

Abstract
P-glycoprotein (P-gp) is a plasma membrane efflux pump that is commonly associated with therapy resistances in cancers and infectious diseases. P-gp can lower the intracellular concentrations of many drugs to subtherapeutic levels by translocating them out of the cell. Because of the broad range of substrates transported by P-gp, overexpression of P-gp causes multidrug resistance. We reported previously on dynamic transitions of P-gp as it moved through conformations based on crystal structures of homologous ABCB1 proteins using in silico targeted molecular dynamics techniques. We expanded these studies here by docking transport substrates to drug binding sites of P-gp in conformations open to the cytoplasm, followed by cycling the pump through conformations that opened to the extracellular space. We observed reproducible transport of two substrates, daunorubicin and verapamil, by an average of 11 to 12 Å through the plane of the membrane as P-gp progressed through a catalytic cycle. Methyl-pyrophosphate, a ligand that should not be transported by P-gp, did not show this movement through P-gp. Drug binding to either of two subsites on P-gp appeared to determine the initial pathway used for drug movement through the membrane. The specific side-chain interactions with drugs within each pathway seemed to be, at least in part, stochastic. The docking and transport properties of a P-gp inhibitor, tariquidar, were also studied. A mechanism of inhibition by tariquidar is presented that involves stabilization of an outward open conformation with tariquidar bound in intracellular loops or at the drug binding domain of P-gp.
Keywords: ABC transporter, ATP-binding cassette transporter, ABCB1, drug resistance, multidrug transporter, multidrug resistance, P-glycoprotein, P-gp, targeted molecular dynamics, transporter mechanism, tariquidar inhibition
INTRODUCTION
The mammalian ATP binding cassette transporter, P-glycoprotein (P-gp), is present in many tissues1 and helps in detoxifying cells by pumping xenobiotics across the plasma membrane2. P-gp is able to bind and transport a diverse range of cationic amphipathic molecules ranging in size from 100 to 4,000 Da3. This broad transport substrate spectrum is used to remove cytotoxic compounds from cells of the intestinal epithelium, adrenal glands, placenta, the brush border of the renal tubule, the canalicular membrane of hepatocytes, pancreatic ductile cells, and capillary endothelial cells of the brain and testes4, 5. Because of this broad range of substrates, P-gp is associated with multidrug resistance (MDR) in the chemotherapies of many types of cancers6–8, has been implicated in Alzheimer’s disease9, 10, and has proven problematic in the management of HIV/AIDS11. P-gp is a ~170-kDa protein consisting of two pseudo-symmetrical halves, each containing a nucleotide-binding domain (NBD) and a transmembrane domain (TMD)12. It has been hypothesized that ABC-transporter proteins undergo large conformational changes powered by the binding and hydrolysis of ATP and that these changes alter the transmembrane domains from “open to the cytoplasm” (inward facing) to “open to the extracellular space” (outward facing) conformations (see for example 13–21). These conformational changes are thought to allow the movement of substrates across the membrane from the cytoplasmic to the extracellular phospholipid leaflets19, 22, ultimately resulting in the dissociation of the substrates from the extracellular membrane leaflet into the extracellular space. It is not clear whether such active transport involves driving the substance towards the external space or whether the rearrangement of transmembrane helices simply alters access from the cytoplasmic to the extracellular space. In either case, the removal of cytotoxins by P-gp from the cytoplasm of cells has been suggested to be an important molecular mechanism of multidrug resistance 23, 24. The inward facing structures of P-gp from mouse25, 26, C. elegans27 and some bacterial homologs19 have fully disengaged NBDs with the transmembrane drug binding domains (DBDs) open to the inside of the cell. These open inside conformations of P-gp have been demonstrated to be remarkably flexible28. The highest resolution X-ray structural models of ABCB1 transporters were obtained from a multidrug resistance pump from the bacterium, S. aureus22, 29, a prokaryotic homolog that shares some drug transport substrates, pumping activities and inhibitors with P-gp30. The S. aureus structures exhibit fully engaged NBDs with nucleotides bound and DBDs that are open to the outside 22, 29. We previously reported the simulation of conformational changes in models of human P-gp as the transporter transitions though a putative catalytic cycle31. We used targeted molecular dynamics techniques32–35 and structures of human P-gp homologs that had drug binding domains positioned “wide open” to the cytoplasm (mouse 3G60 structure36, no nucleotides bound), “partially open” to the cytoplasm (Vibrio cholerae 3B5X structure19, no nucleotides bound), “open” to the extracellular space (S. aureus SAV1866 2HYD structure, ADP bound to the nucleotide binding sites22) and “wide open” to the extracellular space (Salmonella typhimurium MsbA structure 3B5Z with an ADP-vanadate transition-state analog bound at the nucleotide binding site19) to simulate the conformational transitions of P-gp during a putative catalytic cycle31. These dynamic models of P-gp proved useful in the identification of several novel inhibitors of the transporter that target its nucleotide binding domains37. In more recent work from our lab using these targeted molecular dynamics techniques, corrected versions of the mouse structures as reported in 25, 26 and new structures of eukaryotic P-gp from C. elegans27 have been employed. In a recent report38 using TMD techniques that were similar to those reported earlier by us31, drug transport simulations were attempted. These simulations, however, did not show significant movement of transport substrates through P-gp38.
In the work reported here, an in depth look at the movement of two transport substrates through P-gp from one side of the membrane to the other was investigated during targeted molecular dynamics simulations of putative transport cycles as developed previously31. We used the chemotherapeutic, daunorubicin, a known transport substrate of P-gp and the antihypertensive agent, verapamil, a known modulator of P-gp-conferred multidrug resistances of cancers39–41 in these simulations. The simulation experiments were replicated six times each. We observed large transport substrate movements from cytoplasmic to extracellular positions in all of the simulations where daunorubicin and verapamil were used. To simulate the conformational changes of P-gp during a catalytic cycle, forces were applied to selected Cα atoms of the protein to induce the targeted changes in the P-gp structures. No forces were directed at the transport ligands other than those applied by the moving protein. The conformational changes were chosen so P-gp would transition the drug binding sites from wide open to the cytoplasm to wide open to the extracellular space. These studies have allowed the investigation of protein-driven movement of transport substrates through the membrane. In additional experiments, we also examined the behavior of a potent inhibitor of P-gp, tariquidar42.
The simulations presented here show that transport substrates docked into different initial binding sites within the cytoplasmic leaflet of the drug binding domains of P-gp were transported through the membrane as P-gp transitioned from inside open to outside open conformations. Repeated simulations with these transport substrates revealed associations with different parts of the membrane embedded transmembrane helices of P-gp. The results suggest that once bound, movement of substrates through P-gp occurs through at least two general pathways via stochastic mechanisms.
MATERIALS AND METHODS
Materials
The Visual Molecular Dynamics program suite (VMD)43 was extensively used in this work. Molecular dynamics experiments were performed with NAMD v. 2.944 using the CHARMM27 force field45. AutoDock 4.246 was used for initial docking of transport ligands to the drug binding domains of P-gp. Computational resources of the SMU Center for Scientific Computation were used.
Ligand docking to P-gp
Prior to the start of molecular dynamic simulations, AutoDock 4.246 or AutoDock Vina47 was used to dock daunorubicin or verapamil using a human P-gp structure equivalent to the mouse 4KSB crystal structure25 as described previously31. Ligand interactions were limited to the cytoplasmic extensions of the transmembrane helices and transmembrane sections of P-gp. Ligand binding to NBDs was not investigated. For AutoDock 4.2 experiments, grids were calculated using 0.375 Å spacing in a 126 Å3 cube. For each ligand, 100 genetic algorithm experiments were performed using 3,000,000 energy evaluations, a population size of 300, and 27,000 generations. The ligand docking positions resulting from these experiments were ranked by predicted affinities. The conformational pose of ligand that was predicted to have the highest affinity was used as a starting point in the molecular dynamics simulations, except where indicated in the text. For AutoDock Vina experiments, 300 replicates (exhaustiveness of 300) was performed using a cube having lengths of 56 Å per side, unless otherwise noted.
Molecular dynamics simulations
Daunorubicin and verapamil were parameterized as previously described by us for other nonstandard molecules48. Equilibrium geometries were obtained from ab initio quantum mechanical simulations using GAMESS and an unrestricted Hartree-Fock 6–31 basis set49. Parameter and topology files were created using this data and as described in work of Fajer and coworkers50. We also used the antechamber module51 from the AMBER14 suite of programs52 together with the general AMBER force field 53 and the AM1-BCC charge estimation methods54, 55 to parameterize daunorubicin, verapamil, and tariquidar. To test the influence of the parameterization methods used on the results of the simulations, results using the AMBER-produced parameters for molecular dynamics simulations of verapamil were compared to those produced by the methods of Fajer and coworkers50 in experiments that were otherwise identical to those shown in Figure 3A (see below). No significant difference in the behavior of the verapamil parameterized using the AMBER General Force Field for organic molecules (Version 1.7, Nov 2013) compared to verapamil parameterized with GAMESS as described in 50 was observed (data not shown).
Figure 3. Movement of daunorubicin and verapamil though P-glycoprotein.
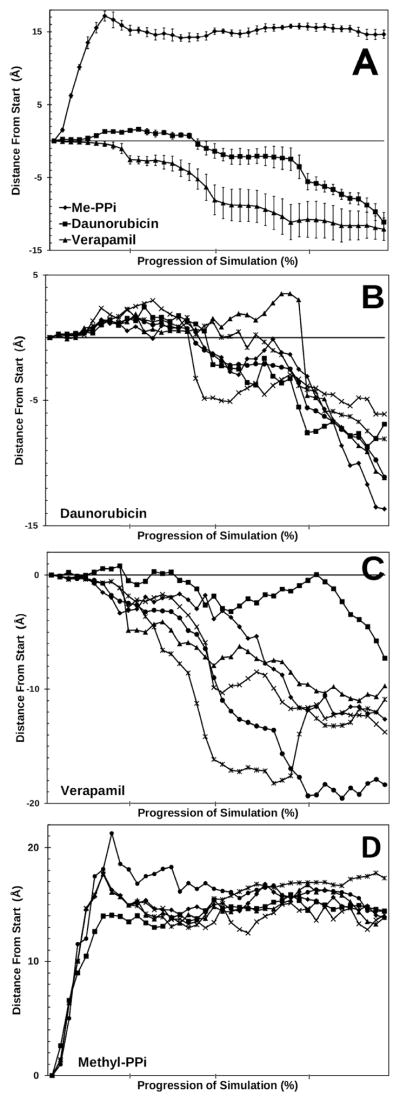
Panel A: The center of mass of daunorubicin, verapamil, or methylpyrophosphate was calculated for each step of the simulation relative to the distance from their respective starting locations. The first quarter of the graphs represents unconstrained molecular dynamics before the application of the targeted forces. The remaining data represents the movements between targeted structures achieved by TMD techniques. The structures were oriented with the plane of the membrane in the X and Y direction, so that movement through the membrane is oriented on the Z-axis. Movement towards the extracellular space is represented by negative directions on the Z-axes, while movement towards the cytoplasm is in positive Z-axis directions. Distances are presented in Å and show the movement of the centers of mass of the ligands in the Z-axis. Six simulations were performed for daunorubicin (squares), verapamil (triangles), or methylpyrophosphate (circles) each. Average movement of the centers of mass of each ligand are presented. Error bars represent one standard error of the mean. Movement through the membrane towards the extracellular space is equivalent to movement from positive values to negative values in these plots. Panel B : Same as panel A except that results for the six individual daunorubicin simulations are presented. Panel C: Same as B except that individual results for verapamil are shown. Panel D: Same as B except that individual results for methylpyrophosphate are shown.
Models for human P-glycoprotein inserted into a POPC phospholipid bilayer, and including nucleotides, Mg2+ ions, Na+ and Cl− counter ions and water were created as described31. Molecular dynamics (MD) simulations were performed with constant temperature and pressure (NPT ensemble) in a periodic cell using Langevin temperature and pressure control, as well as particle-mesh Ewald electrostatics calculations at 310 K. Targeted molecular dynamics (TMD) simulations56, 57 on P-gp were performed using target coordinates derived from mouse Pgp25 (PDB accession 4KSB), the Sav1866 transporter (PDB accession 2HYD)22, and MsbA structures 3B5X and 3B5Z19 as previously described31. Briefly, the target structures were aligned using STAMP58 as implemented in the Multiple Alignment59 module of VMD43 and the coordinates of homologous Cα atoms for each structure were used as target coordinates of the respective Cα atoms of the human P-gp model. TMD was performed with each target in sequential simulations. Forces were applied using tcl scripts in NAMD to the selected Cα atoms of the human P-gp model in order to gently move the atoms towards the respective target coordinates. The magnitudes of these forces were calculated to be inversely proportional to the RMSD of the distances separating the selected Cα atoms and the target coordinates. The final systems used in TMD simulations shown in Figure 1 contained 157,540 atoms for P-gp with bound verapamil and 157,518 atoms for the system with daunorubicin bound to P-gp. In control experiments, a ligand that should not be transported by P-gp, the methyl ester of pyrophosphate (CH3O7P2) was added to P-gp in the same location where verapamil had docked in experiments as described above. The methylpyrophosphate system had 157,482 total atoms. Experiments with tariquidar had a similar number of atoms. Each ligand bound system was minimized, heated and equilibrated for 2 ns before the targeted molecular dynamics scripts were initiated.
Figure 1. Human P-glycoprotein conformations extracted at the start and end of targeted molecular dynamics simulations.

Human P-gp model structures are shown at the beginning of the targeted molecular dynamics simulations (left) and at the end of the TMD simulations (right). The right-hand structure shows the protein rotated about the Y-axis by 90° to better visualize the opening of the transmembrane helices to the outside. Human P-gp was pushed to each structure in TMD simulations as described in 31 and Methods. The structures shown have calculated RMSD values for targeted Cα atoms of human P-gp to target structures of < 0.3 Å. The target structure at the beginning of the simulations was mouse P-gp (4KSB)25 and the ending target structure was the transition state MsbA structure (3B5Z)19. The beginning structure (left) has the drug binding sites wide open to the cytoplasm (top of the membrane) and disengaged nucleotide binding domains. The ending structure has fully engaged nucleotide binding domains with the drug binding domain wide open to the extracellular space (below the membrane). verapamil (blue space filling representations) and daunorubicin (red space filling representations) are shown at their highest affinity binding sites in the starting structure and what may be the release sites in the ending structure. Starting positions for the transport substrates were determined by docking experiments (Methods). The N-terminal half of the protein is shown in both ribbon and surface representations in blue, while the C-terminal half of the transporter is shown in gold. The black bar shows the direction of the Z-axis of the system and measures approximately 150 Å.
Analyses of the overall movement of verapamil, daunorubicin, methylpyrophosphate, or tariquidar during each simulation were made by calculating the centers of mass of the ligands over the course of the simulations using tcl programming scripts in VMD. Analyses of the interactions of individual transmembrane helices with each one of the transport substrates and the negative control over the course of several simulations were also accomplished with the help of tcl scripts and VMD. Briefly, at equal intervals throughout a complete simulation, the numbers of contacts between substrate and protein were determined for each transmembrane helix as well as any contacts outside of the transmembrane regions. The results were summed for each simulation frame analyzed and plotted in 3-dimensional plots for each helix versus values of the progress of the simulation. Data were also collected on whether these interactions between transport substrate and protein were mediated by nonpolar, aromatic, polar, acidic or basic amino acids. Results are reported either for individual simulations or as the averages of all contacts for each drug at equivalent stages of simulation.
RESULTS
Molecular simulations of the conformational changes in human P-glycoprotein during a transport cycle
The work presented here builds on our previous work that aimed at understanding the catalytic transitions that P-gp undergoes during a putative transport cycle. To simulate the actual molecular transport, we added transport substrates into the drug binding domain (DBD) of P-gp by ligand docking methods and then simulated the transport cycle using targeted molecular dynamics (TMD) simulation techniques. We reasoned that if the putative catalytic transitions presented earlier31 were relevant approximations of the normally occurring P-gp-catalyzed drug transport, then freely mobile, completely unrestrained ligands known to be transport substrates of P-gp should move from the cytoplasmic side of the membrane to the extracellular side of the membrane during the course of the simulations. Minimally, if the conformational changes shown previously31 were relevant to P-gp drug transport, the ligands should at the very least gain access to the extracellular membrane leaflet.
To detect possible effects on ligands bound to the drug binding sites of P-gp, we first performed TMD experiments with either daunorubicin or verapamil bound to the transporter. These ligands are known to be transported by P-gp60, 61. daunorubicin or verapamil were docked in separate experiments to models of human P-gp in the starting conformation (with drug binding domains wide open to the cytoplasmic side of the membrane, corresponding to the mouse 4KSB structure). Figure 1 (left panel) shows a composite figure of the two transport ligands on a single protein (verapamil in blue; daunorubicin in red). It is interesting that the highest predicted affinity docking poses for daunorubicin and verapamil had the substrates bound to opposite sides of this open to the cytoplasm conformation of P-gp.
Figure 2 shows a close-up of the initial starting positions of daunorubicin (left) and verapamil (right) in the two drug binding subsites of P-glycoprotein that are equivalent to the poses shown in the composite depiction of Figure 1 (left). While the predictive ability of docking tools are limited, especially for large search areas, docking was used as a way to generate a plausible starting point for molecular dynamics simulations. After docking and assembly of a complete system including P-gp, lipids, water and ions, the docking poses for each ligand were allowed to relax in molecular dynamics simulations after being brought to 310 K. This relaxation of the docking poses varied the starting positions of verapamil and daunorubicin before the targeted molecular dynamics simulations were initiated (see below). Quantification of the resultant variation in initial ligand positions before the start of targeted molecular dynamics simulations for the six independent equilibrations per ligand showed RMSD values relative to the initial docking position of 3.2 Å +/− 0.4 Å with a range of 2.6 to 3.8 Å for daunorubicin and 4.9 Å +/− 1.0 Å with a range of 3.9 to 6.7 Å for verapamil. Figure 2 shows unique interactions of each drug in its different subsite on P-gp. While daunorubicin interacted with transmembrane helices (TM) 1, 2, 3, 6, and 11, verapamil predominantly interacted with TM 4–8 and 12. Supplemental Table 1 identifies the amino acid residues and the corresponding transmembrane helices that were found to be within 4.5 Å of the highest affinity docking poses of verapamil and daunorubicin in this initial conformation of the transporter after the docked ligands were allowed to relax in the equilibration phase of the MD simulations. Supplemental Table 1 also points out residues that have been experimentally identified 26, 62–69 or computationally identified 70 to be part of the drug binding sites of P-gp. Each drug binding subsite had a variety of interactions with different types of amino acid residues, but as expected, nonpolar and aromatic interactions dominated both the daunorubicin and the verapamil binding sites. Also of interest in Figure 2 are the polar interactions of P-gp with the polar portions of daunorubicin. Verapamil has a computed octanol/water partition coefficient (xlogP3) of 3.8, and is much less polar than daunorubicin (xlogP3 of 1.8, both values were obtained from the NCBI PubChem database). It therefore seems reasonable that verapamil interacted mainly with nonpolar and aromatic residues of P-gp with only an occasional polar contact, while daunorubicin displayed more frequent interactions with polar and even charged residues (Supplemental Table 1). Daunorubicin was observed in a few instances to contact residues outside of the TM region of P-gp (residues 875–879) during molecular dynamics, but it should be noted that all of these contacts were less than about one helical turn towards the cytosolic side of TM10. The initial docking modes for both daunorubicin and verapamil seemed to be reasonable starting points for the TMD simulations.
Figure 2. Structures of human P-glycoprotein with daunorubicin and verapamil bound.

P-gp is represented in cyan with the labeled α-helices drawn in a cartoon representation. The amino acid side chains within 3Å of daunorubicin (left) and verapamil (right) are drawn in stick format and are labeled. Daunorubicin and verapamil are drawn in green stick format. Linear depth cueing is implemented in these figures.
Movement of daunorubicin and verapamil through P-glycoprotein
During the targeted molecular dynamics simulations presented here, small forces were applied to a set of selected Cα atoms of P-gp as in 31 and Methods. The applied forces were recalculated during each step of the simulations to direct protein movements toward the respective target Cα coordinates. These TMD techniques allowed us to continuously simulate the movement of P-gp domains starting from conformations that had the DBD wide open to the cytoplasm (4KSB) to conformations with the DBD slightly opened to the cytoplasm and NBDs partially engaged (3B5X-target), to those with fully engaged NBDs and DBD opened to the exterior (2HYD-target), to a final conformation with NBDs in an ATP hydrolysis transition state where the DBD are fully opened to the extracellular space (3B5Z-target). All conformational transitions were calculated in an explicit, fully hydrated system that contained a combined aqueous and membrane environment. Figure 1 shows a composite structure of P-gp with both daunorubicin and verapamil at the starting conformations (left panel) and ending conformations (right panel) from two independent simulations. Supplemental Figure 1 shows snapshots of human P-gp in conformations that have Cα atoms within 0.25 Å RMSD of the four respective targets. Supplemental Figures 2 and 3 show video representations of the simulated trajectories of two of the simulations reported here for daunorubicin and verapamil, respectively.
When TMD simulations were performed starting with daunorubicin bound at the drug binding domain of P-gp in the wide open to the cytoplasm conformation (as shown in Figure 1 left and Figure 2), vectorial movement of the bound ligand perpendicular through the plane of the membrane in the direction of the extracellular space was observed in each simulation. Figure 3A (closed squares) shows the average position of the calculated center of mass of daunorubicin that were plotted over the progress of six independent simulation experiments. The error bars represent the standard error of the mean between each of the simulations. In these experiments, the P-glycoprotein – membrane bilayer system was oriented such that the plane of the membrane was in the X – Y coordinate plane. The movement of ligand from one side of the membrane to the other side could therefore be indicated as a vectorial movement on the Z-axes of the systems. As a reference, the black bar in Figure 1 shows the direction of the Z-axis relative to membrane and transporter. In each simulation of daunorubicin bound to P-gp, a vectorial movement of the drug along the Z-axes from the inside surface of the membrane to the outside surface of the membrane was observed (Figure 3A and B). In these experiments, daunorubicin moved an average of approximately 11 Å from the cytoplasmic side to the external side of P-gp. Interestingly, variations in both the timing and the extent of vectorial movement of daunorubicin along the Z-axes were observed during the six simulations (Figure 3B, comparing each individual simulation of daunorubicin transport). This variation in vectorial transport is reflected in the standard errors of the mean shown in Figure 3A. Figure 1 (right panel) presents a composite image of human P-gp at the end of the targeted molecular dynamics simulations performed here.
Similar to the experiments shown for daunorubicin, targeted molecular dynamics simulations of verapamil docked to the drug binding sites in an open to the cytosol conformation of P-gp showed significant movement of the ligand from the cytoplasmic side of the membrane to the external side of the membrane. Figure 3A (closed triangles) presents the average position of the center of mass of verapamil that was obtained from six independent simulation experiments. Results from each individual simulation for verapamil are shown in Figure 3C. Again, as for daunorubicin, no external forces were applied to the transport substrate in these simulations, yet a net vectorial movement of verapamil through the plane of the membrane averaging about 12 Å was observed. As described for the daunorubicin simulations, variations in the extent and the timing of the movements of verapamil were observed (Figure 3C) which was reflected in the errors of the mean as shown in Figure 3A. The variations in the movement of verapamil observed in these simulations were greater than those observed for daunorubicin. It should be noted, however, that verapamil is a much more flexible ligand than is the rather rigid four-ring system of the anthracycline, daunorubicin. This increased flexibility of verapamil may have led to a wider variation in its measured center of mass as compared to daunorubicin.
Despite the variations observed in the timing and to some degree in the extent of movement of the drugs through P-gp, each simulation resulted in a net vectorial transport of either daunorubicin or verapamil through the plane of the membrane (compare Figure 1 left and right panels). The centers of mass of daunorubicin in these simulations were observed to move between 6 and 14 Å towards the external space, while the centers of mass of verapamil were observed to move between 8 and nearly 20 Å towards the outside (Figure 3B and C).
It was also of interest to us to determine whether transport of daunorubicin would occur when the substrate was bound in a non-optimal docking position. To this aim we chose a less preferred docking position for daunorubicin that was in the preferred verapamil docking site. The starting position of daunorubicin in these simulations was one of the top nine poses found in our docking experiments. The ligand in these simulations was not observed to strongly associate with any one residue and did not move towards its preferred docking position. The results of six TMD simulations performed with daunorubicin at the preferred verapamil docking site showed that daunorubicin moved an average of 13 Å through P-gp (Supplemental Figure 4).
Placing verapamil at the preferred daunorubicin docking site was more problematic, since verapamil was not found in any of the best nine docking poses in this position. We therefore performed new docking experiments with a restricted target box at the preferred daunorubicin docking site to restrict verapamil to this starting location. The estimated binding energy for this interaction for verapamil was −6.2 kcal/mol compared to −7.4 kcal/mol for daunorubicin at this site. Results of the six simulations we performed with verapamil starting at the preferred daunorubicin site were mixed. While an average movement of 4.5 Å towards the extracellular space was observed (Supplemental Figure 4), unlike with daunorubicin, two of the simulations showed < 3 Å movement through the membrane and one simulation showed verapamil moving 4 Å away from the extracellular space. The three other simulations showed movement of verapamil that was comparable to that observed when it started from its preferred docking position, moving between 7 and 11 Å towards the extracellular space (Supplemental Figure 4).
Overall, these latter experiments with daunorubicin or verapamil placed in non-optimal positions within P-gp demonstrated that both of the transport substrates could be moved toward the extracellular space during the TMD simulated catalytic cycles.
Movement of a pyrophosphate methyl ester out of the drug binding sites of P-glycoprotein and into the cytoplasm
In order to test whether or not transport would be observed with a compound that clearly does not have the attributes of a good P-gp transport substrate, we placed a negatively charged, hydrophilic substance (pyrophosphate methyl ester) at the initial starting position of verapamil and performed simulations as presented above. In these simulations, an entirely different behavior was observed when P-gp transitioned through the catalytic conformational changes. Pyrophosphate methyl ester does not fall into the category of compounds normally transported by P-gp, which are mostly hydrophobic molecules or hydrophobic cations3. Figure 3A (closed diamonds) shows the average position of methylpyrophosphate in six independent TMD simulations as P-gp moved from open to the inside to open to the outside conformations. As can be seen in Figure 3A, methylpyrophosphate immediately moved away from the drug binding sites of P-glycoprotein and away from the hydrophobic core of the phospholipid bilayer towards the cytoplasm during the simulations. A total of six such simulations were calculated for methylpyrophosphate and each simulation (Figure 3D) showed this immediate movement of the polyanion into the cytoplasmic space.
Movement of tariquidar, a P-glycoprotein inhibitor, during a simulated transport cycle
In further experiments aimed at elucidating important ligand interactions that may occur during simulated transport cycles, we investigated a known inhibitor of P-glycoprotein, tariquidar42. In three different docking studies using tariquidar, we identified three separately located sites on P-gp that had high estimated ligand binding energies as calculated by AutoDock Vina (Figure 4). The highest affinity tariquidar docking site (site 1 of Figure 4) was found using all of P-gp except the nucleotide binding domains as a docking target. The best docked example (from 300 replicates) had an estimated binding energy of −10.1 kcal/mol and was located in the intracellular loop (ICL) regions on TM4 and TM9 (see Supplemental Table II for residues). For comparison, daunorubicin and verapamil docking at preferred sites within the drug binding domains had estimated binding energies of −7.6 and 7.4 kcal/mol, respectively. A second docking experiment targeted tariquidar to the entire drug binding domain, but did not include the ICL regions of P-gp. The best example from this experiment (also 300 replicates) is shown in Figure 4 as site 2. This site was located at the level of the cytoplasmic leaflet in the drug binding sites and had an estimated binding energy of −9.1 kcal/mol. In order to also explore the drug binding site described in 71, a docking experiment was initiated that limited interactions to this area of the P-gp drug binding domain. These experiments resulted in the docking of tariquidar with an estimated affinity of −8.4 kcal/mol (site 3 in Figure 4).
Figure 4. Movement of Tariquidar when bound to P-glycoprotein.
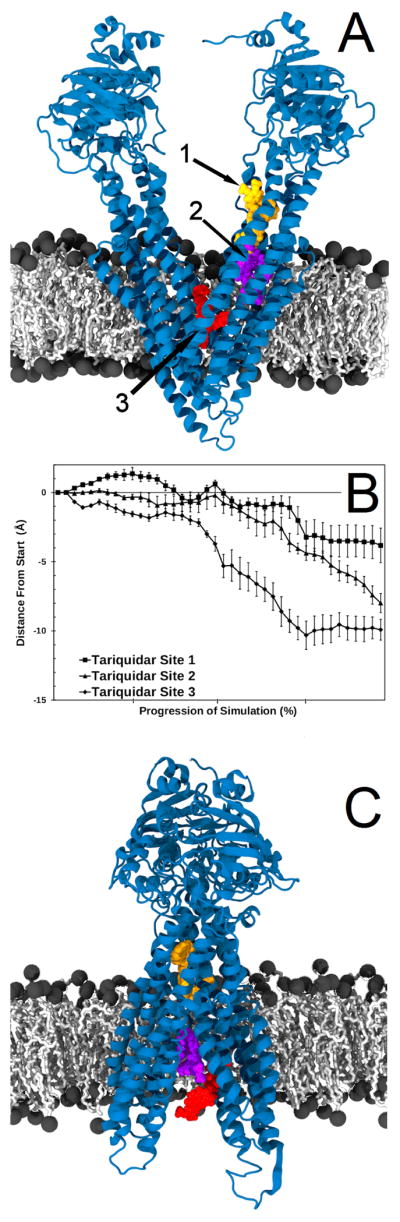
Tariquidar was docked to P-glycoprotein as described in Methods and Results and was found to preferentially bind to three different sites within P-gp. Panel A shows the three tariquidar docking sites in van der Waal’s spheres with site 1 in yellow, site 2 in purple and site 3 in red. Targeted molecular dynamics simulations were performed with tariquidar bound at each of these sites (site 1, 2 and 3, with six replicate simulations for each site) and the average movement of the center of mass of tariquidar is shown in Panel B. Movement was calculated as in Figure 3. Error bars represent one standard error of the mean. Panel C shows the positions of tariquidar at the end of representative simulations with the same color coding as in Panel A.
The results of TMD simulations of transport cycles with these three tariquidar starting positions are presented in Figure 4. Figure 4B shows the average movement of the center of mass of tariquidar for all six replicates with one standard error of the mean represented by the error bars. Figure 4B (squares) shows the movement of tariquidar when starting at the preferred docking site 1. Very little movement of tariquidar towards the extracellular space was observed from this starting position (~3.8 Å). It is also important to note that tariquidar bound at site 1 did not leave the region of P-gp that it initially interacted with in the docking experiments, and that it did not leave the ICL regions of P-gp. Importantly, residues D177 and N820 were observed to interact with tariquidar when the NBDs were in the closed positions in these simulations. These two residues were previously shown experimentally by Loo and Clarke72 to activate ATP hydrolysis 10-fold when crosslinked as cysteine mutations. Stabilization of the close interaction of NBDs (and residues D177 and N820) by tariquidar may be important in its mechanism of inhibition (see Discussion).
When simulated transport cycles began with tariquidar at either site 2 or at site 3 (both within the transmembrane drug binding sites of P-gp), much greater movement through the membrane toward the extracellular space was observed (average of about 8 and 10 Å from site 2 and site 3, respectively, see Figure 4). These results suggest that tariquidar may be transported through the membrane by P-gp, if it is initially bound at a drug binding site within the transmembrane domain.
Two transport substrate pathways through P-gp
Figure 5 shows the average number of contacts made by the individual transmembrane helices of P-gp (1 through 12) with either daunorubicin or verapamil over the course of the replicated simulations. These admittedly complex graphs show that very different patterns of contacts with the transmembrane helices of P-gp were observed for daunorubicin as compared with verapamil. During daunorubicin transport from its preferred docking position (Figure 5 – left), transmembrane helices 1, 2, 3, 11 and 12 showed multiple contacts with the transport substrate over the course of the simulations, while TMs 6 and 10 had one contact or less with the drug. Helices 4, 7, 8 and 9 did not interact at all with daunorubicin. For verapamil transport from its preferred starting position, a different pattern emerged (Figure 5 - right). TMs 5, 6, 7 and 12 appeared to be very important in moving verapamil through the membrane, while TMs 4, 8, 9 and 11 interacted only to a minor extent. Helices 1, 2, 3 and 10 showed no interactions with verapamil over the course of the simulations. Neither daunorubicin nor verapamil significantly interacted with any residues outside of the transmembrane helices (designated as “helix 0” in the figure). Essentially the same results were obtained when 3.5 or 2.5 Å cutoff distances were chosen for these analyses (data not shown). These differences in which particular helices were used to drive either daunorubicin or verapamil through the membrane (as shown in Figure 5) appeared to be predetermined by where in the protein the drug was bound when the simulated transport cycles began. When daunorubicin was bound initially in its preferred binding site, TMs 1, 2, 3, 6, 11 and 12 were involved in its transport. When verapamil was initially bound in its preferred drug binding site, TMs 5, 6, 7, 8 and 12 were involved in its transport. Both sets of simulations had in common only the “last” TMs of the half transporter subunits, helices 6 and 12. These results suggest that there are at least two pathways through P-gp for transported substrates that are predetermined by which of the drug binding subsites is initially occupied by drug.
Figure 5. Average number of contacts made by transmembrane helices of P-glycoprotein during drug transport.
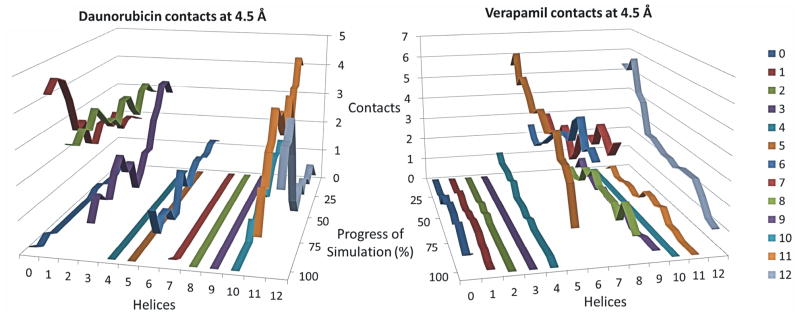
The number of interactions of transport substrate with each of the 12 transmembrane helices of P-gp (helices 1 through 12 – labeled on the X-axes) are plotted versus the progress of the simulations as P-gp cycled from the open to the inside conformations (0% on the “progress of simulation” Y-axes) to fully opened to the exterior conformations (100% on the Y-axes). Helix “0” represents any contact between protein and drug that occurred outside of the 12 transmembrane helices. The Z-axes show the number of contacts made at the various points in the simulation that were within 4.5 Å of any atom of P-gp. The color coding legend for the helices is shown on the right of the figure. The contact values were compiled for each of the six independently replicated simulations for each drug and then averaged. Results for daunorubicin transport are shown on the left panel while those for verapamil are on the right panel.
Stochastic drug contacts with transmembrane helices in the two transport pathways through P-gp
Figures 6 and 7 and Supplemental Figures 5 and 6 show that there is considerable variability in when a particular transmembrane helix interacts with a drug and even which particular TM makes contact with a drug within either of these two pathways. As an example, Supplemental Figures 5 and 6 show the contacts made between P-gp and daunorubicin or verapamil, respectively, in each of the six individual simulations. It is apparent that in the six individual simulations different TMs (1, 2, 3, 6, 11 and 12) were involved to differing extents in contacts that presumably pushed daunorubicin through the membrane portion of P-gp (Supplemental Figure 5). Very similar results indicating large variability in when different TMs interacted with verapamil were obtained when the six individual simulations of verapamil were plotted (Supplemental Figure 6). In the case of verapamil, TMs 5 through 8 and 12 were always involved, but some helices were contacted more often in some of the simulations compared to others.
Figure 6. Residuals of the average number of contacts made by transmembrane helices of P-glycoprotein with daunorubicin for each independent simulation.

These graphs are similar to those presented in Figure 5, except that the number of contacts at each point of progression in the six individual simulations with daunorubicin was subtracted from the average number of contacts at that point in the simulations. The graphs are intended to highlight the variability of contacts made between each transmembrane helix with daunorubicin as P-gp moved from open inside to open outside conformations. Results from six independent simulations are shown.
Figure 7. Residuals of the average number of contacts made by transmembrane helices of P-glycoprotein with verapamil for each independent simulation.

These graphs are similar to those presented in Figure 5, except that the number of contacts at each point of progression in the six individual simulations with verapamil has been subtracted from the average number of contacts at that point in each one of the six independent simulations.
The observations that different helices seemed to dominate the interactions in some of the simulations, while others dominated in other simulations are easier to visualize when the residuals of contacts relative to the average number of contacts between drug and TMs are plotted against the progress of individual simulations (Figures 6 and 7 for daunorubicin and verapamil, respectively). The graphs show the differences between individual simulation runs and the averages of all simulations for each of the drugs. Large differences in contacts with any given transmembrane helix indicate that those helices are being used to different extents in the given simulations compared to average interactions. For example, in the daunorubicin simulations (Figure 6), TM helix 2 (green bars) was used much more extensively in simulation 2 than in simulation 6. Conversely, TM helix 1 was used much less in simulation 1 than in simulation 2. Also, simulations 4 and 6 show very different patterns of daunorubicin contacts with helix 6 (blue). Other differences between individual simulations are also apparent from Figure 6.
Results of residual analyses between the average contacts of each TM with those observed in an individual verapamil transport simulation (Figure 7) led to comparable observations, even though different pathways and associated TM helices were involved. In the verapamil simulations, the effects seem even more dramatic than for the daunorubicin simulations. For example, comparison of the very different contacts of verapamil with helix 12 (light blue) in simulations 1, 2, 4, or 5 with those in simulations 3 or 6 highlights significant differences in the use and timing of the use of this helix in moving verapamil through P-gp. Likewise, TM 5 contacts with verapamil (gold) were drastically different in simulation 2 as compared with simulations 3 or 6. TM 7 also showed dramatically different contacts with verapamil in simulation 5 when compared to 6. As a further example of the stochastic nature of the TM contacts with verapamil, TM 8 appeared heavily involved in drug interactions in simulation 2, but not in 1, 3 or 6. These observations suggest that there is no predefined or predetermined step-by-step sequence of contacts for either of the two pathways for drugs through P-gp.
Nonpolar and aromatic interactions of P-glycoprotein with daunorubicin and verapamil dominate during transport
Figure 8 presents the average number of contacts between transported drug and amino acids of each transmembrane helix for either daunorubicin (left panels) or verapamil (right panels) categorized for nonpolar amino acids (upper panels), aromatic amino acids (middle panels) or polar residues (lower panels). In the case of daunorubicin, nonpolar interactions in TMs 1, 2 and 12 appeared to be most important with minor interactions at TMs 3, 6 and 11. It appeared that aromatic interactions of daunorubicin with TM 11 were especially important when the protein was open to the cytoplasm and that these interactions decreased in number as P-gp progressed to open to the outside conformations. Verapamil showed both nonpolar and aromatic interactions with residues in TMs 5, 6, 7 and 12. Several interactions for both drugs with polar amino acids in the TM regions of P-gp were observed as is expected for these drugs, since both verapamil and daunorubicin have significant numbers of polar functional groups (6 polar groups in verapamil and 11 in daunorubicin). Supplemental Figure 7 shows the results of similar analyses for contacts made between daunorubicin and verapamil with charged amino acid residues of the transmembrane helices of P-gp. Only the more polar daunorubicin was observed to interact with any charged residues (H61, K189, D188 and E875). These interactions were minor, with less than one such charged residue interaction observed at any given stage of simulation. No interactions with charged residues were observed for verapamil.
Figure 8. Interactions between P-gp and daunorubicin or verapamil with nonpolar, aromatic, or polar residues of the transmembrane helices.

The figure presents the average number of contacts in all six replicate simulations for daunorubicin (left panels “Dau”) and verapamil (right panels “Ver”) for nonpolar (top), aromatic (middle) or polar (bottom) amino acid residues of P-gp. Graphs are plotted as described for Figure 5.
DISCUSSION
Binding subsites for daunorubicin and verapamil on P-glycoprotein
Ligand docking experiments were performed here to position the transport substrates, daunorubicin and verapamil, into reasonable positions to analyze potential transport through P-glycoprotein in simulated catalytic cycles. These docking experiments resulted in preferred binding of daunorubicin and verapamil to two different subsites within the drug binding domain of P-gp in an open to the cytoplasm conformation (Figures 1 and 2, Supplemental Table 1). Both drugs bound within the cytoplasmic leaflet part of the cell membrane. Daunorubicin preferentially bound to residues on transmembrane helices 1, 2, 3, 6, and 11 while verapamil docked preferentially to residues on transmembrane helices 4, 5, 6, 7, 8 and 12. Similar observations of different subsites on P-glycoprotein for drug binding have been previously reported 25, 26, 31, 38, 73–78. It is interesting to point out that earlier work 73 suggested the presence of two distinct drug binding sites on P-gp that had different affinities for rhodamine 123 and Hoechst 33342, designated the R and H sites respectively, and that the R site preferentially bound daunorubicin. In even earlier work60, it was observed that daunorubicin at low concentrations noncompetitively inhibited verapamil transport, but at high concentrations it competitively inhibited verapamil transport. As summarized in 73, these observations were taken to mean that daunorubicin and verapamil preferentially bound to different sites with daunorubicin binding at low concentrations at the R-site and with verapamil at the H-site. At high concentrations, daunorubicin appeared to also bind at the H-site competitively inhibiting verapamil transport73. It is tempting to think that the data on the docking of daunorubicin and verapamil to P-gp support these interpretations, since daunorubicin was observed to dock at two of the sites (preferring one site over the other) while verapamil preferentially bound to only one site and had to be forced into docking at the preferred daunorubicin site (the R-site). The transport of verapamil from this latter starting point was also observed to be less consistent than when it started in its preferred position. Unfortunately, the accuracy of estimating binding energies using currently available docking software does not allow us to make this assertion with confidence.
It should be noted that both daunorubicin and verapamil were docked to P-gp under identical conditions. After the initial docking, the complete systems including P-gp, drug, lipid, water, salt, nucleotide and magnesium ion were heated to 310 K and were then equilibrated using molecular dynamics techniques (see Methods). These procedures allowed the docked drugs to independently readjust in each of the replicated simulations. This relaxation step varied the starting position of verapamil and daunorubicin somewhat before the targeted molecular dynamics was initiated. Quantification of this variation showed RMSD relative to the initial docking position of 3.2Å +/− 0.4 Å with a range of 2.6 to 3.8 Å for daunorubicin and 4.9 Å +/− 1.0 Å with a range of 3.9 to 6.7 Å for verapamil. Although a variety of protein-drug interactions were observed within each of the two subsites after equilibration, nonpolar and aromatic interactions clearly dominated for both daunorubicin and verapamil. The presence of many nonpolar and aromatic residues in these subsites (Figure 2, Supplemental Table 1) may help explain some of the broad transport substrate specificities shown by P-gp, since these types of interactions would be available for many different hydrophobic or aromatic compounds. Polar residue interactions with the somewhat more polar daunorubicin were more common than with the more hydrophobic verapamil. Only daunorubicin showed interactions with acidic and basic residues (Supplemental Table 1, Supplemental Figure 7). Daunorubicin also showed a few interactions outside of the transmembrane helices as defined in Uniprot (http://www.uniprot.org/uniprot/P08183), but these residues were all within about one helical turn of TM 10 and were in the lipid head-group region of the cytoplasmic leaflet of the membrane.
The variable interactions of identically docked daunorubicin or verapamil to P-glycoprotein after molecular dynamics “incubation” at 310 K highlights the flexibility and plasticity of transport substrate binding to P-gp as mentioned by us earlier31. The results also support and extend the hypothesis first introduced and tested by Loo and Clarke79 that transport substrate binding to P-gp can induce conformational changes in the transmembrane helices that allow the binding of many different transport substrates. This induced fit model for P-gp79 is extended here to two different subsites that have the flexibility to readjust binding in a dynamic way. These smaller adjustments of protein to ligand are shown here in the different contacts made between identically docked daunorubicin or verapamil after 310 K equilibrations (Supplemental Table 1).
Vectorial transport of verapamil and daunorubicin through the membrane by P-glycoprotein
The studies presented here extend our previous work on the dynamic conformational changes that take place in the drug binding domains of P-gp during a putative catalytic cycle as simulated using targeted molecular dynamics techniques31. It should be reiterated here that these studies used four different targeted P-gp structures that were based on four crystal structures of related ABC transporters. The targeted molecular dynamics technique was used to push P-gp from one structure to the next. A limitation of TMD with only one target structure is that the method is blind to the nuances of movement of the protein in intermediate phases. Here we have employed multiple target structures that comprise intermediates that make biochemical and mechanistic sense. Although we cannot be certain that the lowest energy path between these structures was sampled by the technique, what we can be sure of is that the targets used are close to low energy conformations, since all four were derived from crystallized proteins. The current study strongly suggests and reproducibly showed that the catalytic transport cycle simulated in these TMD experiments resulted in the net vectorial movement of two different transport ligands from binding positions within the cytoplasmic side of the membrane of P-gp to positions equivalent to the exterior leaflet of the membrane bilayer (summarized in Figure 3). During these simulations, the only external forces exerted on the system were directed at the protein, so any net movement from the cytoplasmic to the external side of the membrane must have been a consequence of the drug interacting with the protein. In other words, the vectorial movement of the transport substrates through the membrane was solely caused by the movement of P-gp transmembrane helices and the respective amino acid side chains that contacted the substrate. The movements of transport substrates were from positions in P-glycoprotein equivalent to the cytoplasmic leaflet of the membrane bilayer at the start of the simulations to positions equivalent to the external side of the membrane (see Figures 1 and 3, and Supplemental Figures 2, 3 and 4). The average movement of the calculated centers of mass of both drugs was 11 to 12 Å when starting at the preferred docking positions, see Figure 3. Daunorubicin when placed in the preferred verapamil docking site moved on average a similar distance toward the outside, while verapamil at its non-preferred site moved less well on average, but was observed in half of the simulations to move between 7 and 11 Å outward (Supplemental Figure 4). These results support the hypothesis that P-gp actually drives the drugs from one side of the membrane to the other. The results shown in Figure 3 and Supplemental Figure 4 can be compared to those in Supplemental Figure 8 which shows the results of net drug transport in terms of RMSD changes from the initial drug position to its final state at the end of the simulations.
A report was recently published38 using TMD techniques that were similar to those reported earlier by us31. Despite using similar techniques and protein target structures as those reported here, the authors showed only very small RMSD changes of 1 to 2.5 Å for a series of P-gp transport substrates38, nearly 10-fold less than is presented here. One problem with RMSD measurements when using them to try to show vectorial movements as done in 38 is that a rotation of the molecule about its center of mass will show a large RMSD value when in fact the center of mass does not move. For this reason we have preferred to use center of mass movement measurements instead of RMSD. However, in order to compare our results with those obtained by 38, we also calculated transport substrate RMSDs. The maximal RMSD change for simulations using daunorubicin and verapamil transport were 15.5 and 22.8 Å (see Supplemental Figure 8). These values were significantly larger than those reported by 38. The changes in RMSD reported in 38 may reflect slight movements and changes in conformation of compounds bound to essentially the same subsite on P-gp and that no net vectorial movement of substrates was observed. One important interpretation of the simulations in 38 was that molecules thought to be inhibitors of transport by P-gp stayed associated with residues in the initial docking sites longer than did molecules they listed as transport substrates. Since neither transport substrates nor inhibitors were shown in 38 to significantly move from the initial docking positions, this interpretation should be reevaluated.
Independent of how we measured the transport of daunorubicin or verapamil (either by movement of the center of mass of the drug on the Z-axes as in Figure 3 or by RMSD as in Supplemental Figure 8), all 12 simulations reported here for the ligands bound to preferred docking sites demonstrated very significant movement of either daunorubicin or verapamil. Daunorubicin moved in six replicated simulations between 6 and 14 Å by center of mass in the Z-axes or up to 15.5 Å when measured by RMSD changes. Verapamil moved in six replicates by 8 to 20 Å transport by center of mass in the Z-axes or by up to 22.8 Å by RMSD changes.
Significantly, when we observed the movement of a polyanion, methylpyrophosphate, a compound that should not be transported by P-glycoprotein, no vectorial movement from inside to outside was observed (Figure 3A, D). In these simulations, where the methylpyrophosphate was placed directly into a drug binding site that we showed here was competent for verapamil transport, a movement in the opposite direction to that of verapamil or daunorubicin was observed (Figure 3A). Methylpyrophosphate moved quickly out of the verapamil drug binding subsite and towards the cytoplasmic space, while both verapamil and daunorubicin moved through the transmembrane domain of P-gp toward the extracellular space. In each of six replicated simulations, methylpyrophosphate moved 12 to 18 Å towards the cytoplasm.
These results with methylpyrophosphate are in stark contrast to the movement of either verapamil or daunorubicin during identical targeted molecular dynamics simulations. It should be emphasized that the methylpyrophosphate molecule was not docked in the verapamil subsite of drug binding sites of P-gp, but was manually placed there to test whether an obvious non-transport substrate would also be driven through the membrane as P-gp transitioned from inside open to outside open conformations. The results presented in Figure 3 clearly show that a substance not meeting the criteria of a transport substrate of P-gp was not moved through the membrane by the protein, but instead quickly moved towards the cytosol even when it was inserted into a drug binding site at the start of the simulation. This suggests that not just any substance that can be internalized into a drug binding site of P-gp will be actively transported. These simulations with methylpyrophosphate strongly support the hypothesis that the drug transport observed here with the known P-gp substrates daunorubicin and verapamil as the transporter cycled through a simulated catalytic cycle reflect real drug transport mechanisms by P-glycoprotein.
From the extent of the movements of daunorubicin and verapamil presented in Figure 3, it is clear that not just a re-positioning of helices of P-gp has occurred that granted the drugs access to the outside of the membrane, but that both daunorubicin and verapamil have been driven through the membrane towards the outside of the cell by the conformational changes that occurred during these catalytic cycles. The data strongly suggests that P-glycoprotein possesses a drug pumping activity and not just a differential gating activity that grants access of a drug from one membrane leaflet to the other.
Mechanism of inhibition of P-glycoprotein transport by tariquidar
Docking of tariquidar to P-glycoprotein in the open to the cytoplasm conformation was performed using three different targeting boxes for the ligand. In the first experiment, tariquidar was allowed to dock to any part of P-gp except the NBD structures and this attempt found the docking position with the highest estimated affinity (−10.1 kcal/mol) of the three experiments (site 1 in Figure 4). This site was located in the intracellular loop (ICL) regions on TM4 and TM9. Tariquidar bound at this position was not transported through the membrane during transport cycle simulations (Figure 4) and was not observed to separate from the majority of the residues in its initial starting position.
It is relevant to the following that the initial starting interactions for tariquidar at site 1 included residues around N820 and that these residues stayed in close proximity to tariquidar throughout the entire simulation. In the latter part of these TMD simulations, when the NBDs were in a closed conformation thought competent for ATP binding and hydrolysis, tariquidar was also found to interact with residue D177. These two residues, D177 and N820, when mutated to cysteines and crosslinked, activated ATP hydrolysis rates by P-gp about 10-fold72. It is interesting to speculate that if tariquidar binds to site 1 and stabilizes the closed conformation of the NBDs, then this could explain the observations that tariquidar activates ATP hydrolysis by P-gp in the absence of transport 72, 80. This would also be consistent with tariquidar inhibiting P-gp transitions to open conformations as determined by 81 in work that showed tariquidar inhibited the crosslinking of extracellularly located residues. Finally, if tariquidar binding at site 1 did stabilize conformations of P-gp with closed NBDs, it might also explain the tariquidar-dependent inhibition of the labeling of residues in the drug binding sites as described in 71. It should also be noted that tariquidar was observed to stabilize truncated drug binding domains of P-gp in 81 and that the residues reported here for tariquidar binding site 1 are present in these truncated P-gp peptides.
The fact that others observed inhibition of ATP hydrolysis by tariquidar 42 may perhaps be explained by tariquidar binding to P-gp at an alternative site or sites. When tariquidar was docked into the drug binding sites within P-gp (either site 2 or 3 in Figure 4), significant transport of the inhibitor through the membrane and towards the extracellular space was observed (Figure 4). This suggested that tariquidar could be transported if it binds at drug binding sites on P-gp. Experimental data exists, however, that suggest that tariquidar is not effectively transported by P-gp42, 80. If tariquidar does bind to the drug binding sites of P-gp as implicated in 71, then one might speculate that tariquidar may not readily dissociate from P-gp even when it is exposed to the extracellular space at the end of a transport cycle, thereby inhibiting net turnover of the transporter and perhaps ATP hydrolysis. There is evidence that tariquidar binds significantly to the cell surface of P-gp-expressing cells80 and there is also strong evidence that tariquidar has a much higher affinity to P-gp and slower off-rate from P-gp than does the good transport substrate, vinblastine42.
It is interesting to contrast these proposed mechanisms of inhibition by tariquidar to that proposed for other inhibitors of P-gp like the QZ59 peptides co-crystallized in murine P-gp structural studies26. In recent work, molecular dynamics studies of daunorubicin and the QZ59 inhibitors have been reported 70 that suggest that inhibitors like QZ59-RRR and QZ59-SSS may inhibit P-gp by preventing NBD closure.
Drug transport through P-glycoprotein is both predetermined and stochastic
Figures 6 and 7 show clearly that a different set of transmembrane helices are involved in daunorubicin transport than for verapamil transport through P-glycoprotein. Analyses of residues that contacted the different drugs as they were pushed through the transmembrane region of P-gp clearly showed that daunorubicin used TMs 1,2, 3, 6, 11 and 12 while verapamil mainly used TMs 4, 5, 6, 7, 8, 11 and 12. Only TMs 6 and 12 (and to a minor extent 11) were common to both subsets, and both subsets correlated nearly perfectly with the initial binding subsets for the two substrates. This strongly suggests that the initial site of binding of a drug to P-gp may predetermine which P-gp sub-structures are involved in pushing the drug all the way through to the external side of the membrane. In this sense it appears significant that the general path a drug takes through P-gp is predetermined by which initial subsite in the drug binding sites a given drug binds. This is an important conclusion about the structure and mechanism of drug transport through P-gp that can be inferred from Figure 5 and Supplemental Table 1.
A second important inference that can be made from this work is that within these two general pathways through P-gp, there does not appear to be a single defined path of contacts that drives a transport substrate out of the cell. Figures 6 and 7 (and Supplemental Figures 5 and 6) indicate that within each of the two observed pathways through P-gp, the contacts between TM helices and the drug as any given simulation progressed showed a lot of variation in which TM interacted with a specific drug and even when specific TMs interacted with each drug. These observations suggest that stochastic opportunistic contacts between protein and transport substrate within each of the two general pathways drive the drug through the membrane. We observed that nonpolar and aromatic amino acids contacted transported drugs most often as they were pushed towards the external space by P-gp and that there were significantly more polar and charged amino acid contacts with the more polar daunorubicin compared to verapamil.
Conclusions
The simulations of catalytic transport cycles by P-glycoprotein with different transport substrates presented in this study directly demonstrated that transport drugs can be actively pushed by the protein an average of between 11 and 12 Å from the cytoplasmic to the external side of the membrane. At the same time, access of the transported drug was changed from inside membrane surfaces and cytoplasmic bulk phase solvent to outside membrane surfaces and external bulk solvent. The simulations suggested that there are two pathways for transport substrates through P-gp and that the starting location of the drugs, one of two different drug binding subsites, predetermined which pathway would be used. Additionally, the results suggested that the actual transmembrane helices involved in pushing the drugs through the membrane and the timing of the contacts between protein and drug in either of the two pathways could vary in what appeared to be a stochastic manner. The observations presented here elucidated several characteristics about the mechanism of drug transport through P-glycoprotein: (1) There are at least two different drug binding subsites in the transmembrane domains of P-gp that can preferentially bind transport substrates, (2) it appears that there are at least two different general pathways for a transport substrate to be pushed through the membrane, (3) that the general pathway used by a particular transport substrate appears to be correlated with its initial drug binding subsite, and (4) that within either of the two general pathways for a drug through P-gp, the actual contacts between TMs and drug and the timing of the contacts can be highly variable and stochastic in nature. In additional studies, the strong inhibitor of P-gp, tariquidar, was observed to dock at three plausible sites. The preferred site for tariquidar docking was in the intracellular loop region on TM4 and TM9. Transport of tariquidar through the membrane from this site was not observed. Alternatively, positioning tariquidar within drug binding domains resulted in movement of tariquidar through the membrane. We speculate that in the former case, stabilization of NBD-closed conformations through tariquidar binding explains both the inhibition of transport and stimulation of ATP hydrolysis by P-gp that has been observed. Transport through the membrane in the latter case, coupled with slow dissociation of tariquidar may inhibit both hydrolysis and transport by P-gp.
Supplementary Material
Acknowledgments
Computational resources were provided by the SMU Center for Scientific Computing. A special thanks to Professor Junmei Wang (University of Texas Southwestern Medical Center) for helpful discussions and to Joe Gargiula and Amit Kumar of the SMU Office of Information Technology for their assistance with this work. The authors also thank the reviewers of the paper for their constructive suggestions.
ABBREVIATIONS
- ABCB1
ATP Binding Cassette transporter, type B1
- DBD
drug binding domain – the domain of the transporter made up of the 12 transmembraneous helices and the intracellular extensions of these helices
- ICL
intracellular loop region
- MD
molecular dynamic simulation
- NBD
nucleotide binding domains
- NPT
an isothermic-isobaric molecular simulation ensemble where the number of atoms, pressure and temperature are held constant
- P-gp
P-glycoprotein
- POPC
1-palmitoyl,2-oleoyl-sn-glycero-3-phosphocholine
- RMSD
root mean squared deviation
- TM
transmembrane helix
- TMD
targeted molecular dynamics
Footnotes
The project described was supported by grant number 1R15GM094771 - 01A1 from the National Institute of General Medical Sciences (NIH/NIGMS) to Pia D. Vogel and John G. Wise, and by the Communities Foundation of Texas, the SMU University Research Council, the SMU Dedman College Dean’s Research Council, and the SMU Dedman College Center for Drug Discovery, Design and Delivery. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
The authors declare no competing financial interest.
Supporting information is available. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cascorbi I. P-glycoprotein: tissue distribution, substrates, and functional consequences of genetic variations. Handbook of experimental pharmacology. 2011:261–283. doi: 10.1007/978-3-642-14541-4_6. [DOI] [PubMed] [Google Scholar]
- 2.Eckford PD, Sharom FJ. ABC efflux pump-based resistance to chemotherapy drugs. Chem Rev. 2009;109:2989–3011. doi: 10.1021/cr9000226. [DOI] [PubMed] [Google Scholar]
- 3.Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev. 2003;55:3–29. doi: 10.1016/s0169-409x(02)00169-2. [DOI] [PubMed] [Google Scholar]
- 4.Cordon-Cardo C, O’Brien JP, Boccia J, Casals D, Bertino JR, Melamed MR. Expression of the multidrug resistance gene product (P-glycoprotein) in human normal and tumor tissues. J Histochem Cytochem. 1990;38:1277–1287. doi: 10.1177/38.9.1974900. [DOI] [PubMed] [Google Scholar]
- 5.Lee CA, Cook JA, Reyner EL, Smith DA. P-glycoprotein related drug interactions: clinical importance and a consideration of disease states. Expert opinion on drug metabolism & toxicology. 2010;6:603–619. doi: 10.1517/17425251003610640. [DOI] [PubMed] [Google Scholar]
- 6.Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455:152–162. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- 7.Shen DW, Fojo A, Roninson IB, Chin JE, Soffir R, Pastan I, Gottesman MM. Multidrug resistance of DNA-mediated transformants is linked to transfer of the human mdr1 gene. Molecular and cellular biology. 1986;6:4039–4045. doi: 10.1128/mcb.6.11.4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marquez B, Van Bambeke F. ABC multidrug transporters: target for modulation of drug pharmacokinetics and drug-drug interactions. Current drug targets. 2011;12:600–620. doi: 10.2174/138945011795378504. [DOI] [PubMed] [Google Scholar]
- 9.Vogelgesang S, Kuhnke D, Jedlitschky G, Jucker M, Mosyagin I, Pahnke J, Cascorbi I, Kroemer HK, Walker LC, Warzok RW. P-glycoprotein (ABCB1) mediates transport of Alzheimer’s beta-amyloid peptides. Acta Neuropathol. 2006;112:365–367. [Google Scholar]
- 10.Pahnke J, Langer O, Krohn M. Alzheimer’s and ABC transporters - new opportunities for diagnostics and treatment. Neurobiology of disease. 2014;72(Pt.A):54–60. doi: 10.1016/j.nbd.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varatharajan L, Thomas SA. The transport of anti-HIV drugs across blood-CNS interfaces: summary of current knowledge and recommendations for further research. Antiviral research. 2009;82:A99–109. doi: 10.1016/j.antiviral.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen CJ, Chin JE, Ueda K, Clark DP, Pastan I, Gottesman MM, Roninson IB. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell. 1986;47:381–389. doi: 10.1016/0092-8674(86)90595-7. [DOI] [PubMed] [Google Scholar]
- 13.Senior AE. Catalytic mechanism of P-glycoprotein. Acta Physiol Scand Suppl. 1998;643:213–218. [PubMed] [Google Scholar]
- 14.Sauna ZE, Smith MM, Muller M, Kerr KM, Ambudkar SV. The mechanism of action of multidrug-resistance-linked P-glycoprotein. J Bioenerg Biomembr. 2001;33:481–491. doi: 10.1023/a:1012875105006. [DOI] [PubMed] [Google Scholar]
- 15.Loo TW, Bartlett MC, Clarke DM. Drug binding in human P-glycoprotein causes conformational changes in both nucleotide-binding domains. J Biol Chem. 2003;278:1575–1578. doi: 10.1074/jbc.M211307200. [DOI] [PubMed] [Google Scholar]
- 16.Sharom FJ, Lugo MR, Eckford PD. New insights into the drug binding, transport and lipid flippase activities of the p-glycoprotein multidrug transporter. J Bioenerg Biomembr. 2005;37:481–487. doi: 10.1007/s10863-005-9496-6. [DOI] [PubMed] [Google Scholar]
- 17.Sauna ZE, Ambudkar SV. About a switch: how P-glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work. Mol Cancer Ther. 2007;6:13–23. doi: 10.1158/1535-7163.MCT-06-0155. [DOI] [PubMed] [Google Scholar]
- 18.Dawson RJP, Hollenstein K, Locher KP. Uptake or extrusion: crystal structures of full ABC transporters suggest a common mechanism. Mol Microbiol. 2007;65:250–257. doi: 10.1111/j.1365-2958.2007.05792.x. [DOI] [PubMed] [Google Scholar]
- 19.Ward A, Reyes CL, Yu J, Roth CB, Chang G. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc Natl Acad Sci U S A. 2007;104:19005–19010. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verhalen B, Ernst S, Borsch M, Wilkens S. Dynamic ligand-induced conformational rearrangements in P-glycoprotein as probed by fluorescence resonance energy transfer spectroscopy. J Biol Chem. 2012;287:1112–1127. doi: 10.1074/jbc.M111.301192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verhalen B, Wilkens S. P-glycoprotein retains drug-stimulated ATPase activity upon covalent linkage of the two nucleotide binding domains at their C-terminal ends. J Biol Chem. 2011;286:10476–10482. doi: 10.1074/jbc.M110.193151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dawson RJP, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 23.Hollenstein K, Dawson RJP, Locher KP. Structure and mechanism of ABC transporter proteins. Curr Opin Struct Biol. 2007;17:412–418. doi: 10.1016/j.sbi.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 24.Sauna ZE, Kim IW, Ambudkar SV. Genomics and the mechanism of P-glycoprotein (ABCB1) J Bioenerg Biomembr. 2007;39:481–487. doi: 10.1007/s10863-007-9115-9. [DOI] [PubMed] [Google Scholar]
- 25.Ward AB, Szewczyk P, Grimard V, Lee CW, Martinez L, Doshi R, Caya A, Villaluz M, Pardon E, Cregger C, Swartz DJ, Falson PG, Urbatsch IL, Govaerts C, Steyaert J, Chang G. Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc Natl Acad Sci U S A. 2013;110:13386–13391. doi: 10.1073/pnas.1309275110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J, Jaimes KF, Aller SG. Refined structures of mouse P-glycoprotein. Protein Sci. 2014;23:34–46. doi: 10.1002/pro.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin MS, Oldham ML, Zhang Q, Chen J. Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature. 2012;490:566–569. doi: 10.1038/nature11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wen PC, Verhalen B, Wilkens S, McHaourab HS, Tajkhorshid E. On the origin of large flexibility of P-glycoprotein in the inward-facing state. J Biol Chem. 2013;288:19211–19220. doi: 10.1074/jbc.M113.450114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dawson RJP, Locher KP. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 2007;581:935–938. doi: 10.1016/j.febslet.2007.01.073. [DOI] [PubMed] [Google Scholar]
- 30.Velamakanni S, Yao Y, Gutmann DAP, van Veen HW. Multidrug transport by the ABC transporter Sav1866 from Staphylococcus aureus. Biochemistry. 2008;47:9300–9308. doi: 10.1021/bi8006737. [DOI] [PubMed] [Google Scholar]
- 31.Wise JG. Catalytic transitions in the human MDR1 P-glycoprotein drug binding sites. Biochemistry. 2012;51:5125–5141. doi: 10.1021/bi300299z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amaro RE, Baron R, McCammon JA. An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J Comput Aided Mol Des. 2008;22:693–705. doi: 10.1007/s10822-007-9159-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ivetac A, McCammon JA. Molecular recognition in the case of flexible targets. Curr Pharm Des. 2011;17:1663–1671. doi: 10.2174/138161211796355056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin J-H, Perryman AL, Schames JR, McCammon JA. Computational drug design accommodating receptor flexibility: the relaxed complex scheme. J Am Chem Soc. 2002;124:5632–5633. doi: 10.1021/ja0260162. [DOI] [PubMed] [Google Scholar]
- 35.Roberts E, Eargle J, Wright D, Luthey-Schulten Z. MultiSeq: unifying sequence and structure data for evolutionary analysis. BMC Bioinformatics. 2006;7:382. doi: 10.1186/1471-2105-7-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brewer FK, Follit CA, Vogel PD, Wise JG. In silico Screening for Inhibitors of P-Glycoprotein that Target the Nucleotide Binding Domains. Molecular pharmacology. 2014;86:716–726. doi: 10.1124/mol.114.095414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prajapati R, Sangamwar AT. Translocation mechanism of P-glycoprotein and conformational changes occurring at drug-binding site: Insights from multi-targeted molecular dynamics. Biochim Biophys Acta. 2014;1838:2882–2898. doi: 10.1016/j.bbamem.2014.07.018. [DOI] [PubMed] [Google Scholar]
- 39.Tsuruo T, Iida H, Tsukagoshi S, Sakurai Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer research. 1981;41:1967–1972. [PubMed] [Google Scholar]
- 40.Rogan AM, Hamilton TC, Young RC, Klecker RW, Jr, Ozols RF. Reversal of adriamycin resistance by verapamil in human ovarian cancer. Science. 1984;224:994–996. doi: 10.1126/science.6372095. [DOI] [PubMed] [Google Scholar]
- 41.Ozols RF, Cunnion RE, Klecker RW, Jr, Hamilton TC, Ostchega Y, Parrillo JE, Young RC. Verapamil and adriamycin in the treatment of drug-resistant ovarian cancer patients. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1987;5:641–647. doi: 10.1200/JCO.1987.5.4.641. [DOI] [PubMed] [Google Scholar]
- 42.Martin C, Berridge G, Mistry P, Higgins C, Charlton P, Callaghan R. The molecular interaction of the high affinity reversal agent XR9576 with P-glycoprotein. Br J Pharmacol. 1999;128:403–411. doi: 10.1038/sj.bjp.0702807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 44.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.MacKerell A, Jr, Bashford D, Bellott M, Dunbrack R, Jr, Evanseck J, Field M, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau F, Mattos C, Michnick S, Ngo T, Nguyen D, Prodhom B, Reiher W, III, Roux B, Schlenkrich M, Smith J, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics Studies of proteins. Journal of Physical Chemistry B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 46.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hornung T, Volkov OA, Zaida TM, Delannoy S, Wise JG, Vogel PD. Structure of the cytosolic part of the subunit b-dimer of Escherichia coli F0F1-ATP synthase. Biophys J. 2008;94:5053–5064. doi: 10.1529/biophysj.107.121038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su SJ, Windus TL, Dupuis M, Montgomery JA. General Atomic and Molecular Electronic-Structure System. Journal of Computational Chemistry. 1993;14:1347–1363. [Google Scholar]
- 50.Sale K, Sár C, Sharp KA, Hideg K, Fajer PG. Structural determination of spin label immobilization and orientation: a Monte Carlo minimization approach. J Magn Reson. 2002;156:104–112. doi: 10.1006/jmre.2002.2529. [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 52.Case DA, Cheatham TE, Darden T, 3rd, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 54.Jakalian A, Bush BL, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic Charges. AM1-BCC model: I. Method. Journal of Computational Chemistry. 2000;21:132–146. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 55.Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. Journal of computational chemistry. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 56.Engels M, Jacoby E, Krüger P, Schlitter J, Wollmer A. The T<-->R structural transition of insulin; pathways suggested by targeted energy minimization. Protein Eng. 1992;5:669–677. doi: 10.1093/protein/5.7.669. [DOI] [PubMed] [Google Scholar]
- 57.Schlitter J, Engels M, Krüger P. Targeted molecular dynamics: a new approach for searching pathways of conformational transitions. J Mol Graph. 1994;12:84–89. doi: 10.1016/0263-7855(94)80072-3. [DOI] [PubMed] [Google Scholar]
- 58.Russell RB, Barton GJ. Multiple protein sequence alignment from tertiary structure comparison: assignment of global and residue confidence levels. Proteins. 1992;14:309–323. doi: 10.1002/prot.340140216. [DOI] [PubMed] [Google Scholar]
- 59.Eargle J, Wright D, Luthey-Schulten Z. Multiple Alignment of protein structures and sequences for VMD. Bioinformatics. 2006;22:504–506. doi: 10.1093/bioinformatics/bti825. [DOI] [PubMed] [Google Scholar]
- 60.Spoelstra EC, Westerhoff HV, Pinedo HM, Dekker H, Lankelma J. The multidrug-resistance-reverser verapamil interferes with cellular P-glycoprotein-mediated pumping of daunorubicin as a non-competing substrate. Eur J Biochem. 1994;221:363–373. doi: 10.1111/j.1432-1033.1994.tb18748.x. [DOI] [PubMed] [Google Scholar]
- 61.Omote H, Al-Shawi MK. A novel electron paramagnetic resonance approach to determine the mechanism of drug transport by P-glycoprotein. J Biol Chem. 2002;277:45688–45694. doi: 10.1074/jbc.M206479200. [DOI] [PubMed] [Google Scholar]
- 62.Loo TW, Clarke DM. Do drug substrates enter the common drug-binding pocket of P-glycoprotein through “gates”? Biochem Biophys Res Commun. 2005;329:419–422. doi: 10.1016/j.bbrc.2005.01.134. [DOI] [PubMed] [Google Scholar]
- 63.Loo TW, Bartlett MC, Clarke DM. Transmembrane segment 7 of human P-glycoprotein forms part of the drug-binding pocket. Biochem J. 2006;399:351–359. doi: 10.1042/BJ20060715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Loo TW, Bartlett MC, Clarke DM. Transmembrane segment 1 of human P-glycoprotein contributes to the drug-binding pocket. Biochem J. 2006;396:537–545. doi: 10.1042/BJ20060012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Loo TW, Bartlett MC, Clarke DM. Suppressor mutations in the transmembrane segments of P-glycoprotein promote maturation of processing mutants and disrupt a subset of drug-binding sites. J Biol Chem. 2007;282:32043–32052. doi: 10.1074/jbc.M706175200. [DOI] [PubMed] [Google Scholar]
- 66.Loo TW, Bartlett MC, Clarke DM. Arginines in the first transmembrane segment promote maturation of a P-glycoprotein processing mutant by hydrogen bond interactions with tyrosines in transmembrane segment 11. J Biol Chem. 2008;283:24860–24870. doi: 10.1074/jbc.M803351200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Loo TW, Clarke DM. Mutational analysis of ABC proteins. Arch Biochem Biophys. 2008;476:51–64. doi: 10.1016/j.abb.2008.02.025. [DOI] [PubMed] [Google Scholar]
- 68.Loo TW, Bartlett MC, Clarke DM. Identification of residues in the drug translocation pathway of the human multidrug resistance P-glycoprotein by arginine mutagenesis. J Biol Chem. 2009;284:24074–24087. doi: 10.1074/jbc.M109.023267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Loo TW, Clarke DM. Drug rescue distinguishes between different structural models of human P-glycoprotein. Biochemistry. 2013;52:7167–7169. doi: 10.1021/bi401269m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ma J, Biggin PC. Substrate versus inhibitor dynamics of P-glycoprotein. Proteins. 2013;81:1653–1668. doi: 10.1002/prot.24324. [DOI] [PubMed] [Google Scholar]
- 71.Chufan EE, Kapoor K, Sim HM, Singh S, Talele TT, Durell SR, Ambudkar SV. Multiple transport-active binding sites are available for a single substrate on human P-glycoprotein (ABCB1) PLoS One. 2013;8:e82463. doi: 10.1371/journal.pone.0082463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Loo TW, Bartlett MC, Detty MR, Clarke DM. The ATPase activity of the P-glycoprotein drug pump is highly activated when the N-terminal and central regions of the nucleotide-binding domains are linked closely together. J Biol Chem. 2012;287:26806–26816. doi: 10.1074/jbc.M112.376202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shapiro AB, Ling V. Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur J Biochem. 1997;250:130–137. doi: 10.1111/j.1432-1033.1997.00130.x. [DOI] [PubMed] [Google Scholar]
- 74.Loo TW, Clarke DM. Identification of residues within the drug-binding domain of the human multidrug resistance P-glycoprotein by cysteine-scanning mutagenesis and reaction with dibromobimane. J Biol Chem. 2000;275:39272–39278. doi: 10.1074/jbc.M007741200. [DOI] [PubMed] [Google Scholar]
- 75.Loo TW, Clarke DM. Identification of residues in the drug-binding domain of human P-glycoprotein. Analysis of transmembrane segment 11 by cysteine-scanning mutagenesis and inhibition by dibromobimane. J Biol Chem. 1999;274:35388–35392. doi: 10.1074/jbc.274.50.35388. [DOI] [PubMed] [Google Scholar]
- 76.Loo TW, Clarke DM. Identification of residues in the drug-binding site of human P-glycoprotein using a thiol-reactive substrate. J Biol Chem. 1997;272:31945–31948. doi: 10.1074/jbc.272.51.31945. [DOI] [PubMed] [Google Scholar]
- 77.Loo TW, Clarke DM. Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. J Biol Chem. 2002;277:44332–44338. doi: 10.1074/jbc.M208433200. [DOI] [PubMed] [Google Scholar]
- 78.Loo TW, Bartlett MC, Clarke DM. Simultaneous binding of two different drugs in the binding pocket of the human multidrug resistance P-glycoprotein. J Biol Chem. 2003;278:39706–39710. doi: 10.1074/jbc.M308559200. [DOI] [PubMed] [Google Scholar]
- 79.Loo TW, Bartlett MC, Clarke DM. Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein. Direct evidence for the substrate-induced fit mechanism for drug binding. J Biol Chem. 2003;278:13603–13606. doi: 10.1074/jbc.C300073200. [DOI] [PubMed] [Google Scholar]
- 80.Kannan P, Telu S, Shukla S, Ambudkar SV, Pike VW, Halldin C, Gottesman MM, Innis RB, Hall MD. The “specific” P-glycoprotein inhibitor Tariquidar is also a substrate and an inhibitor for breast cancer resistance protein (BCRP/ABCG2) ACS chemical neuroscience. 2011;2:82–89. doi: 10.1021/cn100078a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Loo TW, Clarke DM. Tariquidar inhibits P-glycoprotein drug efflux but activates ATPase activity by blocking transition to an open conformation. Biochem Pharmacol. 2014;92:558–566. doi: 10.1016/j.bcp.2014.10.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.