bonaventure J, Rousseau F, Legeai-Mallet L, Le Merrer M, Munnich A, Maroteaux P. 1996. Common mutations in the fibroblast growth factor receptor 3 (FGFR 3) gene account for achondroplasia, hypochondroplasia, and thanatophoric dwarfism. Am J Med Genet 63: 148–154. [DOI] [PubMed] [Google Scholar]
- Bosetti M, Leigheb M, Brooks RA, Boccafoschi F, Cannas MF. 2010. Regulation of osteoblast and osteoclast functions by FGF-6. J Cell Physiol 225: 466–471. [DOI] [PubMed] [Google Scholar]
- Britto JA, Evans RD, Hayward RD, Jones BM. 2001. From genotype to phenotype: the differential expression of FGF, FGFR, and TGFβ genes characterizes human cranioskeletal development and reflects clinical presentation in FGFR syndromes. Plast Reconstr Surg 108: 2026–2039. [DOI] [PubMed] [Google Scholar]
- Brodie SG, Kitoh H, Lachman RS, Nolasco LM, Mekikian PB, Wilcox WR. 1999. Platyspondylic lethal skeletal dysplasia, San Diego type, is caused by FGFR3 mutations. Am J Med Genet 84: 476–480. [PubMed] [Google Scholar]
- Browaeys-Poly E, Blanquart C, Perdereau D, Antoine AF, Goenaga D, Luzy JP, Chen H, Garbay C, Issad T, Cailliau K, et al. 2010. Grb14 inhibits FGF receptor signaling through the regulation of PLCγ recruitment and activation. FEBS Lett 584: 4383–4388. [DOI] [PubMed] [Google Scholar]
- Buchtova M, Oralova V, Aklian A, Masek J, Vesela I, Ouyang Z, Obadalova T, Konecna Z, Spoustova T, Pospisilova T, et al. 2015. Fibroblast growth factor and canonical WNT/β-catenin signaling cooperate in suppression of chondrocyte differentiation in experimental models of FGFR signaling in cartilage. Biochim Biophys Acta 1852: 839–850. [DOI] [PubMed] [Google Scholar]
- Camps M, Nichols A, Gillieron C, Antonsson B, Muda M, Chabert C, Boschert U, Arkinstall S. 1998. Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science 280: 1262–1265. [DOI] [PubMed] [Google Scholar]
- Carli A, Gao C, Khayyat-Kholghi M, Li A, Wang H, Ladel C, Harvey EJ, Henderson JE. 2012. FGF18 augments osseointegration of intra-medullary implants in osteopenic FGFR3−/− mice. Eur Cell Mater 24: 107–116. [DOI] [PubMed] [Google Scholar]
- Chen L, Deng CX. 2005. Roles of FGF signaling in skeletal development and human genetic diseases. Front Biosci 10: 1961–1976. [DOI] [PubMed] [Google Scholar]
- Chen Y, Gridley T. 2013. Compensatory regulation of the Snai1 and Snai2 genes during chondrogenesis. J Bone Miner Res 28: 1412–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Li C, Qiao W, Xu X, Deng C. 2001. A Ser(365) → Cys mutation of fibroblast growth factor receptor 3 in mouse downregulates Ihh/PTHrP signals and causes severe achondroplasia. Hum Mol Genet 10: 457–465. [DOI] [PubMed] [Google Scholar]
- Chen L, Li D, Li C, Engel A, Deng CX. 2003. A Ser252Trp [corrected] substitution in mouse fibroblast growth factor receptor 2 (Fgfr2) results in craniosynostosis. Bone 33: 169–178. [DOI] [PubMed] [Google Scholar]
- Chen WJ, Jingushi S, Aoyama I, Anzai J, Hirata G, Tamura M, Iwamoto Y. 2004. Effects of FGF-2 on metaphyseal fracture repair in rabbit tibiae. J Bone Miner Metab 22: 303–309. [DOI] [PubMed] [Google Scholar]
- Chen P, Zhang L, Weng T, Zhang S, Sun S, Chang M, Li Y, Zhang B, Zhang L. 2014. A Ser252Trp mutation in fibroblast growth factor receptor 2 (FGFR2) mimicking human Apert syndrome reveals an essential role for FGF signaling in the regulation of endochondral bone formation. PLoS One 9: e87311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikazu D, Hakeda Y, Ogata N, Nemoto K, Itabashi A, Takato T, Kumegawa M, Nakamura K, Kawaguchi H. 2000. Fibroblast growth factor (FGF)-2 directly stimulates mature osteoclast function through activation of FGF receptor 1 and p42/p44 MAP kinase. J Biol Chem 275: 31444–31450. [DOI] [PubMed] [Google Scholar]
- Chikazu D, Katagiri M, Ogasawara T, Ogata N, Shimoaka T, Takato T, Nakamura K, Kawaguchi H. 2001. Regulation of osteoclast differentiation by fibroblast growth factor 2: stimulation of receptor activator of nuclear factor κB ligand/osteoclast differentiation factor expression in osteoblasts and inhibition of macrophage colony-stimulating factor function in osteoclast precursors. J Bone Miner Res 16: 2074–2081. [DOI] [PubMed] [Google Scholar]
- Choi KY, Kim HJ, Lee MH, Kwon TG, Nah HD, Furuichi T, Komori T, Nam SH, Kim YJ, Ryoo HM. 2005. Runx2 regulates FGF2-induced Bmp2 expression during cranial bone development. Dev Dyn 233: 115–121. [DOI] [PubMed] [Google Scholar]
- Choi SC, Kim SJ, Choi JH, Park CY, Shim WJ, Lim DS. 2008. Fibroblast growth factor-2 and -4 promote the proliferation of bone marrow mesenchymal stem cells by the activation of the PI3K–Akt and ERK1/2 signaling pathways. Stem Cells Dev 17: 725–736. [DOI] [PubMed] [Google Scholar]
- Cobrinik D, Lee MH, Hannon G, Mulligan G, Bronson RT, Dyson N, Harlow E, Beach D, Weinberg RA, Jacks T. 1996. Shared role of the pRB-related p130 and p107 proteins in limb development. Genes Dev 10: 1633–1644. [DOI] [PubMed] [Google Scholar]
- Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. 1996. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet 12: 390–397. [DOI] [PubMed] [Google Scholar]
- Coumoul X, Shukla V, Li C, Wang RH, Deng CX. 2005. Conditional knockdown of Fgfr2 in mice using Cre-LoxP induced RNA interference. Nucleic Acids Res 33: e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutu DL, Francois M, Galipeau J. 2011. Inhibition of cellular senescence by developmentally regulated FGF receptors in mesenchymal stem cells. Blood 117: 6801–6812. [DOI] [PubMed] [Google Scholar]
- Cowan CM, Quarto N, Warren SM, Salim A, Longaker MT. 2003. Age-related changes in the biomolecular mechanisms of calvarial osteoblast biology affect fibroblast growth factor-2 signaling and osteogenesis. J Biol Chem 278: 32005–32013. [DOI] [PubMed] [Google Scholar]
- Dailey L, Laplantine E, Priore R, Basilico C. 2003. A network of transcriptional and signaling events is activated by FGF to induce chondrocyte growth arrest and differentiation. J Cell Biol 161: 1053–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailey L, Ambrosetti D, Mansukhani A, Basilico C. 2005. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev 16: 233–247. [DOI] [PubMed] [Google Scholar]
- Dakouane Giudicelli M, Serazin V, Le Sciellour CR, Albert M, Selva J, Giudicelli Y. 2008. Increased achondroplasia mutation frequency with advanced age and evidence for G1138A mosaicism in human testis biopsies. Fertil Steril 89: 1651–1656. [DOI] [PubMed] [Google Scholar]
- Debiais F, Hott M, Graulet AM, Marie PJ. 1998. The effects of fibroblast growth factor-2 on human neonatal calvaria osteoblastic cells are differentiation stage specific. J Bone Miner Res 13: 645–654. [DOI] [PubMed] [Google Scholar]
- de Frutos CA, Vega S, Manzanares M, Flores JM, Huertas H, Martinez-Frias ML, Nieto MA. 2007. Snail1 is a transcriptional effector of FGFR3 signaling during chondrogenesis and achondroplasias. Dev Cell 13: 872–883. [DOI] [PubMed] [Google Scholar]
- Delezoide AL, Benoist-Lasselin C, Legeai-Mallet L, Le Merrer M, Munnich A, Vekemans M, Bonaventure J. 1998. Spatio–temporal expression of FGFR 1, 2 and 3 genes during human embryo–fetal ossification. Mech Dev 77: 19–30. [DOI] [PubMed] [Google Scholar]
- Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. 1996. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell 84: 911–921. [DOI] [PubMed] [Google Scholar]
- Di Maggio N, Mehrkens A, Papadimitropoulos A, Schaeren S, Heberer M, Banfi A, Martin I. 2012. Fibroblast growth factor-2 maintains a niche-dependent population of self-renewing highly potent non-adherent mesenchymal progenitors through FGFR2c. Stem Cells 30: 1455–1464. [DOI] [PubMed] [Google Scholar]
- Dirckx N, Van Hul M, Maes C. 2013. Osteoblast recruitment to sites of bone formation in skeletal development, homeostasis, and regeneration. Birth Defects Res C Embryo Today 99: 170–191. [DOI] [PubMed] [Google Scholar]
- Di Rocco F, Biosse Duplan M, Heuze Y, Kaci N, Komla-Ebri D, Munnich A, Mugniery E, Benoist-Lasselin C, Legeai-Mallet L. 2014. FGFR3 mutation causes abnormal membranous ossification in achondroplasia. Hum Mol Genet 23: 2914–2925. [DOI] [PubMed] [Google Scholar]
- Du X, Xie Y, Xian CJ, Chen L. 2012. Role of FGFs/FGFRs in skeletal development and bone regeneration. J Cell Physiol 227: 3731–3743. [DOI] [PubMed] [Google Scholar]
- Dufour C, Guenou H, Kaabeche K, Bouvard D, Sanjay A, Marie PJ. 2008. FGFR2–Cbl interaction in lipid rafts triggers attenuation of PI3K/Akt signaling and osteoblast survival. Bone 42: 1032–1039. [DOI] [PubMed] [Google Scholar]
- Dy P, Wang W, Bhattaram P, Wang Q, Wang L, Ballock RT, Lefebvre V. 2012. Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev Cell 22: 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarakumar VP, Schlessinger J. 2007. Skeletal overgrowth is mediated by deficiency in a specific isoform of fibroblast growth factor receptor 3. Proc Natl Acad Sci 104: 3937–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarakumar VP, Monsonego-Ornan E, Pines M, Antonopoulou I, Morriss-Kay GM, Lonai P. 2002. The IIIc alternative of Fgfr2 is a positive regulator of bone formation. Development 129: 3783–3793. [DOI] [PubMed] [Google Scholar]
- Eswarakumar VP, Horowitz MC, Locklin R, Morriss-Kay GM, Lonai P. 2004. A gain-of-function mutation of Fgfr2c demonstrates the roles of this receptor variant in osteogenesis. Proc Natl Acad Sci 101: 12555–12560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarakumar VP, Lax I, Schlessinger J. 2005. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev 16: 139–149. [DOI] [PubMed] [Google Scholar]
- Eswarakumar VP, Ozcan F, Lew ED, Bae JH, Tome F, Booth CJ, Adams DJ, Lax I, Schlessinger J. 2006. Attenuation of signaling pathways stimulated by pathologically activated FGF-receptor 2 mutants prevents craniosynostosis. Proc Natl Acad Sci 103: 18603–18608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhry A, Ratisoontorn C, Vedhachalam C, Salhab I, Koyama E, Leboy P, Pacifici M, Kirschner RE, Nah HD. 2005. Effects of FGF-2/-9 in calvarial bone cell cultures: differentiation stage-dependent mitogenic effect, inverse regulation of BMP-2 and noggin, and enhancement of osteogenic potential. Bone 36: 254–266. [DOI] [PubMed] [Google Scholar]
- Fei Y, Xiao L, Doetschman T, Coffin DJ, Hurley MM. 2011. Fibroblast growth factor 2 stimulation of osteoblast differentiation and bone formation is mediated by modulation of the Wnt signaling pathway. J Biol Chem 286: 40575–40583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei Y, Gronowicz G, Hurley MM. 2013. Fibroblast growth factor-2, bone homeostasis and fracture repair. Curr Pharm Des 19: 3354–3363. [DOI] [PubMed] [Google Scholar]
- Feng JQ, Clinkenbeard EL, Yuan B, White KE, Drezner MK. 2013. Osteocyte regulation of phosphate homeostasis and bone mineralization underlies the pathophysiology of the heritable disorders of rickets and osteomalacia. Bone 54: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foldynova-Trantirkova S, Wilcox WR, Krejci P. 2012. Sixteen years and counting: the current understanding of fibroblast growth factor receptor 3 (FGFR3) signaling in skeletal dysplasias. Hum Mutat 33: 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford-Hutchinson AF, Ali Z, Lines SE, Hallgrimsson B, Boyd SK, Jirik FR. 2007. Inactivation of Pten in osteo-chondroprogenitor cells leads to epiphyseal growth plate abnormalities and skeletal overgrowth. J Bone Miner Res 22: 1245–1259. [DOI] [PubMed] [Google Scholar]
- Fromigue O, Modrowski D, Marie PJ. 2004. Growth factors and bone formation in osteoporosis: roles for fibroblast growth factor and transforming growth factor β. Curr Pharm Des 10: 2593–2603. [DOI] [PubMed] [Google Scholar]
- Fromigue O, Modrowski D, Marie PJ. 2005. Apoptosis in membranous bone formation: role of fibroblast growth factor and bone morphogenetic protein signaling. Crit Rev Eukaryot Gene Expr 15: 75–92. [DOI] [PubMed] [Google Scholar]
- Furdui CM, Lew ED, Schlessinger J, Anderson KS. 2006. Autophosphorylation of FGFR1 kinase is mediated by a sequential and precisely ordered reaction. Mol Cell 21: 711–717. [DOI] [PubMed] [Google Scholar]
- Garcia S, Dirat B, Tognacci T, Rochet N, Mouska X, Bonnafous S, Patouraux S, Tran A, Gual P, Le Marchand-Brustel Y, et al. 2013. Postnatal soluble FGFR3 therapy rescues achondroplasia symptoms and restores bone growth in mice. Sci Transl Med 5: 203ra124. [DOI] [PubMed] [Google Scholar]
- Goetz R, Mohammadi M. 2013. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat Rev Mol Cell Biol 14: 166–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez AM, Hill DJ, Logan A, Maher PA, Baird A. 1996. Distribution of fibroblast growth factor (FGF)-2 and FGF receptor-1 messenger RNA expression and protein presence in the mid-trimester human fetus. Pediatr Res 39: 375–385. [DOI] [PubMed] [Google Scholar]
- Goriely A, Wilkie AO. 2012. Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease. Am J Hum Genet 90: 175–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goriely A, McVean GA, Rojmyr M, Ingemarsson B, Wilkie AO. 2003. Evidence for selective advantage of pathogenic FGFR2 mutations in the male germ line. Science 301: 643–646. [DOI] [PubMed] [Google Scholar]
- Goriely A, McVean GA, van Pelt AM, O'Rourke AW, Wall SA, de Rooij DG, Wilkie AO. 2005. Gain-of-function amino acid substitutions drive positive selection of FGFR2 mutations in human spermatogonia. Proc Natl Acad Sci 102: 6051–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenou H, Kaabeche K, Mee SL, Marie PJ. 2005. A role for fibroblast growth factor receptor-2 in the altered osteoblast phenotype induced by Twist haploinsufficiency in the Saethre-Chotzen syndrome. Hum Mol Genet 14: 1429–1439. [DOI] [PubMed] [Google Scholar]
- Hadari Y, Schlessinger J. 2009. FGFR3-targeted mAb therapy for bladder cancer and multiple myeloma. J Clin Invest 119: 1077–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajihosseini MK. 2008. Fibroblast growth factor signaling in cranial suture development and pathogenesis. Front Oral Biol 12: 160–177. [DOI] [PubMed] [Google Scholar]
- Hall BK, Miyake T. 2000. All for one and one for all: condensations and the initiation of skeletal development. Bioessays 22: 138–147. [DOI] [PubMed] [Google Scholar]
- Hamada T, Suda N, Kuroda T. 1999. Immunohistochemical localization of fibroblast growth factor receptors in the rat mandibular condylar cartilage and tibial cartilage. J Bone Miner Metab 17: 274–282. [DOI] [PubMed] [Google Scholar]
- Hamidouche Z, Fromigue O, Nuber U, Vaudin P, Pages JC, Ebert R, Jakob F, Miraoui H, Marie PJ. 2010. Autocrine fibroblast growth factor 18 mediates dexamethasone-induced osteogenic differentiation of murine mesenchymal stem cells. J Cell Physiol 224: 509–515. [DOI] [PubMed] [Google Scholar]
- Han X, Xiao Z, Quarles LD. 2015. Membrane and integrative nuclear fibroblastic growth factor receptor (FGFR) regulation of FGF-23. J Biol Chem 290: 10447–10459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanafusa H, Torii S, Yasunaga T, Nishida E. 2002. Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat Cell Biol 4: 850–858. [DOI] [PubMed] [Google Scholar]
- Hannema SE, van Duyvenvoorde HA, Premsler T, Yang RB, Mueller TD, Gassner B, Oberwinkler H, Roelfsema F, Santen GW, Prickett T, et al. 2013. An activating mutation in the kinase homology domain of the natriuretic peptide receptor-2 causes extremely tall stature without skeletal deformities. J Clin Endocrinol Metab 98: E1988–E1998. [DOI] [PubMed] [Google Scholar]
- Harada M, Murakami H, Okawa A, Okimoto N, Hiraoka S, Nakahara T, Akasaka R, Shiraishi Y, Futatsugi N, Mizutani-Koseki Y, et al. 2009. FGF9 monomer/dimer equilibrium regulates extracellular matrix affinity and tissue diffusion. Nat Genet 41: 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch NE, Hudson M, Seto ML, Cunningham ML, Bothwell M. 2006. Intracellular retention, degradation, and signaling of glycosylation-deficient FGFR2 and craniosynostosis syndrome-associated FGFR2C278F. J Biol Chem 281: 27292–27305. [DOI] [PubMed] [Google Scholar]
- Haupt LM, Murali S, Mun FK, Teplyuk N, Mei LF, Stein GS, van Wijnen AJ, Nurcombe V, Cool SM. 2009. The heparan sulfate proteoglycan (HSPG) glypican-3 mediates commitment of MC3T3-E1 cells toward osteogenesis. J Cell Physiol 220: 780–791. [DOI] [PubMed] [Google Scholar]
- Havens BA, Velonis D, Kronenberg MS, Lichtler AC, Oliver B, Mina M. 2008. Roles of FGFR3 during morphogenesis of Meckel's cartilage and mandibular bones. Dev Biol 316: 336–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuze Y, Singh N, Basilico C, Jabs EW, Holmes G, Richtsmeier JT. 2014. Morphological comparison of the craniofacial phenotypes of mouse models expressing the Apert FGFR2 S252W mutation in neural crest- or mesoderm-derived tissues. Bone 63: 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinoi E, Bialek P, Chen YT, Rached MT, Groner Y, Behringer RR, Ornitz DM, Karsenty G. 2006. Runx2 inhibits chondrocyte proliferation and hypertrophy through its expression in the perichondrium. Genes Dev 20: 2937–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes G, Rothschild G, Roy UB, Deng CX, Mansukhani A, Basilico C. 2009. Early onset of craniosynostosis in an Apert mouse model reveals critical features of this pathology. Dev Biol 328: 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homer-Bouthiette C, Doetschman T, Xiao L, Hurley MM. 2014. Knockout of nuclear high molecular weight FGF2 isoforms in mice modulates bone and phosphate homeostasis. J Biol Chem 289: 36303–36314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton WA, Hall JG, Hecht JT. 2007. Achondroplasia. Lancet 370: 162–172. [DOI] [PubMed] [Google Scholar]
- House SL, Branch K, Newman G, Doetschman T, Schultz Jel J. 2005. Cardioprotection induced by cardiac-specific overexpression of fibroblast growth factor-2 is mediated by the MAPK cascade. Am J Physiol Heart Circ Physiol 289: H2167–H2175. [DOI] [PubMed] [Google Scholar]
- Hung IH, Yu K, Lavine KJ, Ornitz DM. 2007. FGF9 regulates early hypertrophic chondrocyte differentiation and skeletal vascularization in the developing stylopod. Dev Biol 307: 300–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunziker EB, Schenk RK. 1989. Physiological mechanisms adopted by chondrocytes in regulating longitudinal bone growth in rats. J Physiol 414: 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunziker EB, Schenk RK, Cruz-Orive LM. 1987. Quantitation of chondrocyte performance in growth-plate cartilage during longitudinal bone growth. J Bone Joint Surg Am 69: 162–173. [PubMed] [Google Scholar]
- Hurley MM, Tetradis S, Huang YF, Hock J, Kream BE, Raisz LG, Sabbieti MG. 1999. Parathyroid hormone regulates the expression of fibroblast growth factor-2 mRNA and fibroblast growth factor receptor mRNA in osteoblastic cells. J Bone Miner Res 14: 776–783. [DOI] [PubMed] [Google Scholar]
- Hutchison MR. 2012. BDNF alters ERK/p38 MAPK activity ratios to promote differentiation in growth plate chondrocytes. Mol Endocrinol 26: 1406–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahimi OA, Eliseenkova AV, Plotnikov AN, Yu K, Ornitz DM, Mohammadi M. 2001. Structural basis for fibroblast growth factor receptor 2 activation in Apert syndrome. Proc Natl Acad Sci 5: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignelzi MA Jr, Wang W, Young AT. 2003. Fibroblast growth factors lead to increased Msx2 expression and fusion in calvarial sutures. J Bone Miner Res 18: 751–759. [DOI] [PubMed] [Google Scholar]
- Ikegami D, Akiyama H, Suzuki A, Nakamura T, Nakano T, Yoshikawa H, Tsumaki N. 2011. Sox9 sustains chondrocyte survival and hypertrophy in part through Pik3ca–Akt pathways. Development 138: 1507–1519. [DOI] [PubMed] [Google Scholar]
- Iwata T, Chen L, Li C, Ovchinnikov DA, Behringer RR, Francomano CA, Deng CX. 2000. A neonatal lethal mutation in FGFR3 uncouples proliferation and differentiation of growth plate chondrocytes in embryos. Hum Mol Genet 9: 1603–1613. [DOI] [PubMed] [Google Scholar]
- Iwata T, Li CL, Deng CX, Francomano CA. 2001. Highly activated Fgfr3 with the K644M mutation causes prolonged survival in severe dwarf mice. Hum Mol Genet 10: 1255–1264. [DOI] [PubMed] [Google Scholar]
- Jackson RA, Murali S, van Wijnen AJ, Stein GS, Nurcombe V, Cool SM. 2007. Heparan sulfate regulates the anabolic activity of MC3T3-E1 preosteoblast cells by induction of Runx2. J Cell Physiol 210: 38–50. [DOI] [PubMed] [Google Scholar]
- Jacob AL, Smith C, Partanen J, Ornitz DM. 2006. Fibroblast growth factor receptor 1 signaling in the osteo-chondrogenic cell lineage regulates sequential steps of osteoblast maturation. Dev Biol 296: 315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon E, Yun YR, Kang W, Lee S, Koh YH, Kim HW, Suh CK, Jang JH. 2012. Investigating the role of FGF18 in the cultivation and osteogenic differentiation of mesenchymal stem cells. PLoS One 7: e43982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D, Wilkie AO. 2011. Craniosynostosis. Eur J Hum Genet 19: 369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaabeche K, Lemonnier J, Le Mee S, Caverzasio J, Marie PJ. 2004. Cbl-mediated degradation of Lyn and Fyn induced by constitutive fibroblast growth factor receptor-2 activation supports osteoblast differentiation. J Biol Chem 279: 36259–36267. [DOI] [PubMed] [Google Scholar]
- Kaabeche K, Guenou H, Bouvard D, Didelot N, Listrat A, Marie PJ. 2005. Cbl-mediated ubiquitination of α5 integrin subunit mediates fibronectin-dependent osteoblast detachment and apoptosis induced by FGFR2 activation. J Cell Sci 118: 1223–1232. [DOI] [PubMed] [Google Scholar]
- Kalinina J, Byron SA, Makarenkova HP, Olsen SK, Eliseenkova AV, Larochelle WJ, Dhanabal M, Blais S, Ornitz DM, Day LA, et al. 2009. Homodimerization controls the fibroblast growth factor 9 subfamily's receptor binding and heparan sulfate-dependent diffusion in the extracellular matrix. Mol Cell Biol 29: 4663–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanazawa H, Tanaka H, Inoue M, Yamanaka Y, Namba N, Seino Y. 2003. Efficacy of growth hormone therapy for patients with skeletal dysplasia. J Bone Miner Metab 21: 307–310. [DOI] [PubMed] [Google Scholar]
- Kanazawa S, Fujiwara T, Matsuzaki S, Shingaki K, Taniguchi M, Miyata S, Tohyama M, Sakai Y, Yano K, Hosokawa K, et al. 2010. bFGF regulates PI3-kinase–Rac1–JNK pathway and promotes fibroblast migration in wound healing. PLoS One 5: e12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kant SG, Cervenkova I, Balek L, Trantirek L, Santen GW, de Vries MC, van Duyvenvoorde HA, van der Wielen MJ, Verkerk AJ, Uitterlinden AG, et al. 2015. A novel variant of FGFR3 causes proportionate short stature. Eur J Endocrinol 172: 763–770. [DOI] [PubMed] [Google Scholar]
- Karlsson C, Thornemo M, Henriksson HB, Lindahl A. 2009. Identification of a stem cell niche in the zone of Ranvier within the knee joint. J Anat 215: 355–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolak MR, Yang X, Elefteriou F. 2015. FGFR1 signaling in hypertrophic chondrocytes is attenuated by the Ras-GAP neurofibromin during endochondral bone formation. Hum Mol Genet 24: 2552–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi H, Pilbeam CC, Gronowicz G, Abreu C, Fletcher BS, Herschman HR, Raisz LG, Hurley MM. 1995. Transcriptional induction of prostaglandin G/H synthase-2 by basic fibroblast growth factor. J Clin Invest 96: 923–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi H, Chikazu D, Nakamura K, Kumegawa M, Hakeda Y. 2000. Direct and indirect actions of fibroblast growth factor 2 on osteoclastic bone resorption in cultures. J Bone Miner Res 15: 466–473. [DOI] [PubMed] [Google Scholar]
- Kawaguchi H, Oka H, Jingushi S, Izumi T, Fukunaga M, Sato K, Matsushita T, Nakamura K. 2010. A local application of recombinant human fibroblast growth factor-2 for tibial shaft fractures: a randomized, placebo-controlled trial. J Bone Miner Res 25: 2735–2743. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Rice DP, Kettunen PJ, Thesleff I. 1998. FGF-, BMP- and Shh-mediated signalling pathways in the regulation of cranial suture morphogenesis and calvarial bone development. Development 125: 1241–1251. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Kim JH, Bae SC, Choi JY, Kim HJ, Ryoo HM. 2003. The protein kinase C pathway plays a central role in the fibroblast growth factor-stimulated expression and transactivation activity of Runx2. J Biol Chem 278: 319–326. [DOI] [PubMed] [Google Scholar]
- Kim JM, Shin HI, Cha SS, Lee CS, Hong BS, Lim S, Jang HJ, Kim J, Yang YR, Kim YH, et al. 2012. DJ-1 promotes angiogenesis and osteogenesis by activating FGF receptor-1 signaling. Nat Commun 3: 1296. [DOI] [PubMed] [Google Scholar]
- Kodama N, Nagata M, Tabata Y, Ozeki M, Ninomiya T, Takagi R. 2009. A local bone anabolic effect of rhFGF2-impregnated gelatin hydrogel by promoting cell proliferation and coordinating osteoblastic differentiation. Bone 44: 699–707. [DOI] [PubMed] [Google Scholar]
- Kolupaeva V, Laplantine E, Basilico C. 2008. PP2A-mediated dephosphorylation of p107 plays a critical role in chondrocyte cell cycle arrest by FGF. PLoS One 3: e3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolupaeva V, Daempfling L, Basilico C. 2013. The B55α regulatory subunit of protein phosphatase 2A mediates fibroblast growth factor-induced p107 dephosphorylation and growth arrest in chondrocytes. Mol Cell Biol 33: 2865–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komaki H, Tanaka T, Chazono M, Kikuchi T. 2006. Repair of segmental bone defects in rabbit tibiae using a complex of β-tricalcium phosphate, type I collagen, and fibroblast growth factor-2. Biomaterials 27: 5118–5126. [DOI] [PubMed] [Google Scholar]
- Kosher RA, Kulyk WM, Gay SW. 1986. Collagen gene expression during limb cartilage differentiation. J Cell Biol 102: 1151–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, Schlessinger J. 1997. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell 89: 693–702. [DOI] [PubMed] [Google Scholar]
- Kozhemyakina E, Lassar AB, Zelzer E. 2015. A pathway to bone: signaling molecules and transcription factors involved in chondrocyte development and maturation. Development 142: 817–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejci P. 2014. The paradox of FGFR3 signaling in skeletal dysplasia: why chondrocytes growth arrest while other cells over proliferate. Mutat Res Rev Mutat Res 759: 40–48. [DOI] [PubMed] [Google Scholar]
- Krejci P, Salazar L, Goodridge HS, Kashiwada TA, Schibler MJ, Jelinkova P, Thompson LM, Wilcox WR. 2008a. STAT1 and STAT3 do not participate in FGF-mediated growth arrest in chondrocytes. J Cell Sci 121: 272–281. [DOI] [PubMed] [Google Scholar]
- Krejci P, Salazar L, Kashiwada TA, Chlebova K, Salasova A, Thompson LM, Bryja V, Kozubik A, Wilcox WR. 2008b. Analysis of STAT1 activation by six FGFR3 mutants associated with skeletal dysplasia undermines dominant role of STAT1 in FGFR3 signaling in cartilage. PLoS One 3: e3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejci P, Aklian A, Kaucka M, Sevcikova E, Prochazkova J, Masek JK, Mikolka P, Pospisilova T, Spoustova T, Weis M, et al. 2012. Receptor tyrosine kinases activate canonical WNT/β-catenin signaling via MAP kinase/LRP6 pathway and direct β-catenin phosphorylation. PLoS One 7: e35826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn LT, Ou G, Charles L, Hurley MM, Rodner CM, Gronowicz G. 2013. Fibroblast growth factor-2 and bone morphogenetic protein-2 have a synergistic stimulatory effect on bone formation in cell cultures from elderly mouse and human bone. J Gerontol A Biol Sci Med Sci 68: 1170–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D, Lassar AB. 2014. Fibroblast growth factor maintains chondrogenic potential of limb bud mesenchymal cells by modulating DNMT3A recruitment. Cell Rep 8: 1419–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimchak A, Haines DS, Garriga J, Wu S, De Luca F, Sweredoski MJ, Deshaies RJ, Hess S, Grana X. 2013. Activation of p107 by fibroblast growth factor, which is essential for chondrocyte cell cycle exit, is mediated by the protein phosphatase 2A/B55α holoenzyme. Mol Cell Biol 33: 3330–3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuro-o M. 2008. Endocrine FGFs and Klothos: emerging concepts. Trends Endocrinol Metab 19: 239–245. [DOI] [PubMed] [Google Scholar]
- Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, et al. 2006. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 281: 6120–6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan MD, Sellmyer MA, Quarto N, Ho AM, Wandless TJ, Longaker MT. 2011. Chemical control of FGF-2 release for promoting calvarial healing with adipose stem cells. J Biol Chem 286: 11307–11313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyono A, Avishai N, Ouyang Z, Landreth GE, Murakami S. 2012. FGF and ERK signaling coordinately regulate mineralization-related genes and play essential roles in osteocyte differentiation. J Bone Miner Metab 30: 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laederich MB, Horton WA. 2012. FGFR3 targeting strategies for achondroplasia. Expert Rev Mol Med 14: e11. [DOI] [PubMed] [Google Scholar]
- Lamothe B, Yamada M, Schaeper U, Birchmeier W, Lax I, Schlessinger J. 2004. The docking protein Gab1 is an essential component of an indirect mechanism for fibroblast growth factor stimulation of the phosphatidylinositol 3-kinase/Akt antiapoptotic pathway. Mol Cell Biol 24: 5657–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplantine E, Rossi F, Sahni M, Basilico C, Cobrinik D. 2002. FGF signaling targets the pRb-related p107 and p130 proteins to induce chondrocyte growth arrest. J Cell Biol 158: 741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus JE, Hegde A, Andrade AC, Nilsson O, Baron J. 2007. Fibroblast growth factor expression in the postnatal growth plate. Bone 40: 577–586. [DOI] [PubMed] [Google Scholar]
- Legeai-Mallet L, Benoist-Lasselin C, Delezoide AL, Munnich A, Bonaventure J. 1998. Fibroblast growth factor receptor 3 mutations promote apoptosis but do not alter chondrocyte proliferation in thanatophoric dysplasia. J Biol Chem 273: 13007–13014. [DOI] [PubMed] [Google Scholar]
- Legeai-Mallet L, Benoist-Lasselin C, Munnich A, Bonaventure J. 2004. Overexpression of FGFR3, Stat1, Stat5 and p21Cip1 correlates with phenotypic severity and defective chondrocyte differentiation in FGFR3-related chondrodysplasias. Bone 34: 26–36. [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J. 2010. Cell signaling by receptor tyrosine kinases. Cell 141: 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemonnier J, Hay E, Delannoy P, Fromigue O, Lomri A, Modrowski D, Marie PJ. 2001a. Increased osteoblast apoptosis in apert craniosynostosis: role of protein kinase C and interleukin-1. Am J Pathol 158: 1833–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemonnier J, Hay E, Delannoy P, Lomri A, Modrowski D, Caverzasio J, Marie PJ. 2001b. Role of N-cadherin and protein kinase C in osteoblast gene activation induced by the S252W fibroblast growth factor receptor 2 mutation in Apert craniosynostosis. J Bone Miner Res 16: 832–845. [DOI] [PubMed] [Google Scholar]
- Lenton KA, Nacamuli RP, Wan DC, Helms JA, Longaker MT. 2005. Cranial suture biology. Curr Top Dev Biol 66: 287–328. [DOI] [PubMed] [Google Scholar]
- Li C, Xu X, Nelson DK, Williams T, Kuehn MR, Deng CX. 2005. FGFR1 function at the earliest stages of mouse limb development plays an indispensable role in subsequent autopod morphogenesis. Development 132: 4755–4764. [DOI] [PubMed] [Google Scholar]
- Li M, Seki Y, Freitas PH, Nagata M, Kojima T, Sultana S, Ubaidus S, Maeda T, Shimomura J, Henderson JE, et al. 2010. FGFR3 down-regulates PTH/PTHrP receptor gene expression by mediating JAK/STAT signaling in chondrocytic cell line. J Electron Microsc (Tokyo) 59: 227–236. [DOI] [PubMed] [Google Scholar]
- Li Y, Ahrens MJ, Wu A, Liu J, Dudley AT. 2011. Calcium/calmodulin-dependent protein kinase II activity regulates the proliferative potential of growth plate chondrocytes. Development 138: 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian JB, Stein GS, Javed A, van Wijnen AJ, Stein JL, Montecino M, Hassan MQ, Gaur T, Lengner CJ, Young DW. 2006. Networks and hubs for the transcriptional control of osteoblastogenesis. Rev Endocr Metab Disord 7: 1–16. [DOI] [PubMed] [Google Scholar]
- Liao E. 2013. FGF23 associated bone diseases. Front Med 7: 65–80. [DOI] [PubMed] [Google Scholar]
- Liao S, Porter D, Scott A, Newman G, Doetschman T, Schultz Jel J. 2007. The cardioprotective effect of the low molecular weight isoform of fibroblast growth factor-2: the role of JNK signaling. J Mol Cell Cardiol 42: 106–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Tu X, Choi K, Akiyama H, Mishina Y, Long F. 2015. BMP-Smad4 signaling is required for precartilaginous mesenchymal condensation independent of Sox9 in the mouse. Dev Biol 400: 132–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Olsen BR. 2014. Distinct VEGF functions during bone development and homeostasis. Arch Immunol Ther Exp (Warsz) 62: 363–368. [DOI] [PubMed] [Google Scholar]
- Liu Z, Xu J, Colvin JS, Ornitz DM. 2002. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev 16: 859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Lavine KJ, Hung IH, Ornitz DM. 2007. FGF18 is required for early chondrocyte proliferation, hypertrophy and vascular invasion of the growth plate. Dev Biol 302: 80–91. [DOI] [PubMed] [Google Scholar]
- Liu J, Kwon TG, Nam HK, Hatch NE. 2013a. Craniosynostosis-associated Fgfr2(C342Y) mutant bone marrow stromal cells exhibit cell autonomous abnormalities in osteoblast differentiation and bone formation. Biomed Res Int 2013: 292506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Nam HK, Wang E, Hatch NE. 2013b. Further analysis of the Crouzon mouse: effects of the FGFR2(C342Y) mutation are cranial bone-dependent. Calcif Tissue Int 92: 451–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomri A, Lemonnier J, Hott M, de Parseval N, Lajeunie E, Munnich A, Renier D, Marie PJ. 1998. Increased calvaria cell differentiation and bone matrix formation induced by fibroblast growth factor receptor 2 mutations in Apert syndrome. J Clin Invest 101: 1310–1317. [PMC free article] [PubMed] [Google Scholar]
- Lomri A, Lemonnier J, Delannoy P, Marie PJ. 2001. Increased expression of protein kinase Cα, interleukin-1α, and RhoA guanosine 5′-triphosphatase in osteoblasts expressing the Ser252Trp fibroblast growth factor 2 Apert mutation: identification by analysis of complementary DNA microarray. J Bone Miner Res 16: 705–712. [DOI] [PubMed] [Google Scholar]
- Long F. 2012. Building strong bones: molecular regulation of the osteoblast lineage. Nat Rev Mol Cell Biol 13: 27–38. [DOI] [PubMed] [Google Scholar]
- Long F, Ornitz DM. 2013. Development of the endochondral skeleton. Cold Spring Harb Perspect Biol 5: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorget F, Kaci N, Peng J, Benoist-Lasselin C, Mugniery E, Oppeneer T, Wendt DJ, Bell SM, Bullens S, Bunting S, et al. 2012. Evaluation of the therapeutic potential of a CNP analog in a Fgfr3 mouse model recapitulating achondroplasia. Am J Hum Genet 91: 1108–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Su N, Yang J, Huang W, Li C, Zhao L, He Q, Du X, Shen Y, Chen B, et al. 2009. Fibroblast growth factor receptor 1 regulates the differentiation and activation of osteoclasts through Erk1/2 pathway. Biochem Biophys Res Commun 390: 494–499. [DOI] [PubMed] [Google Scholar]
- Maes C, Kobayashi T, Selig MK, Torrekens S, Roth SI, Mackem S, Carmeliet G, Kronenberg HM. 2010. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev Cell 19: 329–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makrythanasis P, Temtamy S, Aglan MS, Otaify GA, Hamamy H, Antonarakis SE. 2014. A novel homozygous mutation in FGFR3 causes tall stature, severe lateral tibial deviation, scoliosis, hearing impairment, camptodactyly, and arachnodactyly. Hum Mutat 35: 959–963. [DOI] [PubMed] [Google Scholar]
- Mansukhani A, Bellosta P, Sahni M, Basilico C. 2000. Signaling by fibroblast growth factors (FGF) and fibroblast growth factor receptor 2 (FGFR2)-activating mutations blocks mineralization and induces apoptosis in osteoblasts. J Cell Biology 149: 1297–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansukhani A, Ambrosetti D, Holmes G, Cornivelli L, Basilico C. 2005. Sox2 induction by FGF and FGFR2 activating mutations inhibits Wnt signaling and osteoblast differentiation. J Cell Biol 168: 1065–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani FV, Ahn CP, Martin GR. 2008. Genetic evidence that FGFs have an instructive role in limb proximal-distal patterning. Nature 453: 401–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie PJ. 2008. Transcription factors controlling osteoblastogenesis. Arch Biochem Biophys 473: 98–105. [DOI] [PubMed] [Google Scholar]
- Marie PJ. 2015. Osteoblast dysfunctions in bone diseases: from cellular and molecular mechanisms to therapeutic strategies. Cell Mol Life Sci 72: 1347–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie PJ, Coffin JD, Hurley MM. 2005. FGF and FGFR signaling in chondrodysplasias and craniosynostosis. J Cell Biochem 96: 888–896. [DOI] [PubMed] [Google Scholar]
- Marie PJ, Kaabeche K, Guenou H. 2008. Roles of FGFR2 and twist in human craniosynostosis: insights from genetic mutations in cranial osteoblasts. Front Oral Biol 12: 144–159. [DOI] [PubMed] [Google Scholar]
- Martin A, Liu S, David V, Li H, Karydis A, Feng JQ, Quarles LD. 2011. Bone proteins PHEX and DMP1 regulate fibroblastic growth factor Fgf23 expression in osteocytes through a common pathway involving FGF receptor (FGFR) signaling. FASEB J 25: 2551–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Torrecuadrada J, Cifuentes G, Lopez-Serra P, Saenz P, Martinez A, Casal JI. 2005. Targeting the extracellular domain of fibroblast growth factor receptor 3 with human single-chain Fv antibodies inhibits bladder carcinoma cell line proliferation. Clin Cancer Res 11: 6280–6290. [DOI] [PubMed] [Google Scholar]
- Maruyama T, Mirando AJ, Deng CX, Hsu W. 2010. The balance of WNT and FGF signaling influences mesenchymal stem cell fate during skeletal development. Sci Signal 3: ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita T, Chan YY, Kawanami A, Balmes G, Landreth GE, Murakami S. 2009a. Extracellular signal-regulated kinase 1 (ERK1) and ERK2 play essential roles in osteoblast differentiation and in supporting osteoclastogenesis. Mol Cell Biol 29: 5843–5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita T, Wilcox WR, Chan YY, Kawanami A, Bukulmez H, Balmes G, Krejci P, Mekikian PB, Otani K, Yamaura I, et al. 2009b. FGFR3 promotes synchondrosis closure and fusion of ossification centers through the MAPK pathway. Hum Mol Genet 18: 227–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita M, Kitoh H, Ohkawara B, Mishima K, Kaneko H, Ito M, Masuda A, Ishiguro N, Ohno K. 2013. Meclozine facilitates proliferation and differentiation of chondrocytes by attenuating abnormally activated FGFR3 signaling in achondroplasia. PLoS One 8: e81569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita M, Hasegawa S, Kitoh H, Mori K, Ohkawara B, Yasoda A, Masuda A, Ishiguro N, Ohno K. 2015. Meclozine promotes longitudinal skeletal growth in transgenic mice with achondroplasia carrying a gain-of-function mutation in the FGFR3 gene. Endocrinology 156: 548–554. [DOI] [PubMed] [Google Scholar]
- McDowell LM, Frazier BA, Studelska DR, Giljum K, Chen J, Liu J, Yu K, Ornitz DM, Zhang L. 2006. Inhibition or activation of Apert syndrome FGFR2 (S252W) signaling by specific glycosaminoglycans. J Biol Chem 281: 6924–6930. [DOI] [PubMed] [Google Scholar]
- McEwen DG, Green RP, Naski MC, Towler DA, Ornitz DM. 1999. Fibroblast growth factor receptor 3 gene transcription is suppressed by cyclic adenosine 3′,5′-monophosphate: identification of a chondrocytic regulatory element. J Biol Chem 274: 30934–30942. [DOI] [PubMed] [Google Scholar]
- Melville H, Wang Y, Taub PJ, Jabs EW. 2010. Genetic basis of potential therapeutic strategies for craniosynostosis. Am J Med Genet A 152A: 3007–3015. [DOI] [PubMed] [Google Scholar]
- Merrill AE, Sarukhanov A, Krejci P, Idoni B, Camacho N, Estrada KD, Lyons KM, Deixler H, Robinson H, Chitayat D, et al. 2012. Bent bone dysplasia-FGFR2 type, a distinct skeletal disorder, has deficient canonical FGF signaling. Am J Hum Genet 90: 550–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miraoui H, Marie PJ. 2010. Fibroblast growth factor receptor signaling crosstalk in skeletogenesis. Sci Signal 3: re9. [DOI] [PubMed] [Google Scholar]
- Miraoui H, Oudina K, Petite H, Tanimoto Y, Moriyama K, Marie PJ. 2009. Fibroblast growth factor receptor 2 promotes osteogenic differentiation in mesenchymal cells via ERK1/2 and protein kinase C signaling. J Biol Chem 284: 4897–4904. [DOI] [PubMed] [Google Scholar]
- Miraoui H, Ringe J, Haupl T, Marie PJ. 2010a. Increased EGF- and PDGFα-receptor signaling by mutant FGF-receptor 2 contributes to osteoblast dysfunction in Apert craniosynostosis. Hum Mol Genet 19: 1678–1689. [DOI] [PubMed] [Google Scholar]
- Miraoui H, Severe N, Vaudin P, Pages JC, Marie PJ. 2010b. Molecular silencing of Twist1 enhances osteogenic differentiation of murine mesenchymal stem cells: implication of FGFR2 signaling. J Cell Biochem 110: 1147–1154. [DOI] [PubMed] [Google Scholar]
- Miura K, Kim OH, Lee HR, Namba N, Michigami T, Yoo WJ, Choi IH, Ozono K, Cho TJ. 2014. Overgrowth syndrome associated with a gain-of-function mutation of the natriuretic peptide receptor 2 (NPR2) gene. Am J Med Genet A 164A: 156–163. [DOI] [PubMed] [Google Scholar]
- Moenning A, Jager R, Egert A, Kress W, Wardelmann E, Schorle H. 2009. Sustained platelet-derived growth factor receptor α signaling in osteoblasts results in craniosynostosis by overactivating the phospholipase C-γ pathway. Mol Cell Biol 29: 881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molteni A, Modrowski D, Hott M, Marie PJ. 1999a. Alterations of matrix- and cell-associated proteoglycans inhibit osteogenesis and growth response to fibroblast growth factor-2 in cultured rat mandibular condyle and calvaria. Cell Tissue Res 295: 523–536. [DOI] [PubMed] [Google Scholar]
- Molteni A, Modrowski D, Hott M, Marie PJ. 1999b. Differential expression of fibroblast growth factor receptor-1, -2, and -3 and syndecan-1, -2, and -4 in neonatal rat mandibular condyle and calvaria during osteogenic differentiation in vitro. Bone 24: 337–347. [DOI] [PubMed] [Google Scholar]
- Montero A, Okada Y, Tomita M, Ito M, Tsurukami H, Nakamura T, Doetschman T, Coffin JD, Hurley MM. 2000. Disruption of the fibroblast growth factor-2 gene results in decreased bone mass and bone formation. J Clin Invest 105: 1085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon AM, Guris DL, Seo JH, Li L, Hammond J, Talbot A, Imamoto A. 2006. Crkl deficiency disrupts Fgf8 signaling in a mouse model of 22q11 deletion syndromes. Dev Cell 10: 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore R, Ferretti P, Copp A, Thorogood P. 2002. Blocking endogenous FGF-2 activity prevents cranial osteogenesis. Dev Biol 243: 99–114. [DOI] [PubMed] [Google Scholar]
- Morita J, Nakamura M, Kobayashi Y, Deng CX, Funato N, Moriyama K. 2014. Soluble form of FGFR2 with S252W partially prevents craniosynostosis of the apert mouse model. Dev Dyn 243: 560–567. [DOI] [PubMed] [Google Scholar]
- Motch Perrine SM, Cole TM III, Martinez-Abadias N, Aldridge K, Jabs EW, Richtsmeier JT. 2014. Craniofacial divergence by distinct prenatal growth patterns in Fgfr2 mutant mice. BMC Dev Biol 14: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugniery E, Dacquin R, Marty C, Benoist-Lasselin C, de Vernejoul MC, Jurdic P, Munnich A, Geoffroy V, Legeai-Mallet L. 2012. An activating Fgfr3 mutation affects trabecular bone formation via a paracrine mechanism during growth. Hum Mol Genet 21: 2503–2513. [DOI] [PubMed] [Google Scholar]
- Murakami S, Kan M, McKeehan WL, de Crombrugghe B. 2000. Up-regulation of the chondrogenic Sox9 gene by fibroblast growth factors is mediated by the mitogen-activated protein kinase pathway. Proc Natl Acad Sci 97: 1113–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Balmes G, McKinney S, Zhang Z, Givol D, De Crombrugghe B. 2004. Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev 18: 290–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganawa T, Xiao L, Coffin JD, Doetschman T, Sabbieti MG, Agas D, Hurley MM. 2008. Reduced expression and function of bone morphogenetic protein-2 in bones of Fgf2 null mice. J Cell Biochem 103: 1975–1988. [DOI] [PubMed] [Google Scholar]
- Nah HD, Rodgers BJ, Kulyk WM, Kream BE, Kosher RA, Upholt WB. 1988. In situ hybridization analysis of the expression of the type II collagen gene in the developing chicken limb bud. Coll Relat Res 8: 277–294. [DOI] [PubMed] [Google Scholar]
- Nakajima F, Nakajima A, Ogasawara A, Moriya H, Yamazaki M. 2007. Effects of a single percutaneous injection of basic fibroblast growth factor on the healing of a closed femoral shaft fracture in the rat. Calcif Tissue Int 81: 132–138. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Hara Y, Tagawa M, Tamura M, Yuge T, Fukuda H, Nigi H. 1998. Recombinant human basic fibroblast growth factor accelerates fracture healing by enhancing callus remodeling in experimental dog tibial fracture. J Bone Miner Res 13: 942–949. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Tensho K, Nakaya H, Nawata M, Okabe T, Wakitani S. 2005. Low dose fibroblast growth factor-2 (FGF-2) enhances bone morphogenetic protein-2 (BMP-2)-induced ectopic bone formation in mice. Bone 36: 399–407. [DOI] [PubMed] [Google Scholar]
- Nakao K, Osawa K, Yasoda A, Yamanaka S, Fujii T, Kondo E, Koyama N, Kanamoto N, Miura M, Kuwahara K, et al. 2015. The Local CNP/GC-B system in growth plate is responsible for physiological endochondral bone growth. Sci Rep 5: 10554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naski MC, Wang Q, Xu J, Ornitz DM. 1996. Graded activation of fibroblast growth factor receptor 3 by mutations causing achondroplasia and thanatophoric dysplasia. Nat Genet 13: 233–237. [DOI] [PubMed] [Google Scholar]
- Naski MC, Colvin JS, Coffin JD, Ornitz DM. 1998. Repression of hedgehog signaling and BMP4 expression in growth plate cartilage by fibroblast growth factor receptor 3. Development 125: 4977–4988. [DOI] [PubMed] [Google Scholar]
- Neben CL, Idoni B, Salva JE, Tuzon CT, Rice JC, Krakow D, Merrill AE. 2014. Bent bone dysplasia syndrome reveals nucleolar activity for FGFR2 in ribosomal DNA transcription. Hum Mol Genet 23: 5659–5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niger C, Luciotti MA, Buo AM, Hebert C, Ma V, Stains JP. 2013. The regulation of runt-related transcription factor 2 by fibroblast growth factor-2 and connexin43 requires the inositol polyphosphate/protein kinase Cδ cascade. J Bone Miner Res 28: 1468–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noonan KJ, Hunziker EB, Nessler J, Buckwalter JA. 1998. Changes in cell, matrix compartment, and fibrillar collagen volumes between growth-plate zones. J Orthop Res 16: 500–508. [DOI] [PubMed] [Google Scholar]
- Oh CD, Lu Y, Liang S, Mori-Akiyama Y, Chen D, de Crombrugghe B, Yasuda H. 2014. SOX9 regulates multiple genes in chondrocytes, including genes encoding ECM proteins, ECM modification enzymes, receptors, and transporters. PLoS One 9: e107577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohbayashi N, Shibayama M, Kurotaki Y, Imanishi M, Fujimori T, Itoh N, Takada S. 2002. FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev 16: 870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Montero A, Zhang X, Sobue T, Lorenzo J, Doetschman T, Coffin JD, Hurley MM. 2003. Impaired osteoclast formation in bone marrow cultures of Fgf2 null mice in response to parathyroid hormone. J Biol Chem 278: 21258–21266. [DOI] [PubMed] [Google Scholar]
- Olney RC, Prickett TC, Espiner EA, Mackenzie WG, Duker AL, Ditro C, Zabel B, Hasegawa T, Kitoh H, Aylsworth AS, et al. 2015. C-type natriuretic peptide plasma levels are elevated in subjects with achondroplasia, hypochondroplasia, and thanatophoric dysplasia. J Clin Endocrinol Metab 100: E355–E359. [DOI] [PubMed] [Google Scholar]
- Ono N, Ono W, Nagasawa T, Kronenberg HM. 2014. A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones. Nat Cell Biol 16: 1157–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opperman LA. 2000. Cranial sutures as intramembranous bone growth sites [Review]. Dev Dyn 219: 472–485. [DOI] [PubMed] [Google Scholar]
- Ornitz DM. 2005. FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor Rev 16: 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM, Itoh N. 2015. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol 4: 215–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM, Marie PJ. 2002. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev 16: 1446–1465. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Givol D, Yayon A, Yarden Y, Lonai P. 1991. Developmental expression of two murine fibroblast growth factor receptors, flg and bek. Development 113: 1419–1434. [DOI] [PubMed] [Google Scholar]
- Ozasa A, Komatsu Y, Yasoda A, Miura M, Sakuma Y, Nakatsuru Y, Arai H, Itoh N, Nakao K. 2005. Complementary antagonistic actions between C-type natriuretic peptide and the MAPK pathway through FGFR-3 in ATDC5 cells. Bone 36: 1056–1064. [DOI] [PubMed] [Google Scholar]
- Pannier S, Couloigner V, Messaddeq N, Elmaleh-Berges M, Munnich A, Romand R, Legeai-Mallet L. 2009. Activating Fgfr3 Y367C mutation causes hearing loss and inner ear defect in a mouse model of chondrodysplasia. Biochim Biophys Acta 1792: 140–147. [DOI] [PubMed] [Google Scholar]
- Park OJ, Kim HJ, Woo KM, Baek JH, Ryoo HM. 2010. FGF2-activated ERK mitogen-activated protein kinase enhances Runx2 acetylation and stabilization. J Biol Chem 285: 3568–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peake NJ, Hobbs AJ, Pingguan-Murphy B, Salter DM, Berenbaum F, Chowdhury TT. 2014. Role of C-type natriuretic peptide signalling in maintaining cartilage and bone function. Osteoarthritis Cartilage 22: 1800–1807. [DOI] [PubMed] [Google Scholar]
- Perlyn CA, Morriss-Kay G, Darvann T, Tenenbaum M, Ornitz DM. 2006. A model for the pharmacological treatment of crouzon syndrome. Neurosurgery 59: 210–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters KG, Werner S, Chen G, Williams LT. 1992. Two FGF receptor genes are differentially expressed in epithelial and mesenchymal tissues during limb formation and organogenesis in the mouse. Development 114: 233–243. [DOI] [PubMed] [Google Scholar]
- Peters K, Ornitz D, Werner S, Williams L. 1993. Unique expression pattern of the FGF receptor 3 gene during mouse organogenesis. Dev Biol 155: 423–430. [DOI] [PubMed] [Google Scholar]
- Priore R, Dailey L, Basilico C. 2006. Downregulation of Akt activity contributes to the growth arrest induced by FGF in chondrocytes. J Cell Physiol 207: 800–808. [DOI] [PubMed] [Google Scholar]
- Purcell P, Joo BW, Hu JK, Tran PV, Calicchio ML, O'Connell DJ, Maas RL, Tabin CJ. 2009. Temporomandibular joint formation requires two distinct hedgehog-dependent steps. Proc Natl Acad Sci 106: 18297–18302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, Jin M, Duan Y, Du X, Zhang Y, Ren F, Wang Y, Tian Q, Wang X, Wang Q, et al. 2014. FGFR3 induces degradation of BMP type I receptor to regulate skeletal development. Biochim Biophys Acta 1843: 1237–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing J, Du X, Chen Y, Chan P, Li H, Wu P, Marsters S, Stawicki S, Tien J, Totpal K, et al. 2009. Antibody-based targeting of FGFR3 in bladder carcinoma and t(4;14)-positive multiple myeloma in mice. J Clin Invest 119: 1216–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarles LD. 2012. Skeletal secretion of FGF-23 regulates phosphate and vitamin D metabolism. Nat Rev Endocrinol 8: 276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarto N, Behr B, Li S, Longaker MT. 2009. Differential FGF ligands and FGF receptors expression pattern in frontal and parietal calvarial bones. Cells Tissues Organs 190: 158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raucci A, Laplantine E, Mansukhani A, Basilico C. 2004. Activation of the ERK1/2 and p38 mitogen-activated protein kinase pathways mediates fibroblast growth factor-induced growth arrest of chondrocytes. J Biol Chem 279: 1747–1756. [DOI] [PubMed] [Google Scholar]
- Reinhold MI, Naski MC. 2007. Direct interactions of Runx2 and canonical Wnt signaling induce FGF18. J Biol Chem 282: 3653–3663. [DOI] [PubMed] [Google Scholar]
- Reinhold MI, Abe M, Kapadia RM, Liao Z, Naski MC. 2004. FGF18 represses noggin expression and is induced by calcineurin. J Biol Chem 279: 38209–38219. [DOI] [PubMed] [Google Scholar]
- Rice DP, Rice R. 2008. Locate, condense, differentiate, grow and confront: developmental mechanisms controlling intramembranous bone and suture formation and function. Front Oral Biol 12: 22–40. [DOI] [PubMed] [Google Scholar]
- Rice DP, Kim HJ, Thesleff I. 1999. Apoptosis in murine calvarial bone and suture development. Eur J Oral Sci 107: 265–275. [DOI] [PubMed] [Google Scholar]
- Rice DP, Aberg T, Chan Y, Tang Z, Kettunen PJ, Pakarinen L, Maxson RE, Thesleff I. 2000. Integration of FGF and TWIST in calvarial bone and suture development. Development 127: 1845–1855. [DOI] [PubMed] [Google Scholar]
- Rice DP, Rice R, Thesleff I. 2003. Fgfr mRNA isoforms in craniofacial bone development. Bone 33: 14–27. [DOI] [PubMed] [Google Scholar]
- Robinson D, Hasharoni A, Cohen N, Yayon A, Moskowitz RM, Nevo Z. 1999. Fibroblast growth factor receptor-3 as a marker for precartilaginous stem cells. Clin Orthop Relat Res: S163–S175. [DOI] [PubMed] [Google Scholar]
- Sabbieti MG, Agas D, Xiao L, Marchetti L, Coffin JD, Doetschman T, Hurley MM. 2009. Endogenous FGF-2 is critically important in PTH anabolic effects on bone. J Cell Physiol 219: 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbieti MG, Agas D, Marchetti L, Coffin JD, Xiao L, Hurley MM. 2013. BMP-2 differentially modulates FGF-2 isoform effects in osteoblasts from newborn transgenic mice. Endocrinology 154: 2723–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni M, Ambrosetti DC, Mansukhani A, Gertner R, Levy D, Basilico C. 1999. FGF signaling inhibits chondrocyte proliferation and regulates bone development through the STAT-1 pathway. Genes Dev 13: 1361–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saran U, Gemini Piperni S, Chatterjee S. 2014. Role of angiogenesis in bone repair. Arch Biochem Biophys 561: 109–117. [DOI] [PubMed] [Google Scholar]
- Schlessinger J. 2000. Cell signaling by receptor tyrosine kinases. Cell 103: 211–225. [DOI] [PubMed] [Google Scholar]
- Schlessinger J. 2003. Signal transduction. Autoinhibition control. Science 300: 750–752. [DOI] [PubMed] [Google Scholar]
- Schmidt MH, Dikic I. 2005. The Cbl interactome and its functions. Nat Rev Mol Cell Biol 6: 907–918. [DOI] [PubMed] [Google Scholar]
- Schmid GJ, Kobayashi C, Sandell LJ, Ornitz DM. 2009. Fibroblast growth factor expression during skeletal fracture healing in mice. Dev Dyn 238: 766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian A, Matsushita T, Kawanami A, Mackem S, Landreth GE, Murakami S. 2011. Genetic inactivation of ERK1 and ERK2 in chondrocytes promotes bone growth and enlarges the spinal canal. J Orthop Res 29: 375–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senarath-Yapa K, Chung MT, McArdle A, Wong VW, Quarto N, Longaker MT, Wan DC. 2012. Craniosynostosis: molecular pathways and future pharmacologic therapy. Organogenesis 8: 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severe N, Dieudonne FX, Marie PJ. 2013. E3 ubiquitin ligase-mediated regulation of bone formation and tumorigenesis. Cell Death Dis 4: e463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheeba CJ, Andrade RP, Duprez D, Palmeirim I. 2010. Comprehensive analysis of fibroblast growth factor receptor expression patterns during chick forelimb development. Int J Dev Biol 54: 1517–1526. [DOI] [PubMed] [Google Scholar]
- Shiang R, Thompson LM, Zhu YZ, Church DM, Fielder TJ, Bocian M, Winokur ST, Wasmuth JJ. 1994. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell 78: 335–342. [DOI] [PubMed] [Google Scholar]
- Shimoaka T, Ogasawara T, Yonamine A, Chikazu D, Kawano H, Nakamura K, Itoh N, Kawaguchi H. 2002. Regulation of osteoblast, chondrocyte, and osteoclast functions by fibroblast growth factor (FGF)-18 in comparison with FGF-2 and FGF-10. J Biol Chem 277: 7493–7500. [DOI] [PubMed] [Google Scholar]
- Shimokawa K, Kimura-Yoshida C, Nagai N, Mukai K, Matsubara K, Watanabe H, Matsuda Y, Mochida K, Matsuo I. 2011. Cell surface heparan sulfate chains regulate local reception of FGF signaling in the mouse embryo. Dev Cell 21: 257–272. [DOI] [PubMed] [Google Scholar]
- Shinde DN, Elmer DP, Calabrese P, Boulanger J, Arnheim N, Tiemann-Boege I. 2013. New evidence for positive selection helps explain the paternal age effect observed in achondroplasia. Hum Mol Genet 22: 4117–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla V, Coumoul X, Wang RH, Kim HS, Deng CX. 2007. RNA interference and inhibition of MEK–ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat Genet 39: 1145–1150. [DOI] [PubMed] [Google Scholar]
- Shung CY, Ota S, Zhou ZQ, Keene DR, Hurlin PJ. 2012. Disruption of a Sox9–β-catenin circuit by mutant Fgfr3 in thanatophoric dysplasia type II. Hum Mol Genet 21: 4628–4644. [DOI] [PubMed] [Google Scholar]
- Smith LB, Dally MR, Sainz RD, Rodrigue KL, Oberbauer AM. 2006. Enhanced skeletal growth of sheep heterozygous for an inactivated fibroblast growth factor receptor 3. J Anim Sci 84: 2942–2949. [DOI] [PubMed] [Google Scholar]
- Smith BN, Burton LJ, Henderson V, Randle DD, Morton DJ, Smith BA, Taliaferro-Smith L, Nagappan P, Yates C, Zayzafoon M, et al. 2014. Snail promotes epithelial mesenchymal transition in breast cancer cells in part via activation of nuclear ERK2. PLoS One 9: e104987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su WC, Kitagawa M, Xue N, Xie B, Garofalo S, Cho J, Deng C, Horton WA, Fu XY. 1997. Activation of Stat1 by mutant fibroblast growth-factor receptor in thanatophoric dysplasia type II dwarfism. Nature 386: 288–292. [DOI] [PubMed] [Google Scholar]
- Su N, Sun Q, Li C, Lu X, Qi H, Chen S, Yang J, Du X, Zhao L, He Q, et al. 2010. Gain-of-function mutation in FGFR3 in mice leads to decreased bone mass by affecting both osteoblastogenesis and osteoclastogenesis. Hum Mol Genet 19: 1199–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Suda N, Shiga M, Kobayashi Y, Nakamura M, Iseki S, Moriyama K. 2012. Apert syndrome mutant FGFR2 and its soluble form reciprocally alter osteogenesis of primary calvarial osteoblasts. J Cell Physiol 227: 3267–3277. [DOI] [PubMed] [Google Scholar]
- Szebenyi G, Savage MP, Olwin BB, Fallon JF. 1995. Changes in the expression of fibroblast growth factor receptors mark distinct stages of chondrogenesis in vitro and during chick limb skeletal patterning. Dev Dyn 204: 446–456. [DOI] [PubMed] [Google Scholar]
- Takai S, Tokuda H, Hanai Y, Harada A, Yasuda E, Matsushima-Nishiwaki R, Kato H, Ogura S, Ohta T, Kozawa O. 2007. Negative regulation by p70 S6 kinase of FGF-2-stimulated VEGF release through stress-activated protein kinase/c-Jun N-terminal kinase in osteoblasts. J Bone Miner Res 22: 337–346. [DOI] [PubMed] [Google Scholar]
- Tan Y, Rouse J, Zhang A, Cariati S, Cohen P, Comb MJ. 1996. FGF and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. EMBO J 15: 4629–4642. [PMC free article] [PubMed] [Google Scholar]
- Tanimoto Y, Yokozeki M, Hiura K, Matsumoto K, Nakanishi H, Matsumoto T, Marie PJ, Moriyama K. 2004. A soluble form of fibroblast growth factor receptor 2 (FGFR2) with S252W mutation acts as an efficient inhibitor for the enhanced osteoblastic differentiation caused by FGFR2 activation in Apert syndrome. J Biol Chem 279: 45926–45934. [DOI] [PubMed] [Google Scholar]
- Tavormina PL, Shiang R, Thompson LM, Zhu YZ, Wilkin DJ, Lachman RS, Wilcox WR, Rimoin DL, Cohn DH, Wasmuth JJ. 1995. Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nat Genet 9: 321–328. [DOI] [PubMed] [Google Scholar]
- Tavormina PL, Bellus GA, Webster MK, Bamshad MJ, Fraley AE, McIntosh I, Szabo J, Jiang W, Jabs EW, Wilcox WR, et al. 1999. A novel skeletal dysplasia with developmental delay and acanthosis nigricans is caused by a Lys650Met mutation in the fibroblast growth factor receptor 3 gene. Am J Hum Genet 64: 722–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten Berge D, Brugmann SA, Helms JA, Nusse R. 2008. Wnt and FGF signals interact to coordinate growth with cell fate specification during limb development. Development 135: 3247–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teplyuk NM, Haupt LM, Ling L, Dombrowski C, Mun FK, Nathan SS, Lian JB, Stein JL, Stein GS, Cool SM, et al. 2009. The osteogenic transcription factor Runx2 regulates components of the fibroblast growth factor/proteoglycan signaling axis in osteoblasts. J Cell Biochem 107: 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timsah Z, Ahmed Z, Lin CC, Melo FA, Stagg LJ, Leonard PG, Jeyabal P, Berrout J, O'Neil RG, Bogdanov M, et al. 2014. Competition between Grb2 and Plcγ1 for FGFR2 regulates basal phospholipase activity and invasion. Nat Struct Mol Biol 21: 180–188. [DOI] [PubMed] [Google Scholar]
- Torii S, Kusakabe M, Yamamoto T, Maekawa M, Nishida E. 2004. Sef is a spatial regulator for Ras/MAP kinase signaling. Dev Cell 7: 33–44. [DOI] [PubMed] [Google Scholar]
- Toydemir RM, Brassington AE, Bayrak-Toydemir P, Krakowiak PA, Jorde LB, Whitby FG, Longo N, Viskochil DH, Carey JC, Bamshad MJ. 2006. A novel mutation in FGFR3 causes camptodactyly, tall stature, and hearing loss (CATSHL) syndrome. Am J Hum Genet 79: 935–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang M, Dawid IB. 2004. Promotion and attenuation of FGF signaling through the Ras–MAPK pathway. Sci STKE 2004: pe17. [DOI] [PubMed] [Google Scholar]
- Tsuji T, Kunieda T. 2005. A loss-of-function mutation in natriuretic peptide receptor 2 (Npr2) gene is responsible for disproportionate dwarfism in cn/cn mouse. J Biol Chem 280: 14288–14292. [DOI] [PubMed] [Google Scholar]
- Twigg SR, Healy C, Babbs C, Sharpe JA, Wood WG, Sharpe PT, Morriss-Kay GM, Wilkie AO. 2009. Skeletal analysis of the Fgfr3(P244R) mouse, a genetic model for the Muenke craniosynostosis syndrome. Dev Dyn 238: 331–342. [DOI] [PubMed] [Google Scholar]
- Twigg SR, Vorgia E, McGowan SJ, Peraki I, Fenwick AL, Sharma VP, Allegra M, Zaragkoulias A, Sadighi Akha E, Knight SJ, et al. 2013. Reduced dosage of ERF causes complex craniosynostosis in humans and mice and links ERK1/2 signaling to regulation of osteogenesis. Nat Genet 45: 308–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. 2006. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444: 770–774.17086194 [Google Scholar]
- Vajo Z, Francomano CA, Wilkin DJ. 2000. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans. Endocr Rev 21: 23–39. [DOI] [PubMed] [Google Scholar]
- Valverde-Franco G, Liu H, Davidson D, Chai S, Valderrama-Carvajal H, Goltzman D, Ornitz DM, Henderson JE. 2004. Defective bone mineralization and osteopenia in young adult FGFR3−/− mice. Hum Mol Genet 13: 271–284. [DOI] [PubMed] [Google Scholar]
- Verheyden JM, Lewandoski M, Deng C, Harfe BD, Sun X. 2005. Conditional inactivation of Fgfr1 in mouse defines its role in limb bud establishment, outgrowth and digit patterning. Development 132: 4235–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Spatz MK, Kannan K, Hayk H, Avivi A, Gorivodsky M, Pines M, Yayon A, Lonai P, Givol D. 1999. A mouse model for achondroplasia produced by targeting fibroblast growth factor receptor 3. Proc Natl Acad Sci 96: 4455–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Green RP, Zhao G, Ornitz DM. 2001. Differential regulation of endochondral bone growth and joint development by FGFR1 and FGFR3 tyrosine kinase domains. Development 128: 3867–3876. [DOI] [PubMed] [Google Scholar]
- Wang Y, Xiao R, Yang F, Karim BO, Iacovelli AJ, Cai J, Lerner CP, Richtsmeier JT, Leszl JM, Hill CA, et al. 2005. Abnormalities in cartilage and bone development in the Apert syndrome FGFR2(+/S252W) mouse. Development 132: 3537–3548. [DOI] [PubMed] [Google Scholar]
- Wang Y, Sun M, Uhlhorn VL, Zhou X, Peter I, Martinez-Abadias N, Hill CA, Percival CJ, Richtsmeier JT, Huso DL, et al. 2010. Activation of p38 MAPK pathway in the skull abnormalities of Apert syndrome Fgfr2(+P253R) mice. BMC Dev Biol 10: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Nyman JS, Ono K, Stevenson DA, Yang X, Elefteriou F. 2011. Mice lacking Nf1 in osteochondroprogenitor cells display skeletal dysplasia similar to patients with neurofibromatosis type I. Hum Mol Genet 20: 3910–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren SM, Brunet LJ, Harland RM, Economides AN, Longaker MT. 2003. The BMP antagonist noggin regulates cranial suture fusion. Nature 422: 625–629. [DOI] [PubMed] [Google Scholar]
- Wendt DJ, Dvorak-Ewell M, Bullens S, Lorget F, Bell SM, Peng J, Castillo S, Aoyagi-Scharber M, O'Neill CA, Krejci P, et al. 2015. Neutral endopeptidase-resistant C-type natriuretic peptide variant represents a new therapeutic approach for treatment of fibroblast growth factor receptor 3-related dwarfism. J Pharmacol Exp Ther 353: 132–149. [DOI] [PubMed] [Google Scholar]
- Weng T, Yi L, Huang J, Luo F, Wen X, Du X, Chen Q, Deng C, Chen D, Chen L. 2012. Genetic inhibition of FGFR1 in cartilage attenuates articular cartilage degeneration in adult mice. Arthritis Rheum 64: 3982–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KE, Cabral JM, Davis SI, Fishburn T, Evans WE, Ichikawa S, Fields J, Yu X, Shaw NJ, McLellan NJ, et al. 2005. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am J Hum Genet 76: 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KE, Hum JM, Econs MJ. 2014. Hypophosphatemic rickets: revealing novel control points for phosphate homeostasis. Curr Osteoporos Rep 12: 252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann M, Trueb B. 2000. Characterization of a novel protein (FGFRL1) from human cartilage related to FGF receptors. Genomics 69: 275–279. [DOI] [PubMed] [Google Scholar]
- Wilkie AO. 2007. Cancer drugs to treat birth defects. Nat Genet 39: 1057–1059. [DOI] [PubMed] [Google Scholar]
- Wilkin DJ, Szabo JK, Cameron R, Henderson S, Bellus GA, Mack ML, Kaitila I, Loughlin J, Munnich A, Sykes B, et al. 1998. Mutations in fibroblast growth-factor receptor 3 in sporadic cases of achondroplasia occur exclusively on the paternally derived chromosome. Am J Hum Genet 63: 711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohrle S, Henninger C, Bonny O, Thuery A, Beluch N, Hynes NE, Guagnano V, Sellers WR, Hofmann F, Kneissel M, et al. 2013. Pharmacological inhibition of fibroblast growth factor (FGF) receptor signaling ameliorates FGF23-mediated hypophosphatemic rickets. J Bone Miner Res 28: 899–911. [DOI] [PubMed] [Google Scholar]
- Wong A, Lamothe B, Lee A, Schlessinger J, Lax I. 2002. FRS2α attenuates FGF receptor signaling by Grb2-mediated recruitment of the ubiquitin ligase Cbl. Proc Natl Acad Sci 99: 6684–6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZL, Zhang L, Yabe T, Kuberan B, Beeler DL, Love A, Rosenberg RD. 2003. The involvement of heparan sulfate (HS) in FGF1/HS/FGFR1 signaling complex. J Biol Chem 278: 17121–17129. [DOI] [PubMed] [Google Scholar]
- Wu XL, Gu MM, Huang L, Liu XS, Zhang HX, Ding XY, Xu JQ, Cui B, Wang L, Lu SY, et al. 2009. Multiple synostoses syndrome is due to a missense mutation in exon 2 of FGF9 gene. Am J Hum Genet 85: 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu AL, Feng B, Chen MZ, Kolumam G, Zavala-Solorio J, Wyatt SK, Gandham VD, Carano RA, Sonoda J. 2013. Antibody-mediated activation of FGFR1 induces FGF23 production and hypophosphatemia. PLoS One 8: e57322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao G, Jiang D, Gopalakrishnan R, Franceschi RT. 2002. Fibroblast growth factor 2 induction of the osteocalcin gene requires MAPK activity and phosphorylation of the osteoblast transcription factor, Cbfa1/Runx2. J Biol Chem 277: 36181–36187. [DOI] [PubMed] [Google Scholar]
- Xiao L, Naganawa T, Obugunde E, Gronowicz G, Ornitz DM, Coffin JD, Hurley MM. 2004. Stat1 controls postnatal bone formation by regulating fibroblast growth factor signaling in osteoblasts. J Biol Chem 279: 27743–27752. [DOI] [PubMed] [Google Scholar]
- Xiao L, Liu P, Li X, Doetschman T, Coffin JD, Drissi H, Hurley MM. 2009. Exported 18-kDa isoform of fibroblast growth factor-2 is a critical determinant of bone mass in mice. J Biol Chem 284: 3170–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Sobue T, Esliger A, Kronenberg MS, Coffin JD, Doetschman T, Hurley MM. 2010. Disruption of the Fgf2 gene activates the adipogenic and suppresses the osteogenic program in mesenchymal marrow stromal stem cells. Bone 47: 360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Esliger A, Hurley MM. 2013. Nuclear fibroblast growth factor 2 (FGF2) isoforms inhibit bone marrow stromal cell mineralization through FGF23/FGFR/MAPK in vitro. J Bone Miner Res 28: 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Huang J, Cao L, Liang Y, Han X, Quarles LD. 2014. Osteocyte-specific deletion of Fgfr1 suppresses FGF23. PLoS One 9: e104154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Su N, Jin M, Qi H, Yang J, Li C, Du X, Luo F, Chen B, Shen Y, et al. 2012. Intermittent PTH (1–34) injection rescues the retarded skeletal development and postnatal lethality of mice mimicking human achondroplasia and thanatophoric dysplasia. Hum Mol Genet 21: 3941–3955. [DOI] [PubMed] [Google Scholar]
- Xu D, Esko JD. 2014. Demystifying heparan sulfate-protein interactions. Annu Rev Biochem 83: 129–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Morioka M, Kishi H, Kimura T, Yahara Y, Okada M, Fujita K, Sawai H, Ikegawa S, Tsumaki N. 2014. Statin treatment rescues FGFR3 skeletal dysplasia phenotypes. Nature 513: 507–511. [DOI] [PubMed] [Google Scholar]
- Yan D, Chen D, Cool SM, van Wijnen AJ, Mikecz K, Murphy G, Im HJ. 2011. Fibroblast growth factor receptor 1 is principally responsible for fibroblast growth factor 2-induced catabolic activities in human articular chondrocytes. Arthritis Res Ther 13: R130. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yang F, Wang Y, Zhang Z, Hsu B, Jabs EW, Elisseeff JH. 2008. The study of abnormal bone development in the Apert syndrome Fgfr2+/S252W mouse using a 3D hydrogel culture model. Bone 43: 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Tsang KY, Tang HC, Chan D, Cheah KS. 2014. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc Natl Acad Sci 111: 12097–12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasoda A, Komatsu Y, Chusho H, Miyazawa T, Ozasa A, Miura M, Kurihara T, Rogi T, Tanaka S, Suda M, et al. 2004. Overexpression of CNP in chondrocytes rescues achondroplasia through a MAPK-dependent pathway. Nat Med 10: 80–86. [DOI] [PubMed] [Google Scholar]
- Yeh E, Atique R, Ishiy FA, Fanganiello RD, Alonso N, Matushita H, da Rocha KM, Passos-Bueno MR. 2012. FGFR2 mutation confers a less drastic gain of function in mesenchymal stem cells than in fibroblasts. Stem Cell Rev 8: 685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung Tsang K, Wa Tsang S, Chan D, Cheah KS. 2014. The chondrocytic journey in endochondral bone growth and skeletal dysplasia. Birth Defects Res C Embryo Today 102: 52–73. [DOI] [PubMed] [Google Scholar]
- Yin L, Du X, Li C, Xu X, Chen Z, Su N, Zhao L, Qi H, Li F, Xue J, et al. 2008. A Pro253Arg mutation in fibroblast growth factor receptor 2 (Fgfr2) causes skeleton malformation mimicking human Apert syndrome by affecting both chondrogenesis and osteogenesis. Bone 42: 631–643. [DOI] [PubMed] [Google Scholar]
- Yokota M, Kobayashi Y, Morita J, Suzuki H, Hashimoto Y, Sasaki Y, Akiyoshi K, Moriyama K. 2014. Therapeutic effect of nanogel-based delivery of soluble FGFR2 with S252W mutation on craniosynostosis. PLoS One 9: e101693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM. 2005. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci 102: 5062–5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon WJ, Cho YD, Kim WJ, Bae HS, Islam R, Woo KM, Baek JH, Bae SC, Ryoo HM. 2014. Prolyl isomerase Pin1-mediated conformational change and subnuclear focal accumulation of Runx2 are crucial for fibroblast growth factor 2 (FGF2)-induced osteoblast differentiation. J Biol Chem 289: 8828–8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida CA, Yamamoto H, Fujita T, Furuichi T, Ito K, Inoue K, Yamana K, Zanma A, Takada K, Ito Y, et al. 2004. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev 18: 952–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Ornitz DM. 2001. Uncoupling fibroblast growth factor receptor 2 ligand binding specificity leads to Apert syndrome-like phenotypes. Proc Natl Acad Sci 98: 3641–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Ornitz DM. 2008. FGF signaling regulates mesenchymal differentiation and skeletal patterning along the limb bud proximodistal axis. Development 135: 483–491. [DOI] [PubMed] [Google Scholar]
- Yu K, Herr AB, Waksman G, Ornitz DM. 2000. Loss of fibroblast growth factor receptor 2 ligand-binding specificity in Apert syndrome. Proc Natl Acad Sci 97: 14536–14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. 2003. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 130: 3063–3074. [DOI] [PubMed] [Google Scholar]
- Zeller R, Lopez-Rios J, Zuniga A. 2009. Vertebrate limb bud development: moving towards integrative analysis of organogenesis. Nat Rev Genet 10: 845–858. [DOI] [PubMed] [Google Scholar]
- Zhang K, Corsa CA, Ponik SM, Prior JL, Piwnica-Worms D, Eliceiri KW, Keely PJ, Longmore GD. 2013. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat Cell Biol 15: 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YX, Xu X, Chen L, Li C, Brodie SG, Deng CX. 2000. A Pro250Arg substitution in mouse Fgfr1 causes increased expression of Cbfa1 and premature fusion of calvarial sutures. Hum Mol Genet 9: 2001–2008. [DOI] [PubMed] [Google Scholar]
- Zhou X, von der Mark K, Henry S, Norton W, Adams H, de Crombrugghe B. 2014. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet 10: e1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou ZQ, Ota S, Deng C, Akiyama H, Hurlin PJ. 2015. Mutant activated FGFR3 impairs endochondral bone growth by preventing SOX9 downregulation in differentiating chondrocytes. Hum Mol Genet 24: 1764–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]