

Abstract
Neutrophil extracellular traps (NETs) are web-like structures released by activated neutrophils. Recent studies suggest that NETs play an active role in driving autoimmunity and tissue injury in diseases including rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). The purpose of this study was to investigate if celastrol, a triterpenoid compound, can inhibit NET formation induced by inflammatory stimuli associated with RA and SLE. We found that celastrol can completely inhibit neutrophil oxidative burst and NET formation induced by tumor necrosis factor alpha (TNFα) with an IC50 of 0.34 µM and by ovalbumin:anti-ovalbumin immune complexes (Ova IC) with an IC50 of 1.53 µM. Celastrol also completely inhibited neutrophil oxidative burst and NET formation induced by immunoglobulin G (IgG) purified from RA and SLE patient sera. Further investigating into the mechanisms, we found that celastrol treatment downregulated the activation of spleen tyrosine kinase (SYK) and the concomitant phosphorylation of mitogen-activated protein kinase kinase (MAPKK/MEK), extracellular-signal-regulated kinase (ERK), and NFκB inhibitor alpha (IκBα), as well as citrullination of histones. Our data reveals that celastrol potently inhibits neutrophil oxidative burst and NET formation induced by different inflammatory stimuli, possibly through downregulating the SYK-MEK-ERK-NFκB signaling cascade. These results suggest that celastrol may have therapeutic potentials for the treatment of inflammatory and autoimmune diseases involving neutrophils and NETs.
Keywords: Arthritis, celastrol, inflammation, lupus, neutrophil extracellular trap.
INTRODUCTION
Neutrophils are the most abundant immune cells in human blood and play an essential role in host defense against invading pathogens. After recruitment to the inflammatory site, neutrophils attack and destroy pathogens by phagocytosis, along with releasing antimicrobial peptides, proteolytic enzymes, reactive oxygen species (ROS), and the more recently described neutrophil extracellular traps (NETs) [1-3]. NETs are web-like structures released by activated neutrophils through a process called “NETosis” [1, 4]. NETs are composed of decondensed chromatin backbones with embedded antimicrobial granular molecules, and some cytoplasmic proteins [5]. It is believed that NETs provide an important mechanism for host cells to entrap and kill extracellular microorganisms [1].
Despite their beneficial effects in innate immune response, NETs appear to contribute to the pathogenesis of various inflammatory and autoimmune diseases [6], such as sepsis [7], inflammatory lung diseases [8], vascular disorders [9-11], systemic lupus erythematosus (SLE) [12-18], and rheumatoid arthritis (RA) [19]. NET structures have been identified in kidney lesions of SLE and vasculitis patients and synovial tissues of RA patients, suggesting that prolonged exposure of NET components (histones, bactericidal peptides and proteases, and ROS) may directly cause host tissue damage [11, 14, 18, 19]. In addition, NET contents (such as dsDNA, citrullinated histones and vimentin, myeloid peroxidase, and LL37) may serve as a source of autoantigens that exacerbate the autoimmune responses in these patients [19, 20]. Indeed, it was shown that some SLE patients displayed decreased ability to degrade NETs in their sera and the patients with defective NET degradation had higher levels of anti-dsDNA and anti-NET autoantibodies as well as a higher frequency of developing lupus nephritis [18, 21]. Furthermore, SLE is an immune complex-mediated autoimmune disease with enhanced serum type 1 interferon (IFN) activity (the so called “IFN signature”) [22]. It has been shown that netting neutrophils are a major inducer of type I IFN production by plasmacytoid dendritic cells in SLE [12, 13]. Interestingly, recent studies show that netting neutrophils have the capacity to produce IFNα themselves [23], further supporting a pathogenic role of NETs in SLE. Taken together, NETs may play a prominent role in the pathogenesis of a range of inflammatory and rheumatic diseases. Thus, inhibition of NET formation holds great promise for improved treatment of these diseases.
Celastrol (also called tripterine or tripdiolide) is one of the two main active components in Chinese herbal medicine Tripterygium wilfordii Hook F (TwHF). TwHF extracts have been widely used in East Asia for the treatment of autoimmune and inflammatory diseases for centuries [24]. Phase I and II clinical trials in the United States and China have shown the safety and efficacy of TwHF extracts in the treatment of patients with RA [25-28]. The anti-inflammatory effects of celastrol have been demonstrated in animal models of different diseases, including SLE [29], inflammatory arthritis [30], Alzheimer’s disease [31], and asthma [32]. It has been shown that celastrol downregulates the expression of pro-inflammatory cytokines and modulates the activity of many inflammation-associated molecules such as Janus kinase 2 (JAK2), transcription factor NF-κB, NADPH oxidase, MHC II, and proteasome [24].
The purpose of this study was to determine the effect of celastrol on NET formation and to investigate the potential signaling mechanisms involved. Our results demonstrated that celastrol was a potent inhibitor of NET formation induced by inflammatory stimuli which are known to contribute to the pathogenesis of RA and SLE. Our study also identified spleen tyrosine kinase (SYK) as a new molecular target for the action of celastrol.
MATERIALS AND METHODS
Materials
Celastrol (Mr = 450.6, ≥ 98% pure) was purchased from Cayman Chemical (Ann Arbor, MI, USA). A stock solution of celastrol (30 mM in DMSO) was prepared and diluted with phosphate buffered saline (PBS) before using. Ovalbumin:anti-ovalbumin immune complex (Ova IC) was prepared by mixing ovalbumin (Sigma-Aldrich, St. Louis, MO, USA) and rabbit anti-ovalbumin antibodies (Acris Antibodies, San Diego, CA, USA) at a molar ratio of 1:4 and incubated at 37°C for 1 hr. Human tumor necrosis factor alpha (TNFα) was purchased from Pepro Tech Inc. (Rocky Hill, NJ, USA). Total serum IgG was purified using protein A Sepharose following the manufacturer’s instruction (Biovision Inc., Milpitas, California, USA). Bound IgG on protein A Sepharose was washed using a high salt buffer (3M NaCl and 1.5 M glycine, pH 9.0) to eliminate associated antigens before it is eluted from protein A beads.
Study Subjects
Human studies described in this manuscript were approved by the Institutional Review Board (IRB) of University of Nebraska Medical Center under the protocols #447-10-EP and #342-10-FB. Written informed consent was obtained according to procedures approved by the IRB before blood samples were collected from study subjects. Healthy controls were donors without any known autoimmune diseases. Patients with RA or SLE fulfilled the 1987 American College of Rheumatology classification criteria for RA or SLE, respectively.
Neutrophil Isolation
Peripheral blood samples from healthy donors, SLE and RA patients were obtained after informed consent in accordance with institutional review board approved protocols at the University of Nebraska Medical Center (UNMC). Neutrophils were separated by Ficoll gradient centrifugation using Polymorphprep (Axis-Shield Poc, Oslo, Norway) following the manufacturer’s instruction. The neutrophil layer was collected and washed three times with PBS. Residual red blood cells were lyzed by brief incubation with ice-cold hypotonic buffer (0.2% NaCl in dH2O). The purified neutrophils were about 90% alive by trypan blue or propidium iodide (PI) staining and 90% pure confirmed by flow cytometry scatter analysis and cell surface expression of CD16 (data not shown).
Neutrophil Oxidative Burst
Freshly isolated neutrophils were resuspended in Hank’s Balanced Salt Solution (HBSS) supplemented with 1.09 mM CaCl2,1.62 mM MgCl2, and 5% fetal bovine serum (FBS) at 5x106 cells/ml. Neutrophils were loaded with 1 μg/ml of dihydrorhodamine 123 (DHR123, purchased from Life Technologies, Grand Island, NY, USA) at room temperature (RT) for 15 min, and then stimulated with 100 ng/ml of TNFα,5 μg/ml of Ova IC or 250 μg/ml of purified total IgG from either SLE or RA patient sera at 37°C for 30 min followed by immediate fixation with 2% formaldehyde in PBS. Flow cytometry was performed to measure the fluorescence generated by oxidization of DHR 123. For celastrol treatment, neutrophils were pre-incubated with different doses of celastrol (0.5-20 µM) or vehicle only at RT for 45 min before stimulation. The concentrations of TNFα, Ova IC, total serum IgG, and celastrol in the above assays are within the most commonly used range for in vitro studies in the literature.
NET Formation and Quantification
Freshly isolated neutrophils were resuspended in RPMI medium at 2x106 cells/ml, and seeded into 96-well plates at 50 μl/well. NET formation was stimulated with 100 ng/ml of TNFα, 5 μg/ml of Ova IC, or 250 μg/ml of purified serum IgG for 3 hr at 37°C in a CO2 incubator. SYTOX Green (Life Technologies, Grand Island, NY, USA), a cell non-permeable nucleic acid binding dye, was added at 350 nM final concentrations to detect extracellular chromatin DNA released in NETs. NETs were visualized using fluorescence microscopy and quantified by POLARstar Omega fluorescence polarization microplate reader (BMG Labtech, Ortenberg, Germany).
NET Immunofluorescence Staining
Freshly isolated neutrophils (2x106 cells/ml in RPMI medium) were seeded in poly-L-lysine coated coverslips and incubated at 37°C, 5% CO2 for 3 hr. Cells were then fixed with 4% paraformaldehyde. After permeabilization with 0.2% Triton X-100 for 10 min at RT, cells were blocked with PBS plus 2% BSA at RT for 2 hr. Cells were then stained with mouse anti-human myeloperoxidase antibody (AbD Serotec, Raleigh, NC, USA) for 2 hr, followed by incubation with secondary Rhodamine Red-X-conjugated rat anti-mouse IgG secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for 1 hr at RT. Nuclear DNA was detected by staining with SYTOX Green. Coverslips were mounted onto glass slides and analyzed using an upright fluorescent microscope (Olympus IX81).
Phospho-Specific Flow Cytometry
Neutrophils were stimulated as described above for the examination of neutrophil oxidative burst with the exception that the stimulation was stopped after 5 min at 37°C by fixation with pre-warmed BD PhosflowTM Lyse/Fix Buffer at 37ºC for 10 min. Cells were permeabilized with BD PhosflowTM Perm Buffer II on ice for 30 min and then stained with Alexa Fluor® 488 conjugated anti-phospho SYK antibody (pY348) at RT for 1 hr before flow cytometry analysis. All the above reagents were purchased from BD Biosciences (San Jose, CA, USA).
Western Blot
Neutrophils were stimulated as described in “neutrophil oxidative burst” for 5-10 min at 37°C. Cell lysates were prepared using boiling SDS lysis buffer (2% SDS, 62.5 mM Tris, pH 6.8, 5% β-mercaptoethanol, and 10% glycerol) supplemented with phosphatase and proteinase cocktail inhibitors (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Protein concentration was determined by PierceTM BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of proteins were separated on 10% SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blotted with p-MEK-1 (B-4 for pSer298, #sc-271914), p-ERK 1/2 (pT202/pY204.22A, #sc-136521), or p-IκBα (39A1431, #sc-52943) antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) to determine the activation of MEK, ERK, and NF-êB. The levels of citrullinated histone H3 was determined by western blot analysis with anti-citrullinated H3 antibody (citrulline R2 + R8 + R17, #ab5103) from Abcam (Cambridge, MA, USA) using the similar procedure except that neutrophils were stimulated for 2 hr before processed for western blot. And Western blot analysis with anti-β-actin antibody (#4967, Cell Signaling Technology, Danvers, MA, USA) and histone H3 antibody (#MA5-15150, Thermo Fisher Scientific) served as loading controls. Intensity of protein bands in western blot was quantified by ImageJ.
Statistical Analysis
Ratio paired t-test was used to determine the differences with or without celastrol treatment. All data were presented as mean ± SD. P values less than 0.05 were considered significant. IC50 of celastrol on was obtained by HillSlope analysis using GraphPad Prism software.
RESULTS
Celastrol Inhibited Neutrophil Oxidative Burst
First, we tested the effects of celastrol on the production of reactive oxygen species (ROS, a process called oxidative burst) which normally occurs prior to NET formation [33]. Freshly isolated neutrophils from the blood of healthy donors were pre-incubated with vehicle only or celastrol at the indicated concentrations and then treated with different inflammatory stimuli. TNFα and immune complexes (IC) were first used as the stimuli in the experiments because they have been shown to play a prominent role in the disease pathogenesis of RA and SLE, respectively [34, 35]. TNFα and Ova IC strongly induced neutrophil oxidative burst and this induction was potently inhibited by celastrol in a dose-dependent manner (Fig. 1A-C, ***p < 0.001 and *p < 0.05 respectively, n ≥ 5). Celastrol completely inhibited oxidative burst induced by TNFα and Ova IC at 3 µM (IC50 ≈ 0.34 µM) and 10 µM (IC50 ≈ 1.53 µM), respectively (Fig. 1C). Because autoantibodies, especially the IgG types, play a critical role in the etiology of SLE and RA [36, 37], we then tested if celastrol inhibits neutrophil oxidative burst induced by total IgG purified from the patient sera. Celastrol at 5 µM completely inhibited neutrophil oxidative burst induced by serum IgG purified from an SLE patient (serologically positive for anti-ribonucleoprotein antibodies (anti-RNP+)) and a RA patient (serologically positive for anti-citrullinated protein antibodies (ACPA+)) (Fig. 2A). Similar results were obtained when neutrophils were stimulated with purified serum IgG from 5 other anti-RNP+ SLE patients and 5 other ACPA+ RA patients (Fig. 2B, ****p < 0.0001 and n = 6 for both). These results demonstrated that celastrol potently inhibited neutrophil oxidative burst induced by several inflammatory stimuli relevant to SLE and RA.
Fig. (1).
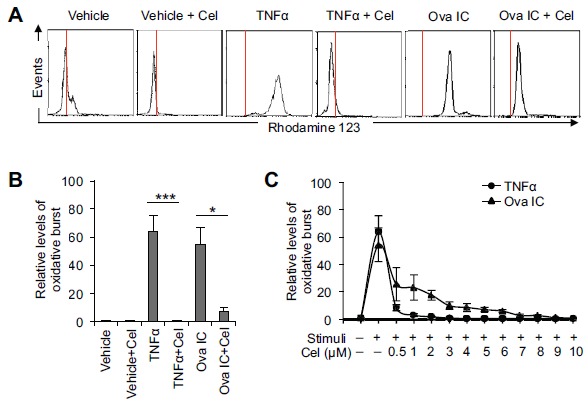
Celastrol inhibits neutrophil oxidative burst induced by TNF#x03B1; or immune complexes. A, Representative flow cytometry histograms showing inhibition of TNFα or Ova IC-induced oxidative burst by celastrol (5 µM). The red vertical lines represent the peak fluorescence intensity of Rhodamine 123 in cells treated with vehicle only (as the base level of oxidative burst). B, A summary showing that celastrol (5 µM) significantly inhibited TNFα or Ova IC-induced oxidative burst. The relative levels of oxidative burst were presented as fold changes of mean fluorescence levels of Rhodamine 123 compared to non-stimulated neutrophils (vehicle only). The data represent at least five independent experiments with neutrophils from different donors. *p<0.05 and ***p< 0.001 by ratio paired t-test. C, A diagram showing dose-dependent inhibition of TNF#x03B1; and Ova IC-induced oxidative burst by celastrol. Data were plotted as the mean ± SD and represented at least three independent experiments.
Fig. (2).
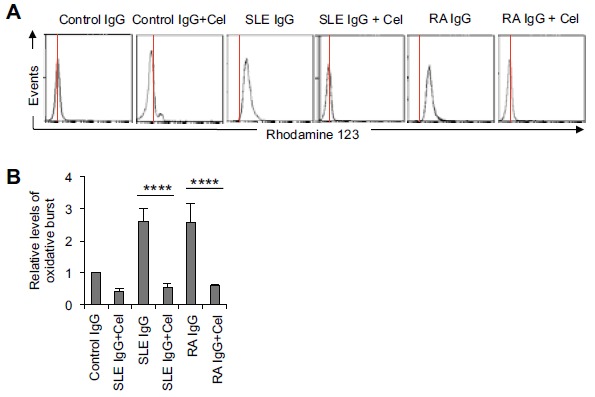
Celastrol inhibits neutrophil oxidative burst induced by total serum IgG from SLE and RA patients. A, Representative flow cytometry histograms showing that celastrol (5 µM) completely inhibited oxidative burst induced by total serum IgG (250 µg/ml) purified from an SLE patient (anti-RNP+) or a RA patient (ACPA+). The red vertical lines represent the peak fluorescence intensity of Rhodamine 123 in cells treated with IgG purified from healthy controls (as the base level of oxidative burst). B, A summary showing that celastrol (5 µM) significantly inhibited SLE or RA IgG-induced oxidative burst. The data were summarized from results obtained with serum IgG derived from 6 different SLE patients (anti-RNP+) and 6 different RA patients (ACPA+). ****p < 0.0001 by ratio paired t-test. Cel: celastrol.
Celastrol Inhibited NET Formation
Enhanced spontaneous NET formation has been reported in neutrophils derived from SLE and RA patients compared to those from healthy donors [12, 13, 19]. In addition, total serum IgG and autoantibodies such as anti-RNP+ from SLE patients and ACPA from RA patients also stimulate the release of NETs [12, 19]. We then tested the effect of celastrol on spontaneous NET formation from neutrophils isolated from SLE and RA patients and NET formation induced by TNFa, IC, and IgG purified from SLE and RA patient sera. The released NETs were confirmed by immunofluorescence staining of DNA (green) and co-localized neutrophil granule molecule myeloid peroxidase (red) (Fig. 3A). For quantification, NETs were examined by staining decondensed fiber-like chromatin DNA using non-cell permeable dye SYTOX Green. By fluorescence microscopy analysis, we found that pre-treatment of celastrol (at 10 µM) decreased the basal levels of NETs from neutrophils derived from healthy donors (Fig. 3B). Pre-treatment with celastrol (at 10 µM) also decreased the spontaneous NET levels from neutrophils derived from SLE and RA patients (data not shown). TNFα and IC induced large amounts of NETs and this robust NET formation was efficiently inhibited by 10 µM of celastrol treatment (Fig. 3B). Quantification of the released NET DNA by fluorescence polarization microplate reader from three independent experiments confirmed that celastrol significantly inhibited the spontaneous as well as the TNFα or IC induced NET formation (Fig. 3C, * p < 0.05, **p < 0.01 and ***p < 0.001, respectively). This observation was further confirmed by counting the percentage of cells releasing NET through fluorescence microscopy (Fig. 3D).
Fig. (3).
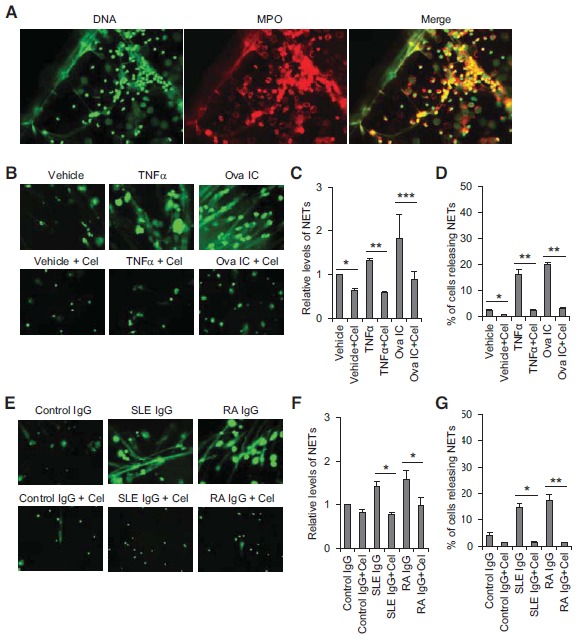
Celastrol inhibits NET formation. A, Representative pictures showing co-localization of NET DNA (stained with DAPI) with myeloperoxidase (stained with myeloperoxidase-specific antibody) by immunofluorescence analysis. B and E, Representative pictures of fluorescence microscopy showing that celastrol (10 µM) inhibited NET formation induced by TNFα and Ova IC (B) or total IgG purified from SLE or RA patient sera (E). Decondensed fiber-like chromatin DNA released in NETs was stained by non-cell permeable nucleic acid dye SYTOX Green. The small round stained dots represent dead cells. C, D, F and G, A summary showing celastrol significantly inhibits NET formation induced by TNFα and Ova IC (C and D) or total IgG purified from SLE or RA patient sera (F and G). In C and F, the relative levels of NET formation were presented by fold changes of SYTOX Green fluorescence intensity compared to vehicle or control IgG-stimulated neutrophils quantified by a fluorescence polarization microplate reader. In D and G, the released NETs were visualized by fluorescence microscopy, and the percentage of neutrophils releasing NETs was calculated as the average of six fields (X20) normalized to the total number of cells. The data was plotted as the mean ± SD and represented at least three independent experiments. *p < 0.05, **p<0.01, and ***p< 0.001 by ratio paired t-test (C and F) or paired t-test (D and G). Cel: celastrol.
Similarly, NET formation induced by total serum IgG purified from an SLE patient (anti-RNP+) and a RA patient (ACPA+) was also efficiently blocked by celastrol (Fig. 3E). The results were repeated when neutrophils were stimulated with total serum IgG purified from 4 other SLE patients (anti-RNP+) and 4 other RA patients (ACPA+) using the two methods of NET quantification as described above (Fig. 3F, 3G, *p < 0.05, **p < 0.01, and n = 5 for each treatment). In summary, our data demonstrated that celastrol was a potent inhibitor for NET formation induced by different stimuli associated with SLE or RA.
Celastrol (≤ 10 µM) Does Not Significantly Affect Cell Viability
We have noticed slightly increased numbers of dead cells in samples treated with 10 ìM of celastrol (Fig. 3A, 3B, 3EE, dead cells were shown as SYTOX green-stained small round dots). We then tested the cytotoxicity of celastrol at different concentrations. Freshly isolated neutrophils from healthy donors were pre-incubated with celastrol and stimulated with TNFα as described above. Neutrophils were then stained with propidium iodide (PI), a membrane impermeable dye which intercalates into double-stranded nucleic acids and is generally used to identify dead cells. Flow cytometry was performed to determine the percentage of dead cells (PI+) out of total cells. Increased concentrations of celastrol (up to 10 ìM) did not significantly affect the cell viability (p > 0.05 by paired T test, compared to without celastrol treatment) although the percentage of dead cells gradually increased from ≈ 2% (without celastrol) to ≈ 5% (with 10 ìM of celastrol) and to ≈ 13.5% (with 20 ìM of celastrol) (Fig. 4A). Fluorescence microscopy analysis of neutrophils stained with cell impermeable DNA-staining dye SYTOX Green confirmed that over 90% of cells were alive (not stained by SYTOX Green) with 10 ìM of celastrol treatment (Fig. 4B). Therefore, the observed inhibitory effects of celastrol on neutrophil activation were most likely not due to the cytotoxicity of celastrol.
Fig. (4).
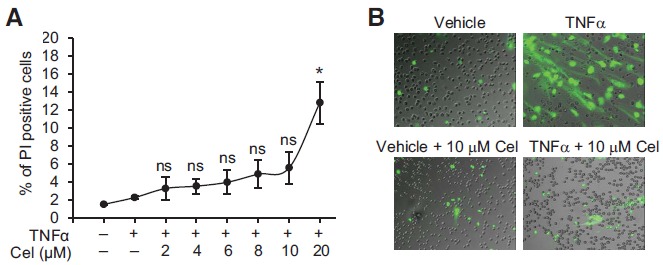
Cytotoxicity test of celastrol. A, A diagram showing percentages of dead neutrophils (PI+) when treated with different doses of celastrol during TNFα-induced oxidative burst. Data were plotted as the mean ± SD and represented at least three independent experiments. B, Representative pictures of fluorescence microscopy showing cytotoxicity of celastrol (10 µM) on neutrophils during TNFα-induced NET formation. Chromatin DNA in NETs or dead cells was stained by non-cell permeable nucleic acid dye SYTOX Green. The graphs shown were fluorescence images merged with differential interference contrast (DIC) images. NS: not significant (p > 0.05 by paired T test); * p < 0.05 by paired T test; Cel: celastrol.
Celastrol Downregulated the Activation of SYK, MEK, ERK, and NF-κB As Well As Citrullination of Histones
It has been reported that the Raf-MEK-ERK signaling cascade is required for NET formation [38]. To investigate the potential molecular mechanisms of celastrol-induced inhibition on NET formation, we first analyzed its effect on the activation of SYK, a kinase which acts upstream of the Raf-MEK-ERK pathway. SYK is also important for neutrophil activation through mediating the signaling of immunoreceptors on neutrophils [39]. Neutrophils derived from a healthy donor were pre-incubated with 5 ìM of celastrol and stimulated with the indicated stimuli for 5 min. Intracellular flow cytometry analysis using phospho-SYK specific antibodies showed that celastrol treatment completely inhibited SYK phosphorylation induced by TNFα, Ova IC, or total IgG purified from SLE or RA patient sera (Fig. 5A, 5B). These experiments were repeated with neutrophils isolated from 5 different healthy donors and purified serum IgG from 6 different SLE or RA patients (Fig. 5C, 5D, * p < 0.05).
Fig. (5).
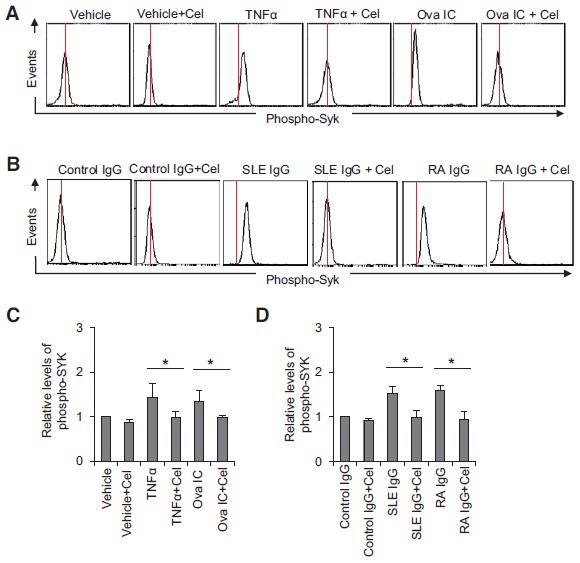
Celastrol inhibits SYK phosphorylation. A and B, Representative histograms of phospho-specific flow cytometry showing that celastrol (5 µM) inhibited SYK phosphorylation induced by TNFα, Ova IC, and purified total IgG from SLE or RA patient sera. C and D, A summary showing that celastrol (5 µM) significantly inhibited SYK phosphorylation induced by TNFα, Ova IC, and purified total IgG from SLE or RA patient sera. The relative levels of phospho-SYK were presented as fold changes of mean fluorescence intensity of phospho-SYK compared to non-stimulated neutrophils. Data in C represent at least five independent experiments with neutrophils from different donors. Data in D were summarized from results obtained with serum IgG derived from 6 different SLE patients (anti-RNP+) and 6 different RA patients (ACPA+). *p<0.05 by ratio paired t-test. Cel: celastrol.
We then tested the effect of celastrol on the activation of MEK (MAPKK) and ERK which act downstream of SYK by western blot analyses using phospho-specific antibodies. Both TNFα and Ova IC-induced phosphorylation of MEK-1, and ERK1/2 which was dramatically suppressed by the pretreatment with 5 µM of celastrol (Fig. 6A, upper two panels). Activation of transcription factor NF-κB has been shown to be involved in the generation of NETs [40]. We then tested if the phosphorylation of IêBα that leads to the activation and nuclear translocation of NF-κB was affected by celastrol treatment. Treatment with 5 µM of celastrol resulted in complete inhibition of IκB( phosphorylation induced by TNFα and Ova IC, while none of the treatments affected the protein levels of constitutively expressed β-actin (Fig. 6A, lower two panels). The results were confirmed by quantification of band intensity on Western blot normalized by the β-actin loading control (Fig. 6B).
Fig. (6).
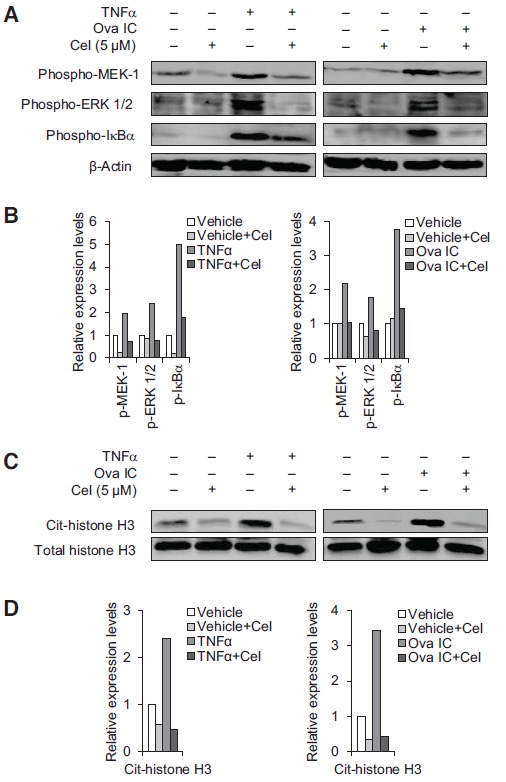
Celastrol inhibits the phosphorylation of MEK-1, ERK 1/2, and IκBα, and citrullination of histone H3. A to D, Western blot analysis using phospho-specific antibodies showing that celastrol (5 µM) diminished TNFα or Ova IC-induced phosphorylation of MEK-1, ERK1/2, and IκBα (A), and citrulllination of histone H3 (C). Western blot using anti-β-actin (A) and total histone H3 (C) antibodies were used as loading controls. The levels of phosphorylated MEK-1, ERK1/2, and IκBα on the Western blot were quantified by ImageJ and normalized by the β-actin loading control (B). The levels of citrullinated histone H3 were quantified by Image J and normalized by the total histone H3 loading control (D). Bar graphs show the relative expression levels of phospho-MEK-1, ERK 1/2, and IκBα, and cit-histone H3 compared to non-stimulated neutrophils (B and D). Cel: celastrol.
Histone hypercitrullination mediates chromatin decondensation during NET formation and citrullinated histones have been shown to be autoantigens in patients with RA and Felty’s syndrome [41]. We then examined if celastrol treatment resulted in decreased histone citrullination. Neutrophils were preincubated with celastrol and stimulated with TNFα or Ova IC for 2 hr. Western blot analysis using antibodies specific for citrullinated histone H3 showed that both TNFα and Ova IC strongly induced histone H3 citrullination and the levels of citrullinated histone H3 were partially decreased by 5 ìM of celastrol treatment (Fig. 6C, upper panel). The levels of total histone H3 remained essentially unchanged by different treatments and served as a loading control (Fig. 6C, lower panel). The results were confirmed by quantification of band intensity on Western blot normalized by the total histone H3 loading control (Fig. 6D).
Taken together, these results suggested that celastrol inhibited the activation of SYK-MEK-ERK-NFκB signaling cascade and the action of celastrol could occur at the early signaling events that activate SYK. Interestingly, celastrol also leads to decreased levels of citrullinated histones, which are autoantigens for patients with RA and Felty’s syndrome [42].
DISCUSSION
NETs play a pivotal role in host defense to microbial infections. However, NETs are also generated upon sterile stimuli during inflammation [6]. Excessive NET formation and/or defective NET clearance may contribute to the initiation and perpetuation of aberrant autoimmune responses and subsequent organ manifestations in inflammatory autoimmune diseases [20]. Therefore, inhibition of NET formation may be beneficial for the treatment of these diseases, such as SLE and RA. In this study, we demonstrate that celastrol potently inhibits neutrophil oxidative burst and NET formation induced by inflammatory stimuli which are known to contribute to the pathogenesis of SLE and RA. Investigating further into the mechanisms, we found that inhibition of NET formation by celastrol involves downregulation of the SYK-MEK-ERK-NFκB signaling cascade and leads to decreased levels of citrullinated histones.
Celastrol is one of the two most pharmacologically active compounds extracted from Chinese herbal medicine TwHF (also known as “thunder god vine”). TwHF has been used in Asia for years in remedies to treat a variety of inflammatory and autoimmune diseases including RA and SLE [24]. In a previous six-month study in the United States, it was shown that RA patients receiving TwHF were almost twice as likely to demonstrate a clinical response as patients receiving sulfasalazine [25]. A recent study in 207 RA patients showed that the combination therapy of TwHF and methotrexate led to a 1.6 fold increase of ACR50 response rate than methotrexate monotherapy (76.8% vs 46.4%), further suggesting the beneficial effect of TwHF in the treatment of RA [28]. Our results add a new mechanism for the anti-inflammatory function of TwHF and also suggest that celastrol, the pure compound derived from TwHF, may be a drug candidate for the treatment of inflammatory autoimmune diseases.
Indeed, animal studies have shown that celastrol treatment reduces inflammation and organ damage in models of rheumatoid arthritis, systemic lupus, asthma, and select neurodegenerative disorders [24]. Interestingly, celastrol can also decrease the titers of anti-dsDNA autoantibodies in NZB/W F1 lupus mice [29] and ACPAs in antigen-induced arthritis models [30]. NETs have recently been suggested to act as a “repository” of autoantigens such as chromatin DNA, modified histones, and citrullinated proteins, for SLE and RA patients [20, 43]. Here, we show that celastrol inhibits the formation of NETs, providing a potential mechanism for the decrease in autoantibody production in SLE and RA. This is particularly noteworthy given the direct pathogenic role that has been proposed for autoantibodies in diseases such as SLE and RA [36, 37].
Celastrol has been shown to directly or indirectly modulate numerous cellular targets including JAK kinase, ERK, c-Jun N-terminal kinase (JNK), and NADPH oxidase [24]. In this study, we showed that celastrol inhibited the phosphorylation of SYK and concomitant phosphorylation of MEK-1, ERK1/2 and IκB( during neutrophil oxidative burst and NET formation. Our data suggest that celastrol could act on the very early signaling events that activate SYK. Because SYK can be autophosphorylated or phosphorylated by other kinases, celastrol may inhibit the kinase activity of SYK itself or kinases that phosphorylate SYK. Future in vitro kinase assays will be performed to determine if SYK is directly targeted by celastrol. Nevertheless, to our knowledge, modulation of SYK activation by celastrol has never been reported before.
The use of TwFH in the treatment of autoimmune diseases is limited by its toxicity at high doses. In this study, we also found that celastrol treatment at concentrations exceeding 10 µM resulted in increased cell death. Therefore, the development of celastrol derivatives with less toxicity and higher solubility may overcome the barrier for its clinical use. Alternatively, targeted delivery of celastrol to inflamed tissues may provide another strategy to decrease its toxicity. Indeed, studies have shown that macromolecular prodrugs have reduced toxicity and enhanced efficacy due to their preferential delivery to the inflamed tissues in animal models of lupus and arthritis [44]. It will be very interesting to develop a macromolecular prodrug of celastrol and test if the prodrug has improved efficacy and reduced toxicity in the treatment of inflammatory autoimmune diseases.
In summary, we report here that celastrol potently inhibits neutrophil oxidative burst and NET formation, which identifies celastrol as a novel inhibitor for NET formation. NETs have recently been suggested to play a pivotal role in autoimmune responses and inflammation-mediated organ damage. Our findings suggest that celastrol may be a promising drug candidate for a range of inflammatory and autoimmune diseases involving neutrophils and NETs.
ACKNOWLEDGEMENTS
Declared none.
ABBREVIATIONS
- ACPA
Anti-citrullinated protein antibodies.
- DHR123
Dihydrorhodamine 123
- ERK
Extracellular-signal-regulated kinase
- FBS
Fetal bovine serum
- HBSS
Hank’s balanced salt solution
- IκBα
NFκB inhibitor alpha
- MAPKK/MEK-1
Mitogen-activated protein kinase kinase
- NETs
Neutrophil extracellular traps
- Ova IC
Ovalbumin:anti-ovalbumin immune complexes
- PBS
Phosphate buffered saline
- RA
Rheumatoid arthritis
- RNP
Ribonucleoprotein
- ROS
Reactive oxygen species
- SLE
Systemic lupus erythematosus
- SYK
Spleen tyrosine kinase
- TNFα
Tumor necrosis factor alpha
- TwHF
Chinese herbal medicine Tripterygium wilfordii Hook F
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
References
- 1.Brinkmann V., Reichard U., Goosmann C., Fauler B., Uhlemann Y., Weiss D.S., Weinrauch Y., Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 2.Nauseef W.M. How human neutrophils kill and degrade microbes: an integrated view. Immunol. Rev. 2007;219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- 3.Almyroudis N.G., Grimm M.J., Davidson B.A., Röhm M., Urban C.F., Segal B.H. NETosis and NADPH oxidase: at the intersection of host defense, inflammation, and injury. Front. Immunol. 2013;4:45. doi: 10.3389/fimmu.2013.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yipp B.G., Kubes P. NETosis: how vital is it? Blood. 2013;122(16):2784–2794. doi: 10.1182/blood-2013-04-457671. [DOI] [PubMed] [Google Scholar]
- 5.Urban C.F., Ermert D., Schmid M., Abu-Abed U., Goosmann C., Nacken W., Brinkmann V., Jungblut P.R., Zychlinsky A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009;5(10):e1000639. doi: 10.1371/journal.ppat.1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaplan M.J., Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. J. Immunol. 2012;189(6):2689–2695. doi: 10.4049/jimmunol.1201719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clark S.R., Ma A.C., Tavener S.A., McDonald B., Goodarzi Z., Kelly M.M., Patel K.D., Chakrabarti S., McAvoy E., Sinclair G.D., Keys E.M., Allen-Vercoe E., Devinney R., Doig C.J., Green F.H., Kubes P. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007;13(4):463–469. doi: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- 8.Cheng O.Z., Palaniyar N. NET balancing: a problem in inflammatory lung diseases. Front. Immunol. 2013;4:1. doi: 10.3389/fimmu.2013.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gardiner E.E., Andrews R.K. Neutrophil extracellular traps (NETs) and infection-related vascular dysfunction. Blood Rev. 2012;26(6):255–259. doi: 10.1016/j.blre.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 10.Kambas K., Mitroulis I., Ritis K. The emerging role of neutrophils in thrombosis-the journey of TF through NETs. Front. Immunol. 2012;3:385. doi: 10.3389/fimmu.2012.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kessenbrock K., Krumbholz M., Schönermarck U., Back W., Gross W.L., Werb Z., Gröne H.J., Brinkmann V., Jenne D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009;15(6):623–625. doi: 10.1038/nm.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia-Romo G.S., Caielli S., Vega B., Connolly J., Allantaz F., Xu Z., Punaro M., Baisch J., Guiducci C., Coffman R.L., Barrat F.J., Banchereau J., Pascual V. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011;3(73):73ra20. doi: 10.1126/scitranslmed.3001201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lande R., Ganguly D., Facchinetti V., Frasca L., Conrad C., Gregorio J., Meller S., Chamilos G., Sebasigari R., Riccieri V., Bassett R., Amuro H., Fukuhara S., Ito T., Liu Y.J., Gilliet M. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 2011;3(73):73ra19. doi: 10.1126/scitranslmed.3001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Villanueva E., Yalavarthi S., Berthier C.C., Hodgin J.B., Khandpur R., Lin A.M., Rubin C.J., Zhao W., Olsen S.H., Klinker M., Shealy D., Denny M.F., Plumas J., Chaperot L., Kretzler M., Bruce A.T., Kaplan M.J. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 2011;187(1):538–552. doi: 10.4049/jimmunol.1100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dörner T. SLE in 2011: Deciphering the role of NETs and networks in SLE. Nat. Rev. Rheumatol. 2012;8(2):68–70. doi: 10.1038/nrrheum.2011.200. [DOI] [PubMed] [Google Scholar]
- 16.Knight J.S., Kaplan M.J. Lupus neutrophils: ‘NET’ gain in understanding lupus pathogenesis. Curr. Opin. Rheumatol. 2012;24(5):441–450. doi: 10.1097/BOR.0b013e3283546703. [DOI] [PubMed] [Google Scholar]
- 17.Yu Y., Su K. Neutrophil Extracellular Traps and Systemic Lupus Erythematosus. J. Clin. Cell. Immunol. 2013;4:139. doi: 10.4172/2155-9899.1000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hakkim A., Fürnrohr B.G., Amann K., Laube B., Abed U.A., Brinkmann V., Herrmann M., Voll R.E., Zychlinsky A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA. 2010;107(21):9813–9818. doi: 10.1073/pnas.0909927107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khandpur R., Carmona-Rivera C., Vivekanandan-Giri A., Gizinski A., Yalavarthi S., Knight J.S., Friday S., Li S., Patel R.M., Subramanian V., Thompson P., Chen P., Fox D.A., Pennathur S., Kaplan M.J. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013;5(178):178ra40. doi: 10.1126/scitranslmed.3005580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knight J.S., Carmona-Rivera C., Kaplan M.J. Proteins derived from neutrophil extracellular traps may serve as self-antigens and mediate organ damage in autoimmune diseases. Front. Immunol. 2012;3:380. doi: 10.3389/fimmu.2012.00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leffler J., Martin M., Gullstrand B., Tydén H., Lood C., Truedsson L., Bengtsson A.A., Blom A.M. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J. Immunol. 2012;188(7):3522–3531. doi: 10.4049/jimmunol.1102404. [DOI] [PubMed] [Google Scholar]
- 22.Mavragani C.P., Niewold T.B., Chatzigeorgiou A., Danielides S., Thomas D., Kirou K.A., Kamper E., Kaltsas G., Crow M.K. Increased serum type I interferon activity in organ-specific autoimmune disorders: clinical, imaging, and serological associations. Front. Immunol. 2013;4:238. doi: 10.3389/fimmu.2013.00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lindau D., Mussard J., Rabsteyn A., Ribon M., Kötter I., Igney A., Adema G.J., Boissier M.C., Rammensee H.G., Decker P. TLR9 independent interferon α production by neutrophils on NETosis in response to circulating chromatin, a key lupus autoantigen. Ann. Rheum. Dis. 2014;73(12):2199–2207. doi: 10.1136/annrheumdis-2012-203041. [DOI] [PubMed] [Google Scholar]
- 24.Kannaiyan R., Shanmugam M.K., Sethi G. Molecular targets of celastrol derived from Thunder of God Vine: potential role in the treatment of inflammatory disorders and cancer. Cancer Lett. 2011;303(1):9–20. doi: 10.1016/j.canlet.2010.10.025. [DOI] [PubMed] [Google Scholar]
- 25.Goldbach-Mansky R, Wilson M, Fleischmann R, et al. Comparison of Tripterygium wilfordii Hook F versus sulfasalazine in the treatment of rheumatoid arthritis: a randomized trial. Ann Intern Med. 2009;151:229–40. doi: 10.7326/0003-4819-151-4-200908180-00005. W49-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tao X., Cush J.J., Garret M., Lipsky P.E. A phase I study of ethyl acetate extract of the chinese antirheumatic herb Tripterygium wilfordii hook F in rheumatoid arthritis. J. Rheumatol. 2001;28(10):2160–2167. [PubMed] [Google Scholar]
- 27.Tao X., Younger J., Fan F.Z., Wang B., Lipsky P.E. Benefit of an extract of Tripterygium Wilfordii Hook F in patients with rheumatoid arthritis: a double-blind, placebo-controlled study. Arthritis Rheum. 2002;46(7):1735–1743. doi: 10.1002/art.10411. [DOI] [PubMed] [Google Scholar]
- 28.Lv QW, Zhang W, Shi Q, et al. Comparison of Tripterygium wilfordii Hook F with methotrexate in the treatment of active rheumatoid arthritis (TRIFRA): a randomised, controlled clinical trial. Ann Rheum Dis. 2014;pii doi: 10.1136/annrheumdis-2013-204807. annrheumdis-2013-204807. [DOI] [PubMed] [Google Scholar]
- 29.Tao X., Fan F., Hoffmann V., Gao C.Y., Longo N.S., Zerfas P., Lipsky P.E. Effective therapy for nephritis in (NZB x NZW)F1 mice with triptolide and tripdiolide, the principal active components of the Chinese herbal remedy Tripterygium wilfordii Hook F. Arthritis Rheum. 2008;58(6):1774–1783. doi: 10.1002/art.23513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Venkatesha S.H., Yu H., Rajaiah R., Tong L., Moudgil K.D. Celastrus-derived celastrol suppresses autoimmune arthritis by modulating antigen-induced cellular and humoral effector responses. J. Biol. Chem. 2011;286(17):15138–15146. doi: 10.1074/jbc.M111.226365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paris D., Ganey N.J., Laporte V., Patel N.S., Beaulieu-Abdelahad D., Bachmeier C., March A., Ait-Ghezala G., Mullan M.J. Reduction of beta-amyloid pathology by celastrol in a transgenic mouse model of Alzheimer’s disease. J. Neuroinflammation. 2010;7:17. doi: 10.1186/1742-2094-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim D.Y., Park J.W., Jeoung D., Ro J.Y. Celastrol suppresses allergen-induced airway inflammation in a mouse allergic asthma model. Eur. J. Pharmacol. 2009;612(1-3):98–105. doi: 10.1016/j.ejphar.2009.03.078. [DOI] [PubMed] [Google Scholar]
- 33.Fuchs T.A., Abed U., Goosmann C., Hurwitz R., Schulze I., Wahn V., Weinrauch Y., Brinkmann V., Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007;176(2):231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Furst D.E., Emery P. Rheumatoid arthritis pathophysiology: update on emerging cytokine and cytokine-associated cell targets. Rheumatology (Oxford) 2014;53(9):1560–1569. doi: 10.1093/rheumatology/ket414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Azevedo P.C., Murphy G., Isenberg D.A. Pathology of systemic lupus erythematosus: the challenges ahead. Methods Mol. Biol. 2014;1134:1–16. doi: 10.1007/978-1-4939-0326-9_1. [DOI] [PubMed] [Google Scholar]
- 36.Dörner T., Lipsky P.E. B cells: depletion or functional modulation in rheumatic diseases. Curr. Opin. Rheumatol. 2014;26(2):228–236. doi: 10.1097/BOR.0000000000000000. [DOI] [PubMed] [Google Scholar]
- 37.Anolik J.H., Looney R.J., Lund F.E., Randall T.D., Sanz I. Insights into the heterogeneity of human B cells: diverse functions, roles in autoimmunity, and use as therapeutic targets. Immunol. Res. 2009;45(2-3):144–158. doi: 10.1007/s12026-009-8096-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hakkim A., Fuchs T.A., Martinez N.E., Hess S., Prinz H., Zychlinsky A., Waldmann H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 2011;7(2):75–77. doi: 10.1038/nchembio.496. [DOI] [PubMed] [Google Scholar]
- 39.Lowell C.A. Src-family and Syk kinases in activating and inhibitory pathways in innate immune cells: signaling cross talk. Cold Spring Harb. Perspect. Biol. 2011;3(3):a002352. doi: 10.1101/cshperspect.a002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lapponi M.J., Carestia A., Landoni V.I., Rivadeneyra L., Etulain J., Negrotto S., Pozner R.G., Schattner M. Regulation of neutrophil extracellular trap formation by anti-inflammatory drugs. J. Pharmacol. Exp. Ther. 2013;345(3):430–437. doi: 10.1124/jpet.112.202879. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y., Li M., Stadler S., Correll S., Li P., Wang D., Hayama R., Leonelli L., Han H., Grigoryev S.A., Allis C.D., Coonrod S.A. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009;184(2):205–213. doi: 10.1083/jcb.200806072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dwivedi N., Radic M. Citrullination of autoantigens implicates NETosis in the induction of autoimmunity. Ann. Rheum. Dis. 2014;73(3):483–491. doi: 10.1136/annrheumdis-2013-203844. [DOI] [PubMed] [Google Scholar]
- 43.Liu C.L., Tangsombatvisit S., Rosenberg J.M., Mandelbaum G., Gillespie E.C., Gozani O.P., Alizadeh A.A., Utz P.J. Specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res. Ther. 2012;14(1):R25. doi: 10.1186/ar3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuan F., Quan L.D., Cui L., Goldring S.R., Wang D. Development of macromolecular prodrug for rheumatoid arthritis. Adv. Drug Deliv. Rev. 2012;64(12):1205–1219. doi: 10.1016/j.addr.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]