

Abstract
Background
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a heritable cardiac disorder characterized by life-threatening ventricular tachycardia caused by exercise or acute emotional stress. The standard diagnostic screening involves Sanger-based sequencing of 45 of the 105 translated exons of the RYR2 gene, and copy number changes of a limited number of exons that are detected using multiplex ligation-dependent probe amplification (MLPA).
Methods
In the current study, a previously validated bespoke array comparative genomic hybridization (aCGH) technique was used to detect copy number changes in the RYR2 gene in a 43-year-old woman clinically diagnosed with CPVT.
Results
The CGH array detected a 1.1 kb deletion encompassing exon 3 of the RYR2 gene. This is the first report using the aCGH technique to screen for mutations causing CPVT.
Conclusions
The aCGH method offers significant advantages over MLPA in genetic screening for heritable cardiac disorders.
Keywords: aCGH, array comparative genomic hybridization, catecholaminergic polymorphic ventricular tachycardia, CPVT, ryanodine receptor 2 gene, RYR2
Introduction
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an inherited cardiac disorder characterized by life-threatening arrhythmias during adrenergic stimulation, such as during exercise or acute emotional stress (1). Those affected by CPVT have structurally normal hearts, and symptoms present at an early age (2); if left untreated the mortality rate of CPVT is between 30%–50% by 35 years of age (2). There are two main forms of the disease: CPVT1 and CPVT2. CPVT1 is an autosomal dominant disorder caused by mutations in the RYR2 gene, which encodes for the cardiac Ca2+ release channel (Ryanodine receptor isoform 2) (3,4). CPVT2 is an autosomal recessive form of the disease, which is caused by mutations in the CASQ2 gene that encodes for a Ca2+ binding protein (Calsequestrin 2) (5,6). Both proteins are located in the cell’s sarcoplasmic reticulum. Mutations in the RYR2 and CASQ2 genes account for 50% and 3%–5%, respectively, of all CPVT cases (1,7). A third (and minor) form of CPVT is caused by mutations in the KCNJ2 gene (8).
The RYR2 gene contains 105 exons and encodes for one of the largest ion channel protein in the human body. CPVT-causing mutations in the RYR2 gene have been found in 45 of the 105 translated exons (8,9); many of these mutations are substitution mutations. The main mutation screening method involves PCR-based amplification of 40–43 exons of the RYR2 gene, and subsequent bidirectional Sanger-based sequencing (tier 1). If no mutations are found in this targeted group of exons then the remaining exons are sequenced (tier 2). In the absence of sequence-detectable mutations, screening methods then move on to those that can detect larger-scale events such as an exon 3 deletion (the deletion size ranges from 1.1 to 37.7 kb; 29 affected individuals) (9-13), a small deletion in exon 99 (one affected individual) (10), and duplication/insertion in exon 97 (one affected individual) (14). These larger mutations are usually detected using multiplex ligation-dependent probe amplification (MLPA), and this method is used to complement the Sanger-based screening; however, this method only screens for deletion and duplication events in a subset of exons of the RYR2 gene (15). In contrast, array comparative genomic hybridization (aCGH) offers enhanced exon coverage to detect exonic duplications and deletions. We have implemented a custom-designed aCGH array in our routine diagnostic testing of cardiac referrals to examine copy number changes in the coding regions of 99 genes associated with inherited cardiac and neuromuscular disorders (15-18).
In the present study, we screened a 43-year-old woman clinically diagnosed with CPVT using an aCGH assay and detected a 1.1-kb deletion encompassing exon 3 of the RYR2 gene. A custom-designed PCR-based assay was developed to screen for this deletion event in her children. The aCGH method has not been previously applied to CPVT screening (15-18).
Materials and methods
Genomic DNA (gDNA) was extracted from peripheral blood EDTA samples using the Gentra Puregene DNA Extraction kit (Qiagen inc., Germantown, Maryland, USA), according to the manufacturer’s instructions.
A Roche NimbleGen 12x135K Custom CGH Array (Roche NimbleGen Inc., Madison, Wisconsin, USA) was used for copy number change analysis. The aCGH was designed to examine the coding regions of 99 genes associated with several cardiac and neuromuscular disorders, including genes responsible for long QT syndrome (LQTS), Brugada syndrome, hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), short QT syndrome (SQT), and CPVT (18). See Supplementary Materials for the full gene list. Details about exonic and intronic probe densities and low-density ‘backbone’ probes that screen the human genome have previously been described (15).
A total of 500 ng of the proband’s gDNA was processed according to the manufacturer’s instructions (NimbleGen Array User’s Guide: CGH and CGH/LOH Arrays v9.1) (19), and has been described previously (15,17). In brief, a patient’s gDNA and Promega control DNA were fluorescently labeled with Cy3 (sample) and Cy5 (control) dyes, and subsequently purified via ethanol precipitation. The fluorescently labeled patient sample and the sex-matched control were combined in equimolar amounts and hybridized to one of 12 arrays on the aCGH slide for approximately 48 h using a Roche NimbleGen Hybridisation Chamber. Subsequently, the slides were washed and scanned using a NimbleGen MS 200 Microarray Scanner. The array image files (.tif) were imported into DEVA v1.2.1 (Roche NimbleGen Inc.) for analysis. The data were filtered using a log2ratio threshold of less than –0.4 over six probes for a deletion event and a log2ratio threshold of greater than 0.4 over 15 probes for a duplication event. All copy number changes that met these criteria were examined further using the UCSC Genome Browser, human genome assembly NCBI36/hg18 (released March 2006) (20), to determine the location and significance of the change.
All significant copy number changes were PCR-verified to determine the exact breakpoint of the deletion. Two sets of primers were designed to flank the deletion site detected by the aCGH approach (RYR_ex3_del_F 5’ GCGTATCAGAGTAAGCTGTGTC 3’; and RYR2_ ex3_del_R, 5’ AACTCTGTGACTTTGGAAAAGGAAT 3’). PCR was performed as follows: 1x FastStart PCR buffer, 2 mM magnesium chloride, 0.8 µM each of the forward and reverse primer, 0.4 mM dNTP, 0.04 U FastStart Taq DNA Polymerase (Roche), and 50 ng of gDNA. The following cycling conditions were used: 95°C for 4 min, 35 cycles of 94°C for 45 s, 60°C for 30 s, 72°C for 2 min 45 s, and a final extension at 72°C for 10 min.
PCR products were purified with ExoSAP-IT (Affymetrix Inc, Santa Clara, California, USA) prior to bi-directional DNA sequencing using BigDye Terminator v3.1 (Applied Biosystems by Life Technologies, Carlsbad, California, USA) to determine the exact deletion breakpoint. The sequenced products were purified using the BigDye XTerminator Purification Kit (Applied Biosystems Ltd) and were then subjected to capillary electrophoresis using the Applied Biosystems model 3130xl Genetic Analyzer. The analysis of sequence traces was performed using Geneious software (Biomatters Ltd, Auckland, New Zealand) (21).
Results
Clinical history
The proband (II-3) was a 43-year-old woman who was suspected to have CPVT, and she was referred to the Cardiac Inherited Disease Group (CIDG) of Auckland City Hospital for further cardiac/genetic investigation (Figure 1). The proband had previously experienced syncope with exertion and multiple ventricular premature complexes (VPCs) whilst monitored (Figure 2A). An exercise tolerance test (ETT) showed runs of polymorphic ventricular tachycardia (VT) with minimal exertion, and echocardiography confirmed a structurally normal heart. She was managed with beta-blockade and an implantable cardioverter-defibrillator (ICD). To control further the VT episodes she later underwent a left thoracoscopic sympathectomy and more recently has responded well to flecainide.
Figure 1.
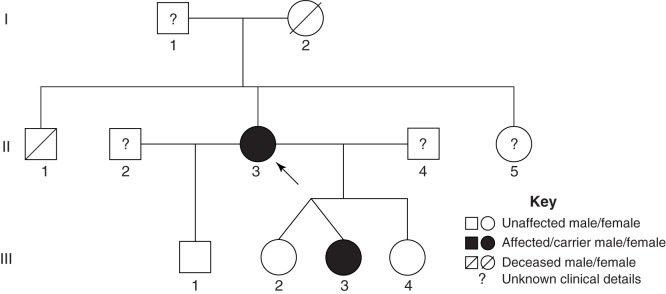
Pedigree of the RYR2 exon 3 deletion carriers. The proband (II-3) is indicated by the black arrow. Family members carrying the RYR2 exon 3 deletion are indicated in solid black. The members with unknown clinical and genetic background information are indicated by ?.
Figure 2.

Rhythm strips of the proband and her daughter. A: Three lead rhythm strip (25 mm/s, 10 mm/mV) taken during exercise of the female proband (II-3), at age 29 years following presentation with exertional syncope. Two minutes into stage 1 of the Bruce protocol, there are runs of polymorphic ventricular ectopy and ventricular tachycardia interrupted with occasional sinus beats. B: 12 lead rhythm strip (25 mm/s, 10 mm/mV) taken during exercise of an asymptomatic 7-year-old daughter of the proband (III-3) in stage 3, 9 min into the Bruce protocol. Frequent left bundle branch, inferior axis monomorphic ventricular extra beats develop into bigeminy.
The family history was significant for the sudden death of the proband’s brother (II-1) at the age of 15 years. He had suffered repeated syncope with documented polymorphic VT, and was taking beta-blockers at the time of his death whilst running. The proband’s mother (I-2) died suddenly at age 52 years, and reportedly had a myocardial infarction on autopsy. Clinical details were not available for II-2, II-4, and II-5. The proband’s four children (III-1 to III-4) also underwent cardiac investigation due to her probable CPVT diagnosis (Figure 1). All had echocardiography Holters and exercise testing. None had evidence of structural inherited heart disease, but one of the symptomatic fraternal twins (III-3) at 7 years of age had ventricular ectopy during exercise both on ETT and Holters. She was managed with beta-blockers and left cardiac sympathetic denervation, and has had no syncope over the ensuing 6 years.
As well as probable CPVT, III-3 also exhibited symptoms of absence seizures. An electroencephalogram confirmed that II-3 suffered from primary generalized epilepsy with absence of seizures. Ethosuximide was administered for the absence seizures. She also exhibited learning difficulties and poor concentration, and was diagnosed with attention deficit hyperactivity disorder (ADHD). ETT (Bruce protocol, see Figure 2B) and Holter conducted eight months post-surgery showed a reduction in VPCs. Four beats of VT were recently documented on exercise, and flecainide has since been added. There may be early signs of sinus node dysfunction, with the resting heart rate having fallen on the same dose of nadolol over recent years; the repolarization pattern has begun to look normal. She is asymptomatic for the bradycardia at present, and there have been no prolonged pauses on Holter.
Molecular genetic analysis
The aCGH analysis of the proband identified a heterozygous deletion on chromosome 1 (1q43) that was approximately 816 bp in size (hg18 co-ordinates chr1:235,560,602-235,561,416) (Figure 3). This deletion encompasses the whole of exon 3 of the RYR2 gene with flanking intronic regions (Figure 4). Sanger-based sequencing was used to confirm the aCGH results and also to determine the exact breakpoints of the deletion. Primers were designed to flank the region of interest (Figure 4A), and subsequent Sanger-based sequencing of the resulting smaller amplicon confirmed the aCGH results but with greater resolution such that the deletion proved to be 1,126 bp in size (Figure 4A). This mutation (c.169-198_c.273+823 del1126) has been previously reported by Ohno et al. (12) and Szentpali et al. (Figure 5) (13). Exon 3 of the RYR2 gene encodes for a highly conserved region of the RYR2 protein, and the mutation is an in-frame deletion of 35 amino acids (p.Asn57-Gly91).
Figure 3.

Copy number changes in chromosome 1q43 of the proband. DEVA software output showing a copy number change (deletion; 10 probes; log2ratio: –0.6726) localized to chromosome 1q43 (235,560,602-235,561,416; hg18 co-ordinates) for the proband (II-3).
Figure 4.

PCR amplification of the region encompassing exon 3 of the RYR2 gene. A: Amplicon sizes of the expected PCR products using DNA from an unaffected individual carrying no deletion of exon 3 of the RYR2 gene (left), and for DNA with the exon 3 deletion (right). The deletion size according to aCGH and the actual deletion size confirmed by Sanger-based sequencing are shown (above red lines). The expected PCR amplicon size of the deletion mutant according to the aCGH data and the actual product size are both shown. B: 2% agarose gel showing the results of PCR amplification of the genomic region encompassing exon 3 of the RYR2 gene for the proband (II-3) and her four children (III-1 to III-4). Chromatogram of the control and the proband showing where the breakpoint is (indicated by the red line).
Figure 5.
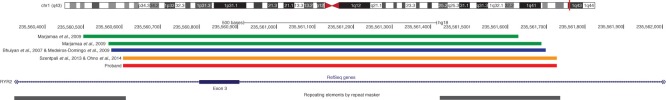
Reported deletions in the RYR2 gene. Ideogram of chromosome 1 showing the location of reported RYR2 gene deletions encompassing exon 3 (shown in green, blue, and yellow bars). The location of the proband’s exon 3 deletion is shown in red, and the location and extent of Alu family repetitive sequences are shown at the bottom of the figure. These graphics were redrawn from the UCSC genome browser by accessing the NCBI36/hg18 assembly (http://genome.uscs.edu/).
The proband’s four children were also tested for the same mutation using a PCR-based screen, with only the daughter (II-3) proving to be a carrier of the deletion event; interestingly, this assay preferentially amplified only the deleted RYR2 gene allele in carrier individuals (Figure 4B). The outcome of this mutation screen correlated with the clinical phenotypes of the proband’s children.
Discussion
The custom-designed NimbleGen 12x135K aCGH confirmed the CPVT diagnosis of the proband, and the targeted PCR-based amplification assay confirmed the CPVT diagnosis of her daughter. The microarray was able to detect the deletion of only one exon of the RYR2 gene. The custom-designed aCGH described here has been used previously in screening for deletion/duplication events in a large number of cardiac genes, but the design criteria have also been more generally applied to allow screening for mutations in non-cardiac-related referrals (15-18).
Missense mutations are common within the RYR2 gene, with many of the mutations located in 45 out of the 105 exons. While deletion and duplication mutations are not common in the RYR2 gene, it is still important to check for copy number changes for this gene as deletion and duplication events have been detected (9-13). These large heterozygous deletions cannot be detected using standard genetic screening such as direct DNA sequencing and high-performance liquid chromatography (9). While MLPA is able to detect exonic deletions, the technique can only be applied to a small number of genes and a limited number of exons. The current aCGH method has made it more efficient in screening for dosage changes in several patient samples simultaneously across a wide range of suspected genes.
The deletion of exon 3 has been reported by five different groups (9-13), and 31 individuals carry this mutation (nine families, including this study). Of the 31 individuals, only three appear to have a deletion of the same location and extent as the one reported here (Figure 5, Table I) (12,13). It is tempting to suggest that the deletion cluster represents recombination between repetitive elements (Alu family of repeats) (Figure 5) that flank exon 3. Interestingly, the three individuals previously reported do not come from the same geographical region as our patients; the two unrelated cases reported by Ohno et al. (12) are ethnically different from our proband. Nineteen individuals have deletions of similar sizes in this region (Figure 5) (10,11), and two individuals have much larger deletions in this region (3.6 kb and 37.7 kb; these two mutations are not shown in Figure 5) (9,12). Approximately 80% of these cases suffer from ventricular tachycardia, and approximately half experience atrial fibrillation and sino-atrial dysfunction. A quarter have received an ICD or pacemaker, and only the two subjects in the current study have received a left thoracoscopic cardiac sympathectomy. The symptoms exhibited by the patients in the study reported here match the other symptoms found in those with the same mutations (i.e. syncope, bradycardia, ventricular tachycardia, and VPCs); however, the two affected members in our study do not yet show clear signs of sino-atrial dysfunction and left ventricular non-compaction cardiomyopathy (LVNC) which were found in the two other studies (12,13).
Table I.
Clinical summary of reported families with RYR2 exon 3 deletion. The rows shaded grey represent patients with the same deletion mutation as the current study.
| Family | Deletion | n | Syn | Brady | AV block | VT | VPC | LVNC | AF | SA dysfunction | LV dysfunction | ICD | Pacemaker | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | c.161-236_c.272+781del1126 | 11 | 3 | 11 | 10 | 7 | 4 | 1 | (10) | |||||
| 2 | c.161-236_c.272+781del1126 | 2 | 1 | 2 | 2 | 1 | 1 | 1 | ||||||
| 3 | 3.6 kb del of exon 3 | 1 | (9) | |||||||||||
| 4 | c.168-301_c.273+722del1128 | 4 | 1 | 2 | 1 | 4 | 1 | 2 | 2 | (11) | ||||
| 5 | c.168-228_c.273+793del1126 | 2 | 1 | 2 | 2 | 1 | 1 | 1 | ||||||
| 6 | c.169-198_273+823del1126 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | (13) | |||||
| 7 | c.169-22924_c.273+14653del37682 | 6 | 2 | 5 | 1 | 2 | 2 | 5 | 1 | 3 | 2 | 1 | (12) | |
| 8 | c.169-198_c.273+823del1126 | 2 | 2 | 2 | 1 | 1 | 1 | 2 | 1 | 1 | ||||
| 9 | c.169-198_c.273+823del1126 | 2 | 1 | 2 | 2 | 2 | 2 | Current study | ||||||
| Total | 31 | 8 | 12 | 7 | 25 | 9 | 9 | 16 | 16 | 5 | 5 | 3 | ||
| (%) | (25.8%) | (38.7%) | (22.6%) | (80.6%) | (29.0%) | (29.0%) | (51.6%) | (51.6%) | (16.1%) | (16.1%) | (9.7%) |
AF = atrial fibrillation; AV = atrioventricular block; Brady = bradycardia; ICD = implantable cardioverter-defibrillator; LV = left ventricular; LVNC = left ventricular non-compaction cardiomyopathy; SA = sino-atrial; Syn = syncope; VPC = ventricular premature complex; VT = ventricular tachycardia.
The RYR2 protein is a major Ca2+ releasing channel of the sarcoplasmic reticulum (SR) in cardiac muscle and plays an essential role in excitation–contraction coupling and Ca2+ homeostasis in SR (22). All mutations found in the RYR2 gene to date have a gain-of-function effect, which causes a lowered threshold for either cytoplasmic Ca2+ or SR Ca2+ levels (23). This increase in cytoplasmic Ca2+ leads to delays after depolarization (DAD) (23). The exon 3 region of the RYR2 gene is highly susceptible to large Alu repeat-mediated genomic rearrangements (9). In vitro studies of the effect of RYR2 lacking the exon 3-encoded domain in HEK293 cells have shown increased Ca2+ release into the cytoplasm, which has been attributed to Ca2+ overload leading to DADs (24).
Of the five members of the family that were tested here, two members were affected by ADHD (one daughter was confirmed to have CPVT, the other was asymptomatic). As the RYR2 gene mutation did not co-segregate with the appearance of ADHD, it could not be concluded that the gene is associated with the disorder. However, there have been two reported cases where CPVT patients (substitution mutations in the RYR2 gene) suffer from ADHD (25,26). The proband’s daughter (II-3) was also affected by generalized epilepsy with absence seizures, which could be caused by the RYR2 exon 3 deletion. Previous reports have found that approximately half of children with CPVT also present with seizures (27), and half of RYR2 gene mutation carriers in a large Dutch cohort suffered from seizures (28). Knock-in mouse studies conducted by Lehnart et al. (29) showed that a RYR2 gene missense mutation found in CPVT patients caused seizures and sudden death in mice, providing evidence that mutations in the RYR2 gene can cause seizures.
Acknowledgements
We thank the family for kindly permitting this report. Funding: This study was financially supported by the National Heart Foundation (New Zealand), and the Maurice and Phyllis Paykel Trust. The Cardiac Inherited Disease Group is supported by Cure Kids, who also partially fund Dr Skinner’s salary. Dr Leong was financially supported during a large part of this work by Cure Kids, but also by The Rutherford Foundation as a New Zealand Postdoctoral Fellow. Jennifer Sucich has been supported by a LabPLUS (Auckland City Hospital) Masters postgraduate scholarship.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia . Circulation. 2002;106:69–74. doi: 10.1161/01.cir.0000020013.73106.d8. [DOI] [PubMed] [Google Scholar]
- 2.Swan H, Piippo K, Viitasalo M, Heikkila P, Paavonen T, Kainulainen K, et al. Arrhythmic disorder mapped to chromosome 1q42-q43 causes malignant polymorphic ventricular tachycardia in structurally normal hearts . J Am Coll Cardiol. 1999;34:2035–42. doi: 10.1016/s0735-1097(99)00461-1. [DOI] [PubMed] [Google Scholar]
- 3.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia . Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 4.Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia . Circulation. 2001;103:485–90. doi: 10.1161/01.cir.103.4.485. [DOI] [PubMed] [Google Scholar]
- 5.Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel . Am J Hum Genet. 2001;69:1378–84. doi: 10.1086/324565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Postma AV, Denjoy I, Hoorntje TM, Lupoglazoff JM, Da Costa A, Sebillon P, et al. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia . Circ Res. 2002;91:e21–6. doi: 10.1161/01.res.0000038886.18992.6b. [DOI] [PubMed] [Google Scholar]
- 7.Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA) . Heart Rhythm. 2011;8:1308–39. doi: 10.1016/j.hrthm.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 8.Vega AL, Tester DJ, Ackerman MJ, Makielski JC. Protein kinase A-dependent biophysical phenotype for V227F-KCNJ2 mutation in catecholaminergic polymorphic ventricular tachycardia . Circ Arrhythm Electrophysiol. 2009;2:540–7. doi: 10.1161/CIRCEP.109.872309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Medeiros-Domingo A, Bhuiyan ZA, Tester DJ, Hofman N, Bikker H, van Tintelen JP, et al. The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: a comprehensive open reading frame mutational analysis . J Am Coll Cardiol. 2009;54:2065–74. doi: 10.1016/j.jacc.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhuiyan ZA, van den Berg MP, van Tintelen JP, Bink-Boelkens MT, Wiesfeld AC, Alders M, et al. Expanding spectrum of human RYR2-related disease: new electrocardiographic, structural, and genetic features . Circulation. 2007;116:1569–76. doi: 10.1161/CIRCULATIONAHA.107.711606. [DOI] [PubMed] [Google Scholar]
- 11.Marjamaa A, Laitinen-Forsblom P, Lahtinen AM, Viitasalo M, Toivonen L, Kontula K, et al. Search for cardiac calcium cycling gene mutation in familial ventricular arrhythmias resembling catecholaminergic polymorphic ventricular tachycardia . BMC Med Genet. 2009;10:12. doi: 10.1186/1471-2350-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohno S, Omura M, Kawamura M, Kimura H, Itoh H, Makiyama T, et al. Exon 3 deletion of RYR2 encoding cardiac ryanodine receptor is associated with left ventricular non-compaction . Europace. 2014;16:1646–54. doi: 10.1093/europace/eut382. [DOI] [PubMed] [Google Scholar]
- 13.Szentpali Z, Szili-Torok T, Caliskan K. Primary electrical disorder or primary cardiomyopathy? A case with a unique association of noncompaction cardiomyopathy and cathecolaminergic polymorphic ventricular tachycardia caused by ryanodine receptor mutation . Circulation. 2013;127:1165–6. doi: 10.1161/CIRCULATIONAHA.112.144949. [DOI] [PubMed] [Google Scholar]
- 14.Tester D, Kopplin L, Will M, Ackerman M. Spectrum and prevalence of cardiac ryanodine receptor (RyR2) mutations in a cohort of unrelated patients referred explicitly for long QT syndrome genetic testing . Heart Rhythm. 2005;2:1099–105. doi: 10.1016/j.hrthm.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 15.Marquis-Nicholson R, Doherty E, Love J, Lan C, George A, Thrush A, et al. Array-based identification of copy number changes in a diagnostic setting: simultaneous gene-focused and low resolution whole human genome analysis . Sultan Qaboos Univ Med J. 2013;13:69–79. doi: 10.12816/0003198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marquis-Nicholson R, Lai D, Lan CC, Love JM, Love DR. A streamlined protocol for molecular testing of the DMD gene within a diagnostic laboratory: a combination of array comparative genomic hybridisation and bidirectional sequence analysis . ISRN Neurol. 2013;2013:908317. doi: 10.1155/2013/908317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marquis-Nicholson R, Prosser D, Love J, Love D. Gene dosage analysis in a clinical environment: gene-targeted microarrays as the platform-of-choice. Microarray. 2013;2:51–62. doi: 10.3390/microarrays2020051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marquis-Nicholson R, Prosser DO, Love JM, Zhang L, Hayes I, George AM, et al. Array comparative genomic hybridization identifies a heterozygous deletion of the entire KCNJ2 gene as a cause of sudden cardiac death . Circ Cardiovasc Genet. 2014;4:17–22. doi: 10.1161/CIRCGENETICS.113.000415. [DOI] [PubMed] [Google Scholar]
- 19.Roche Roche NimbleGen Array User’s Guide: CGH and CGH/LOH Arrays v9.1. http://www.nimblegen.com/downloads/support/05434483001_NG_CGHLOH_UGuide_v9p1.pdf. Available from. [Accessed in October 2013]
- 20.UCSC Genome Browser. http:/genome.ucsc.edu/ Available from. [Accessed in October 2013]
- 21.Biomatters. GeneGene vR6.1.6. http://www.geneious.com/ Available from. [Accessed in October 2013]
- 22.Bers D. Cardiac excitation-contraction coupling. Nature. 2001;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 23.Lobo P, Kimlicka L, Tung C, van Petegem F. The deletion of exon 3 in the cardiac ryanodine receptor is rescued by β strand switching . Structure. 2011;19:790–8. doi: 10.1016/j.str.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 24.Tang Y, Tian X, Wang R, Fill M, Chen SR. Abnormal termination of Ca2+ release is a common defect of RyR2 mutations associated with cardiomyopathies . Circ Res. 2012;110:968–77. doi: 10.1161/CIRCRESAHA.111.256560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beery TA, Shah MJ, Benson DW. Genetic characterization of familial CPVT after 30 years . Biol Res Nurs. 2009;11:66–72. doi: 10.1177/1099800409333369. [DOI] [PubMed] [Google Scholar]
- 26.Creighton W, Virmani R, Kutys R, Burke A. Identification of novel missense mutations of cardiac ryanodine receptor gene in exercise-induced sudden death at autopsy . J Mol Diagn. 2006;8:62–7. doi: 10.2353/jmoldx.2006.050081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients . Circulation. 1995;91:1512–19. doi: 10.1161/01.cir.91.5.1512. [DOI] [PubMed] [Google Scholar]
- 28.Postma AV, Denjoy I, Kamblock J, Alders M, Lupoglazoff JM, Vaksmann G, et al. Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients . J Med Genet. 2005;42:863–70. doi: 10.1136/jmg.2004.028993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lehnart SE, Mongillo M, Bellinger A, Lindegger N, Chen BX, Hsueh W, et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice . J Clin Invest. 2008;118:2230–45. doi: 10.1172/JCI35346. [DOI] [PMC free article] [PubMed] [Google Scholar]