

Abstract
Gerstmann-Sträussler-Scheinker (GSS) disease is a familial neurological disorder pathologically characterized by amyloid deposition in the cerebrum and cerebellum. The GSS amyloid is immunoreactive to antisera raised against the hamster prion protein (PrP) 27-30. This is a proteinase K-resistant glycoprotein of 27-30 kd that is derived from an abnormal isoform of a neuronal glycoprotein of 33-35 kd designated PrPSc and is a molecular marker of amyloid fibrils isolated from animals with scrapie and humans with related disorders. We have purified and characterized proteins extracted from amyloid plaque cores isolated from two patients of the Indiana kindred of GSS disease. We found that the major component of GSS amyloid is an 11 kd degradation product of PrP, whose N-terminus corresponds to the glycine residue at position 58 of the amino acid sequence deduced from the human PrP cDNA. In addition, amyloid fractions contained larger PrP fragments with apparently intact N-termini and amyloid P component. These findings suggest that the disease process leads to proteolytic cleavage of PrP, generating an amyloidogenic peptide that polymerizes into insoluble fibrils. The N-terminal cleavage of PrP in GSS disease occurs at a tryptophan-glycine peptide bond identical to that cleaved by proteinase K in vitro to generate PrP 27-30 from hamster PrPSc at codon 90. Since no mutations of the structural PrP gene have been found in the Indiana family of GSS disease, it is conceivable that factors other than the primary structure of PrP play a crucial role in the process of amyloid formation and the development of clinical neurologic dysfunction.
Full text
PDF


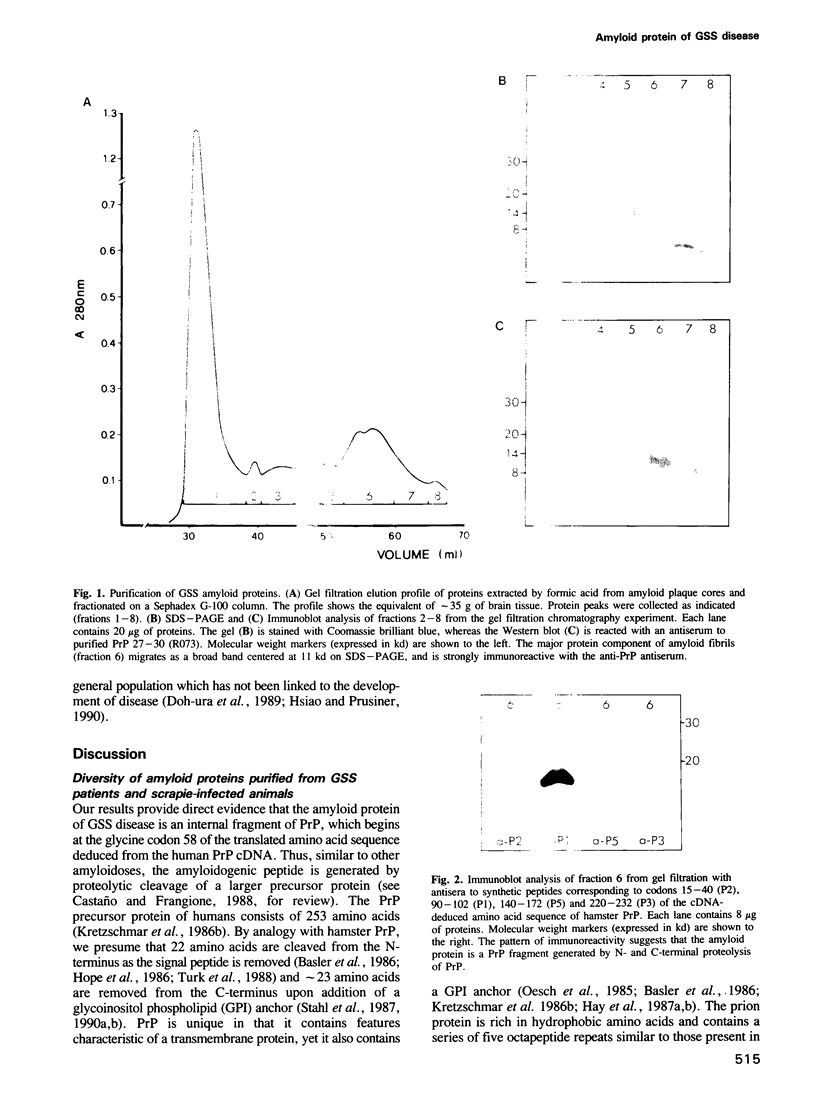
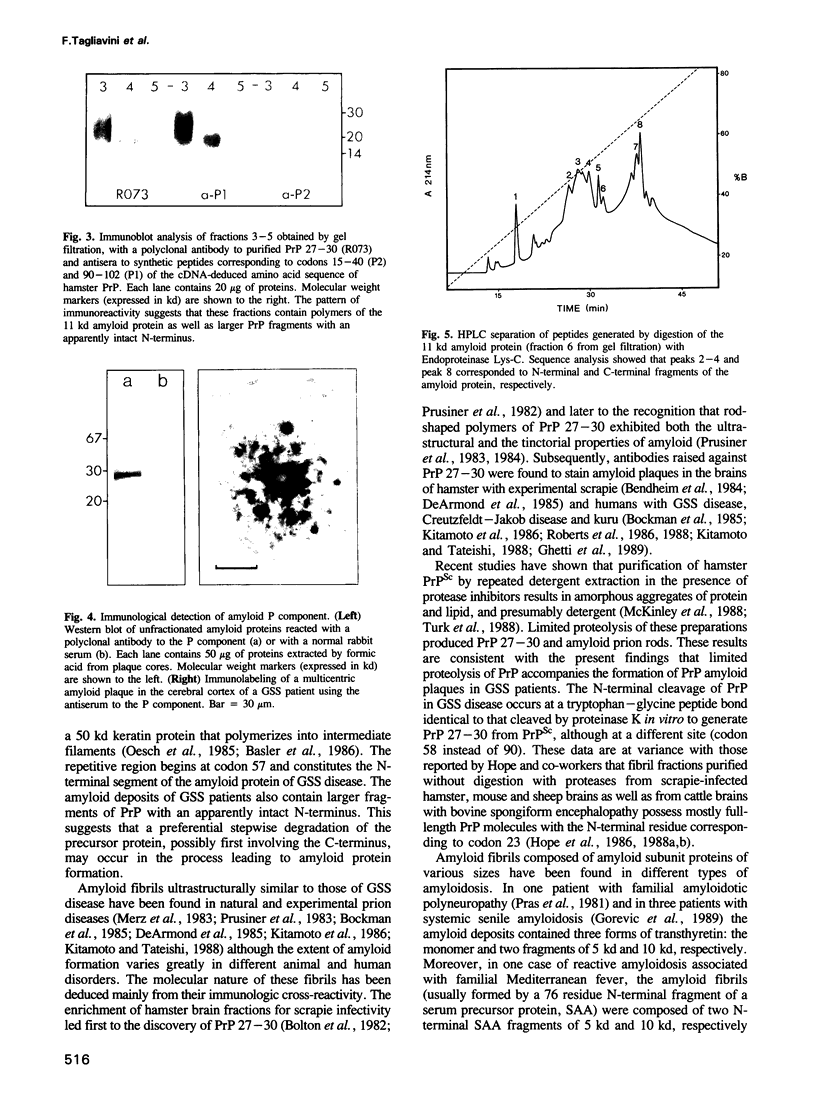



Images in this article


Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Azzarelli B., Muller J., Ghetti B., Dyken M., Conneally P. M. Cerebellar plaques in familial Alzheimer's disease (Gerstmann-Sträussler-Scheinker variant?). Acta Neuropathol. 1985;65(3-4):235–246. doi: 10.1007/BF00687003. [DOI] [PubMed] [Google Scholar]
- Baker H. F., Ridley R. M., Crow T. J. Experimental transmission of an autosomal dominant spongiform encephalopathy: does the infectious agent originate in the human genome? Br Med J (Clin Res Ed) 1985 Aug 3;291(6491):299–302. doi: 10.1136/bmj.291.6491.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry R. A., Vincent M. T., Kent S. B., Hood L. E., Prusiner S. B. Characterization of prion proteins with monospecific antisera to synthetic peptides. J Immunol. 1988 Feb 15;140(4):1188–1193. [PubMed] [Google Scholar]
- Basler K., Oesch B., Scott M., Westaway D., Wälchli M., Groth D. F., McKinley M. P., Prusiner S. B., Weissmann C. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell. 1986 Aug 1;46(3):417–428. doi: 10.1016/0092-8674(86)90662-8. [DOI] [PubMed] [Google Scholar]
- Bendheim P. E., Barry R. A., DeArmond S. J., Stites D. P., Prusiner S. B. Antibodies to a scrapie prion protein. Nature. 1984 Aug 2;310(5976):418–421. doi: 10.1038/310418a0. [DOI] [PubMed] [Google Scholar]
- Bockman J. M., Kingsbury D. T., McKinley M. P., Bendheim P. E., Prusiner S. B. Creutzfeldt-Jakob disease prion proteins in human brains. N Engl J Med. 1985 Jan 10;312(2):73–78. doi: 10.1056/NEJM198501103120202. [DOI] [PubMed] [Google Scholar]
- Boellaard J. W., Schlote W. Subakute spongiforme Encephalopathie mit multiformer Plaquebildung. "Eigenartige familiär-hereditäre Krankheit des Zentralnervensystems [spino-cerebellare Atrophie mit Demenz, Plaques und plaqueähnlichen Ablagerungen im Klein- und Grossirn" (Gerstmann, Sträussler, Scheinker)]. Acta Neuropathol. 1980;49(3):205–212. doi: 10.1007/BF00707108. [DOI] [PubMed] [Google Scholar]
- Bolton D. C., McKinley M. P., Prusiner S. B. Identification of a protein that purifies with the scrapie prion. Science. 1982 Dec 24;218(4579):1309–1311. doi: 10.1126/science.6815801. [DOI] [PubMed] [Google Scholar]
- Castaño E. M., Frangione B. Human amyloidosis, Alzheimer disease and related disorders. Lab Invest. 1988 Feb;58(2):122–132. [PubMed] [Google Scholar]
- Castaño E. M., Ghiso J., Prelli F., Gorevic P. D., Migheli A., Frangione B. In vitro formation of amyloid fibrils from two synthetic peptides of different lengths homologous to Alzheimer's disease beta-protein. Biochem Biophys Res Commun. 1986 Dec 15;141(2):782–789. doi: 10.1016/s0006-291x(86)80241-8. [DOI] [PubMed] [Google Scholar]
- Collinge J., Harding A. E., Owen F., Poulter M., Lofthouse R., Boughey A. M., Shah T., Crow T. J. Diagnosis of Gerstmann-Sträussler syndrome in familial dementia with prion protein gene analysis. Lancet. 1989 Jul 1;2(8653):15–17. doi: 10.1016/s0140-6736(89)90256-0. [DOI] [PubMed] [Google Scholar]
- Coria F., Castaño E., Prelli F., Larrondo-Lillo M., van Duinen S., Shelanski M. L., Frangione B. Isolation and characterization of amyloid P component from Alzheimer's disease and other types of cerebral amyloidosis. Lab Invest. 1988 Apr;58(4):454–458. [PubMed] [Google Scholar]
- DeArmond S. J., McKinley M. P., Barry R. A., Braunfeld M. B., McColloch J. R., Prusiner S. B. Identification of prion amyloid filaments in scrapie-infected brain. Cell. 1985 May;41(1):221–235. doi: 10.1016/0092-8674(85)90076-5. [DOI] [PubMed] [Google Scholar]
- Diringer H., Gelderblom H., Hilmert H., Ozel M., Edelbluth C., Kimberlin R. H. Scrapie infectivity, fibrils and low molecular weight protein. Nature. 1983 Dec 1;306(5942):476–478. doi: 10.1038/306476a0. [DOI] [PubMed] [Google Scholar]
- Doh-ura K., Tateishi J., Sasaki H., Kitamoto T., Sakaki Y. Pro----leu change at position 102 of prion protein is the most common but not the sole mutation related to Gerstmann-Sträussler syndrome. Biochem Biophys Res Commun. 1989 Sep 15;163(2):974–979. doi: 10.1016/0006-291x(89)92317-6. [DOI] [PubMed] [Google Scholar]
- Farlow M. R., Yee R. D., Dlouhy S. R., Conneally P. M., Azzarelli B., Ghetti B. Gerstmann-Sträussler-Scheinker disease. I. Extending the clinical spectrum. Neurology. 1989 Nov;39(11):1446–1452. doi: 10.1212/wnl.39.11.1446. [DOI] [PubMed] [Google Scholar]
- Gabizon R., McKinley M. P., Groth D., Prusiner S. B. Immunoaffinity purification and neutralization of scrapie prion infectivity. Proc Natl Acad Sci U S A. 1988 Sep;85(18):6617–6621. doi: 10.1073/pnas.85.18.6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabizon R., Prusiner S. B. Prion liposomes. Biochem J. 1990 Feb 15;266(1):1–14. doi: 10.1042/bj2660001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghetti B., Tagliavini F., Masters C. L., Beyreuther K., Giaccone G., Verga L., Farlow M. R., Conneally P. M., Dlouhy S. R., Azzarelli B. Gerstmann-Sträussler-Scheinker disease. II. Neurofibrillary tangles and plaques with PrP-amyloid coexist in an affected family. Neurology. 1989 Nov;39(11):1453–1461. doi: 10.1212/wnl.39.11.1453. [DOI] [PubMed] [Google Scholar]
- Giaccone G., Tagliavini F., Verga L., Frangione B., Farlow M. R., Bugiani O., Ghetti B. Neurofibrillary tangles of the Indiana kindred of Gerstmann-Sträussler-Scheinker disease share antigenic determinants with those of Alzheimer disease. Brain Res. 1990 Oct 22;530(2):325–329. doi: 10.1016/0006-8993(90)91304-y. [DOI] [PubMed] [Google Scholar]
- Goldgaber D., Goldfarb L. G., Brown P., Asher D. M., Brown W. T., Lin S., Teener J. W., Feinstone S. M., Rubenstein R., Kascsak R. J. Mutations in familial Creutzfeldt-Jakob disease and Gerstmann-Sträussler-Scheinker's syndrome. Exp Neurol. 1989 Nov;106(2):204–206. doi: 10.1016/0014-4886(89)90095-2. [DOI] [PubMed] [Google Scholar]
- Gorevic P. D., Prelli F. C., Wright J., Pras M., Frangione B. Systemic senile amyloidosis. Identification of a new prealbumin (transthyretin) variant in cardiac tissue: immunologic and biochemical similarity to one form of familial amyloidotic polyneuropathy. J Clin Invest. 1989 Mar;83(3):836–843. doi: 10.1172/JCI113966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay B., Barry R. A., Lieberburg I., Prusiner S. B., Lingappa V. R. Biogenesis and transmembrane orientation of the cellular isoform of the scrapie prion protein [published errratum appears in Mol Cell Biol 1987 May;7(5):2035]. Mol Cell Biol. 1987 Feb;7(2):914–920. doi: 10.1128/mcb.7.2.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay B., Prusiner S. B., Lingappa V. R. Evidence for a secretory form of the cellular prion protein. Biochemistry. 1987 Dec 15;26(25):8110–8115. doi: 10.1021/bi00399a014. [DOI] [PubMed] [Google Scholar]
- Hope J., Morton L. J., Farquhar C. F., Multhaup G., Beyreuther K., Kimberlin R. H. The major polypeptide of scrapie-associated fibrils (SAF) has the same size, charge distribution and N-terminal protein sequence as predicted for the normal brain protein (PrP). EMBO J. 1986 Oct;5(10):2591–2597. doi: 10.1002/j.1460-2075.1986.tb04539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope J., Multhaup G., Reekie L. J., Kimberlin R. H., Beyreuther K. Molecular pathology of scrapie-associated fibril protein (PrP) in mouse brain affected by the ME7 strain of scrapie. Eur J Biochem. 1988 Mar 1;172(2):271–277. doi: 10.1111/j.1432-1033.1988.tb13883.x. [DOI] [PubMed] [Google Scholar]
- Hope J., Reekie L. J., Hunter N., Multhaup G., Beyreuther K., White H., Scott A. C., Stack M. J., Dawson M., Wells G. A. Fibrils from brains of cows with new cattle disease contain scrapie-associated protein. Nature. 1988 Nov 24;336(6197):390–392. doi: 10.1038/336390a0. [DOI] [PubMed] [Google Scholar]
- Hsiao K., Baker H. F., Crow T. J., Poulter M., Owen F., Terwilliger J. D., Westaway D., Ott J., Prusiner S. B. Linkage of a prion protein missense variant to Gerstmann-Sträussler syndrome. Nature. 1989 Mar 23;338(6213):342–345. doi: 10.1038/338342a0. [DOI] [PubMed] [Google Scholar]
- Hsiao K., Prusiner S. B. Inherited human prion diseases. Neurology. 1990 Dec;40(12):1820–1827. doi: 10.1212/wnl.40.12.1820. [DOI] [PubMed] [Google Scholar]
- Ishii T., Haga S., Yagishita S., Tateishi J. The presence of complements in amyloid plaques of Creutzfeldt-Jakob disease and Gerstmann-Straussler-Scheinker disease. Appl Pathol. 1984;2(6):370–379. [PubMed] [Google Scholar]
- Kitamoto T., Tateishi J. Immunohistochemical confirmation of Creutzfeldt-Jakob disease with a long clinical course with amyloid plaque core antibodies. Am J Pathol. 1988 Jun;131(3):435–443. [PMC free article] [PubMed] [Google Scholar]
- Kitamoto T., Tateishi J., Tashima T., Takeshita I., Barry R. A., DeArmond S. J., Prusiner S. B. Amyloid plaques in Creutzfeldt-Jakob disease stain with prion protein antibodies. Ann Neurol. 1986 Aug;20(2):204–208. doi: 10.1002/ana.410200205. [DOI] [PubMed] [Google Scholar]
- Kretzschmar H. A., Prusiner S. B., Stowring L. E., DeArmond S. J. Scrapie prion proteins are synthesized in neurons. Am J Pathol. 1986 Jan;122(1):1–5. [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar H. A., Stowring L. E., Westaway D., Stubblebine W. H., Prusiner S. B., Dearmond S. J. Molecular cloning of a human prion protein cDNA. DNA. 1986 Aug;5(4):315–324. doi: 10.1089/dna.1986.5.315. [DOI] [PubMed] [Google Scholar]
- Levy E., Carman M. D., Fernandez-Madrid I. J., Power M. D., Lieberburg I., van Duinen S. G., Bots G. T., Luyendijk W., Frangione B. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990 Jun 1;248(4959):1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- Masters C. L., Gajdusek D. C., Gibbs C. J., Jr Creutzfeldt-Jakob disease virus isolations from the Gerstmann-Sträussler syndrome with an analysis of the various forms of amyloid plaque deposition in the virus-induced spongiform encephalopathies. Brain. 1981 Sep;104(3):559–588. doi: 10.1093/brain/104.3.559. [DOI] [PubMed] [Google Scholar]
- McKinley M. P., Bolton D. C., Prusiner S. B. A protease-resistant protein is a structural component of the scrapie prion. Cell. 1983 Nov;35(1):57–62. doi: 10.1016/0092-8674(83)90207-6. [DOI] [PubMed] [Google Scholar]
- Merz P. A., Wisniewski H. M., Somerville R. A., Bobin S. A., Masters C. L., Iqbal K. Ultrastructural morphology of amyloid fibrils from neuritic and amyloid plaques. Acta Neuropathol. 1983;60(1-2):113–124. doi: 10.1007/BF00685355. [DOI] [PubMed] [Google Scholar]
- Meyer R. K., McKinley M. P., Bowman K. A., Braunfeld M. B., Barry R. A., Prusiner S. B. Separation and properties of cellular and scrapie prion proteins. Proc Natl Acad Sci U S A. 1986 Apr;83(8):2310–2314. doi: 10.1073/pnas.83.8.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Multhaup G., Diringer H., Hilmert H., Prinz H., Heukeshoven J., Beyreuther K. The protein component of scrapie-associated fibrils is a glycosylated low molecular weight protein. EMBO J. 1985 Jun;4(6):1495–1501. doi: 10.1002/j.1460-2075.1985.tb03808.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesch B., Westaway D., Wälchli M., McKinley M. P., Kent S. B., Aebersold R., Barry R. A., Tempst P., Teplow D. B., Hood L. E. A cellular gene encodes scrapie PrP 27-30 protein. Cell. 1985 Apr;40(4):735–746. doi: 10.1016/0092-8674(85)90333-2. [DOI] [PubMed] [Google Scholar]
- Owen F., Poulter M., Lofthouse R., Collinge J., Crow T. J., Risby D., Baker H. F., Ridley R. M., Hsiao K., Prusiner S. B. Insertion in prion protein gene in familial Creutzfeldt-Jakob disease. Lancet. 1989 Jan 7;1(8628):51–52. doi: 10.1016/s0140-6736(89)91713-3. [DOI] [PubMed] [Google Scholar]
- Pras M., Franklin E. C., Prelli F., Frangione B. A variant of prealbumin from amyloid fibrils in familial polyneuropathy of Jewish origin. J Exp Med. 1981 Sep 1;154(3):989–993. doi: 10.1084/jem.154.3.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prelli F., Pras M., Frangione B. Degradation and deposition of amyloid AA fibrils are tissue specific. Biochemistry. 1987 Dec 15;26(25):8251–8256. doi: 10.1021/bi00399a035. [DOI] [PubMed] [Google Scholar]
- Prusiner S. B., Bolton D. C., Groth D. F., Bowman K. A., Cochran S. P., McKinley M. P. Further purification and characterization of scrapie prions. Biochemistry. 1982 Dec 21;21(26):6942–6950. doi: 10.1021/bi00269a050. [DOI] [PubMed] [Google Scholar]
- Prusiner S. B., Groth D. F., Bolton D. C., Kent S. B., Hood L. E. Purification and structural studies of a major scrapie prion protein. Cell. 1984 Aug;38(1):127–134. doi: 10.1016/0092-8674(84)90533-6. [DOI] [PubMed] [Google Scholar]
- Prusiner S. B., McKinley M. P., Bowman K. A., Bolton D. C., Bendheim P. E., Groth D. F., Glenner G. G. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell. 1983 Dec;35(2 Pt 1):349–358. doi: 10.1016/0092-8674(83)90168-x. [DOI] [PubMed] [Google Scholar]
- Roberts G. W., Lofthouse R., Allsop D., Landon M., Kidd M., Prusiner S. B., Crow T. J. CNS amyloid proteins in neurodegenerative diseases. Neurology. 1988 Oct;38(10):1534–1540. doi: 10.1212/wnl.38.10.1534. [DOI] [PubMed] [Google Scholar]
- Roberts G. W., Lofthouse R., Brown R., Crow T. J., Barry R. A., Prusiner S. B. Prion-protein immunoreactivity in human transmissible dementias. N Engl J Med. 1986 Nov 6;315(19):1231–1233. doi: 10.1056/NEJM198611063151919. [DOI] [PubMed] [Google Scholar]
- Serban D., Taraboulos A., DeArmond S. J., Prusiner S. B. Rapid detection of Creutzfeldt-Jakob disease and scrapie prion proteins. Neurology. 1990 Jan;40(1):110–117. doi: 10.1212/wnl.40.1.110. [DOI] [PubMed] [Google Scholar]
- Stahl N., Baldwin M. A., Burlingame A. L., Prusiner S. B. Identification of glycoinositol phospholipid linked and truncated forms of the scrapie prion protein. Biochemistry. 1990 Sep 25;29(38):8879–8884. doi: 10.1021/bi00490a001. [DOI] [PubMed] [Google Scholar]
- Stahl N., Borchelt D. R., Hsiao K., Prusiner S. B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987 Oct 23;51(2):229–240. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- Stahl N., Borchelt D. R., Prusiner S. B. Differential release of cellular and scrapie prion proteins from cellular membranes by phosphatidylinositol-specific phospholipase C. Biochemistry. 1990 Jun 5;29(22):5405–5412. doi: 10.1021/bi00474a028. [DOI] [PubMed] [Google Scholar]
- Tagliavini F., Giaccone G., Frangione B., Bugiani O. Preamyloid deposits in the cerebral cortex of patients with Alzheimer's disease and nondemented individuals. Neurosci Lett. 1988 Nov 11;93(2-3):191–196. doi: 10.1016/0304-3940(88)90080-8. [DOI] [PubMed] [Google Scholar]
- Tateishi J., Kitamoto T., Hashiguchi H., Shii H. Gerstmann-Sträussler-Scheinker disease: immunohistological and experimental studies. Ann Neurol. 1988 Jul;24(1):35–40. doi: 10.1002/ana.410240108. [DOI] [PubMed] [Google Scholar]
- Tateishi J., Sato Y., Nagara H., Boellaard J. W. Experimental transmission of human subacute spongiform encephalopathy to small rodents. IV. Positive transmission from a typical case of Gerstmann-Sträussler-Scheinker's disease. Acta Neuropathol. 1984;64(1):85–88. doi: 10.1007/BF00695613. [DOI] [PubMed] [Google Scholar]
- Turk E., Teplow D. B., Hood L. E., Prusiner S. B. Purification and properties of the cellular and scrapie hamster prion proteins. Eur J Biochem. 1988 Sep 1;176(1):21–30. doi: 10.1111/j.1432-1033.1988.tb14246.x. [DOI] [PubMed] [Google Scholar]
- Vitek M. P., Rasool C. G., de Sauvage F., Vitek S. M., Bartus R. T., Beer B., Ashton R. A., Macq A. F., Maloteaux J. M., Blume A. J. Absence of mutation in the beta-amyloid cDNAs cloned from the brains of three patients with sporadic Alzheimer's disease. Brain Res. 1988 Sep;464(2):121–131. doi: 10.1016/0169-328x(88)90004-6. [DOI] [PubMed] [Google Scholar]
- Westaway D., Carlson G. A., Prusiner S. B. Unraveling prion diseases through molecular genetics. Trends Neurosci. 1989 Jun;12(6):221–227. doi: 10.1016/0166-2236(89)90126-4. [DOI] [PubMed] [Google Scholar]