

Abstract
The steroidogenic acute regulatory protein-related lipid transfer (START) domain family is defined by a conserved 210-amino acid sequence that folds into an α/β helix-grip structure. Members of this protein family bind a variety of ligands, including cholesterol, phospholipids, sphingolipids, and bile acids, with putative roles in nonvesicular lipid transport, metabolism, and cell signaling. Among the soluble START proteins, STARD4 is expressed in most tissues and has previously been shown to transfer sterol, but the molecular mechanisms of membrane interaction and sterol binding remain unclear. In this work, we use biochemical techniques to characterize regions of STARD4 and determine their role in membrane interaction and sterol binding. Our results show that STARD4 interacts with anionic membranes through a surface-exposed basic patch and that introducing a mutation (L124D) into the Omega-1 (Ω1) loop, which covers the sterol binding pocket, attenuates sterol transfer activity. To gain insight into the attenuating mechanism of the L124D mutation, we conducted structural and biophysical studies of wild-type and L124D STARD4. These studies show that the L124D mutation reduces the conformational flexibility of the protein, resulting in a diminished level of membrane interaction and sterol transfer. These studies also reveal that the C-terminal α-helix, and not the Ω1 loop, partitions into the membrane bilayer. On the basis of these observations, we propose a model of STARD4 membrane interaction and sterol binding and release that requires dynamic movement of both the Ω1 loop and membrane insertion of the C-terminal α-helix.
Sterols are a critical component of eukaryotic cell membranes. In mammalian cells, there is an approximately 7-fold range of cholesterol content in various organelles.1,2 For instance, cholesterol accounts for ∼35% of the total lipids in the plasma membrane3,4 and is highly enriched in the endocytic recycling compartment (ERC).5 In comparison, in the endoplasmic reticulum (ER) where cholesterol is synthesized, cholesterol accounts for ∼5% of the total lipids.6 In part, the distribution of cholesterol among cellular organelles can be attributed to its differential stability in various membranes,7,8 and this distribution is maintained by vesicular and nonvesicular transport mechanisms.8,9
Several lines of evidence indicate that nonvesicular transport mediated by sterol transfer proteins plays an important role in maintaining the correct distribution of cholesterol among organelles.5,7,10 There are several protein families that are classified as lipid transfer proteins that can transfer lipids among membranes.11 One such family is the steroidogenic acute regulatory (StAR) protein-related lipid transfer (START) domain (STARD) family.9,12
Bioinformatic studies have identified START domains in the genomes of plants, protists, bacteria, and animals.13,14 In plants, START domains are highly prevalent and are often found in tandem with homeodomain proteins, suggesting a role in regulating gene expression.14,15 The protein architecture of the START homeodomain is unique to plants.14 In vertebrates, the START domains are often linked to other motifs to form multidomain proteins, and these START domains play a role in protein localization, enzymatic activity, and cellular signaling.13
There are 15 members of the mammalian START family12,16 that can be classified into six subfamilies based on domain architecture and ligand binding.13 In general terms, there are the cholesterol/oxysterol binding proteins (STARD1/3 subfamily), soluble proteins (STARD4/5/6 subfamily), phospholipid and sphingolipid binding proteins {STARD2 [phosphatidylcholine transfer protein (PCTP)]/7/8/10/11 subfamily}, putative Rho-GTPase signaling proteins (STARD8/12/13 subfamily), thioesterase activity-containing proteins (STARD14/15 subfamily), and the STARD9 subfamily composed of a single member whose function and ligand remain unclear.
Among the START domain members, the STARD4 subfamily members are the only START proteins that lack a subcellular localization motif or domain and are thought to be distributed throughout the cytosol,9 with some preference for binding organelles. This family is composed of STARD4, -D5, and -D6. STARD4 and STARD5 are widely expressed, while STARD6 expression is limited to testes, with the highest level being in spermatids.17 Overexpression of STARD4 has been shown to increase the extent of cholesteryl ester accumulation in lipid droplets, in an acetyl-CoA:cholesterol acyl-transferase (ACAT)-dependent manner,18,19 and its expression is controlled at the transcriptional level by sterols.10,17 STARD4 has been implicated as an important sterol transport protein involved in maintaining cholesterol homeostasis.10,19
The crystal structures of human STARD3, murine STARD4, and human phosphatidylcholine transfer protein (PCTP/STARD2) were among the first of the START proteins to be described.20−22 The structures of these proteins showed an α/β helix-grip fold with an internal binding pocket, formed by β-sheets, where ligand binds.23 However, access to the internal binding pocket is occluded by the domain’s C-terminal α-helix and adjacent loops.20 Conformational changes in these regions following membrane interaction may facilitate ligand entry and exit.16
As there are no crystal structures of STARD proteins bound to cholesterol, models of cholesterol in complex with several START domain proteins have been proposed from molecular docking and molecular dynamic simulations.20,24−26 In one model, the C-terminal α-helix can undergo a small and reversible local unfolding that is independent of the rest of the domain.24,27,28 In this intermediate state, the cholesterol binding site would become accessible, facilitating ligand binding and release. When cholesterol is bound in the pocket, the C-terminal α-helix would refold to form a stable protein that could diffuse in the cytosol to deliver cholesterol to the target organelle membrane.28 Consistent with this model, disulfide bridges cross-linking the C-terminal α-helix to the loop between β-strands 1 and 2 result in attenuated cholesterol binding and steroidogenic activity of STARD1.29 Murcia et al.25 proposed an alternative mechanism for START domain cholesterol binding and release based on molecular dynamic simulations. Following docking of cholesterol into the STARD3 structure or STARD1 model, cholesterol was released from the ligand binding pocket through a path created by conformational movement of the Omega-1 (Ω1) loop (Figure 1A, green). Consistent with this model, crystallographic data from STARD4 and STARD11 have showed high B values for the Ω1 loop,21,30 suggesting structural flexibility. However, the precise molecular mechanisms that mediate START domain protein membrane interaction and sterol extraction remain unclear.
Figure 1.

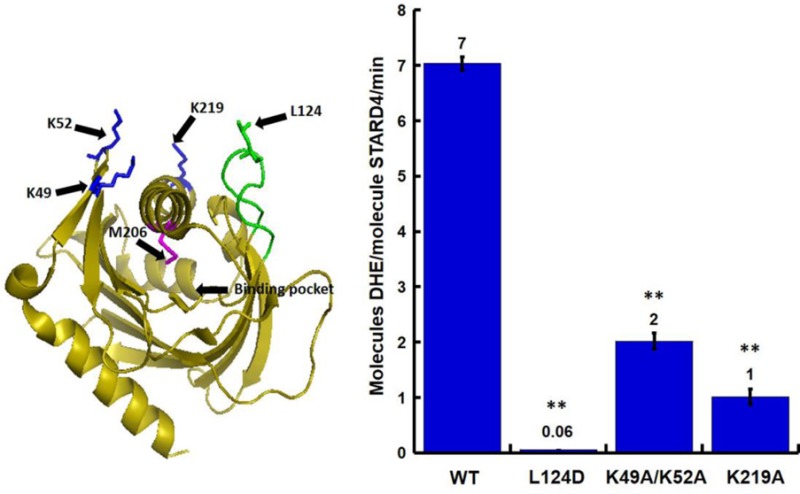
Regions of STARD4 involved in membrane interactions. (A) Ribbon representation of STARD4 in which positively charged K49/52 and K219 (blue), M206 (magenta), and Omega-1 (Ω1) loop (green) are featured. Individual residues are identified according to the sequence of STARD4. (B) Surface representation of STARD4 colored according to the electrostatic potential.21 Structures are rotated 90° about the z-axis for a view of the sterol binding pocket of STARD4. Structures were created in PyMOL (DeLano Scientific, Palo Alto, CA).
In this study, we investigate the mechanism of interaction of STARD4 with membranes and the mechanism by which STARD4 binds and releases sterol. We examined the role of specific regions of STARD4, either by replacement of surface-exposed lysine residues or by replacement of leucine with aspartic acid at the apex of the Ω1 loop to determine if these regions are required for activity. Utilizing structural and biochemical methods, we report that mutation of the Ω1 loop results in a large reduction in the level of membrane interaction and sterol transfer activity. We show that the attenuated activity of this mutation is not the result of a structural alteration of the protein but stems from stabilization of a closed or inactive conformation. Additionally, we show that the Ω1 loop does not insert into the membrane bilayer. Instead, we provide evidence that insertion of the carboxyl-terminal helix into the bilayer is associated with membrane interaction and sterol exchange with the membrane. Our findings suggest a unifying model of interactions of STARD protein with membrane, and sterol binding and release that require dynamic movement of the Ω1 loop and membrane insertion of the C-terminal α-helix.
Materials and Methods
Wild-Type and Mutant cDNA Constructs
The cDNAs encoding wild-type, K49A/K52A, K219A, and L124D STARD4 were subcloned into the pET-SUMO vector (Invitrogen).10,31 L124C and M206C STARD4 were generated using site-directed mutagenesis. Briefly, the resulting STARD4 contains an N-terminally fused hexahistidine (6-his)-tagged yeast SUMO protein for enhanced solubility.
Purification of Wild-Type and Mutant mSTARD4 Constructs
STARD4 and STARD4 mutants (K49A/K52A, K219A, M206C, L124C, and L124D) in the pET-SUMO vector were expressed in Escherichia coli BL21(DE3) cells during overnight incubation at 18 °C. Bacterial pellets were resuspended in 20 mM HEPES (pH 7.2), 100 mM KCl, 20 mM imidazole, 1 mM TCEP, and 0.1% IGEPAL supplemented with an anti-protease cocktail (Roche) and 1 mM phenylmethanesulfonyl fluoride. The resuspended cell pellets were lysed on ice by sonication followed by ultracentrifugation at 100000g for 1 h. The supernatant was incubated with pre-equilibrated Ni-NTA resin under constant agitation at 4 °C for 1 h. Following incubation, the supernatant/resin slurry was passed through a column, and the column was washed with 20 mM HEPES (pH 7.2), 100 mM KCl, 20 mM imidazole, and 1 mM tris(2-carboxyethyl)phosphine (TCEP). The SUMO–STARD4 protein was eluted with 20 mM HEPES (pH 7.2), 100 mM KCl, 0.5 M imidazole, and 1 mM TCEP. The eluted protein was dialyzed overnight at 4 °C in 20 mM HEPES (pH 7.2), 100 mM KCl, and 1 mM DTT in the presence of the Ulp1 protease to remove the His-SUMO tag. STARD4 was further purified using size exclusion chromatography using a Superdex200 column (GE Healthcare) in 20 mM HEPES (pH 7.2), 100 mM KCl, and 1 mM DTT. Purified protein was stored at −80 °C.
Liposomes
Lipids in chloroform were purchased from Avanti Polar Lipids (Alabaster, AL), except dehydroergosterol (DHE) (powder, Sigma). A dried film was prepared by evaporation of a mixture of the indicated lipids in chloroform. Lipids were hydrated in 50 mM HEPES (pH 7.2) and 120 mM potassium acetate by five freeze–thaw cycles. The suspension was extruded sequentially 11 times through 0.4 and 0.1 μm (pore size) polycarbonate filters using a hand extruder (Avanti) at a final lipid concentration of 1 mM. Liposomes were stored at room temperature, protected from light, and used within 4 days. Liposomes termed “donors” or “acceptors” were used in a sterol transfer assay. The composition of donor liposomes was 31 mol % 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (POPC), 23 mol % 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine (POPE), 23 mol % 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylserine (POPS), and 23 mol % DHE. The composition of acceptor liposomes was 70 mol % POPC, 7 mol % POPE, 15 mol % liver phosphatidylinositol (PI), 5 mol % POPS, and 3 mol % dansyl-phosphatidylethanolamine (PE). The composition of brominate donor lipids was 31 mol % 1-palmitoyl-2-(6,7-dibromo)stearoyl-sn-glycero-3-phosphocholine (6,7-Br PC), 23 mol % POPE, 23 mol % POPS, and 23 mol % cholesterol
Sterol Transfer Assay
The sterol transport activity of STARD4 was measured by a Forster resonance energy transfer (FRET) assay, as previously described.10 Experiments were performed in quartz cuvettes (100 μL) in HK buffer [50 mM HEPES (pH 7.2) and 120 mM potassium acetate] equilibrated at 37 °C on a SpectraMax M2 fluorometer (MDS Analytical Technologies, Sunnyvale, CA). FRET traces were fit to a single exponential. Data represents averages [±the standard error of the mean (SEM)] of at least three independent experiments.
Crystallization
Purified L124D STARD4 in 20 mM HEPES (pH 7.2), 100 mM KCl, and 1 mM DTT was concentrated to 10 mg/mL, and crystals were grown using hanging drop vapor diffusion in a 1:1 protein:well solution ratio. Full size crystals were harvested after 2 days in 0.1 M MES (pH 6.2) and 2.2 M ammonium sulfate and flash-frozen in liquid nitrogen with 20% (v/v) glycerol as a cyroprotectant.
Data Collection, Structure Determination, and Refinement
X-ray scattering data were collected at beamline X29 at Brookhaven National Laboratory (Upton, NY). Crystals diffracted to 2.0 Å resolution, and all data were indexed, integrated, and scaled using HKL2000.32 The STARD4 wild-type crystal structure [Protein Data Bank (PDB) entry 1JSS(21)] was used as a molecular replacement model to phase the L124D STARD4 structure using Phaser-MR.33 The initial model was subjected to several rounds of model building and refinement using COOT34 and Phenix.refine,35 respectively. Data collection statistics and the final data refinement statistics are listed in Table 1. The coordinates and structure factors have been deposited as PDB entry 5BRL.
Table 1. Data Collection and Refinement Statistics of L124D mSTARD4.
| Data Collection | |
| space group | P21 |
| cell dimensions | |
| a, b, c (Å) | 47.4, 41.7, 121.5 |
| α, β, γ (deg) | 90, 90.02, 90.00 |
| no. of molecules per asymmetric unit | 2 |
| wavelength (Å) | 1.075 |
| resolution (Å) | 50–2.00 |
| Rmerge (last shell) | 0.065 (0.19) |
| I/σ(I) | 16.2 (5.8) |
| completeness (%) | 80.6 (84.9) |
| redundancy | 2.5 (2.4) |
| Refinement | |
| resolution (Å) | 39.4–2.0 |
| Rwork/Rfree (%) | 18.8/22.6 |
| no. of residues | 424 |
| no. of waters | 195 |
| mean B factor (Å2) | 17.9 |
| no. of residues per backbone | 19.43 |
| root-mean-square deviation | |
| bond lengths (Å) | 0.007 |
| bond angles (deg) | 1.050 |
| Ramachandran plot (%) | |
| favored | 95.9 |
| additional allowed | 4.1 |
| disallowed | 0 |
NMR Spectroscopy
Isotopically labeled wild-type and L124D STARD4 were produced using the media-swap method36 and purified using identical methods. Purification of isotopically labeled protein was the same as for the natural abundance protein, after which the protein was exchanged into 20 mM NaCl, 20 mM Tris, 5 mM DTT, pH 6.4 buffer, supplemented with 10% D2O, which was used for NMR experiments. Backbone amide assignments for the wild-type protein were transferred from previously published assignments for the L124D protein31 using an HNCA experiment.
Wild-type and L124D StARD4 (final concentration of 50 μM) were incubated with 6 mM total lipids meant to mimic a cholesterol-rich donor membrane (31 mol % POPC, 23 mol % POPE, 23 mol % POPS, and 23 mol % cholesterol). 1H–15N HSQC spectra were collected using a 600 MHz Varian Inova instrument with a cryogenically cooled probe using the same acquisition parameters on concentration-matched samples with and without liposomes. The peak intensity was measured for 25 well-resolved cross-peaks arising from residues 13, 24, 32, 41, 50, 56, 66, 76, 80, 93, 99, 109, 119, 128, 139, 146, 150, 160, 173, 181, 184, 194, 200, 208, and 216 in all spectra, and the ratio of peak intensity in lipid-containing spectra to peak intensity in lipid-free spectra was calculated. NMR data were processed with NMRpipe37 and analyzed with NMRViewJ.38
Membrane Interaction by Size Exclusion Chromatography
Wild-type and L124D STARD4 at final concentrations of 20 μM were examined in the absence and presence of liposomes by using size exclusion chromatography using a Superdex200 column (GE Healthcare) in 20 mM HEPES (pH 7.2), 100 mM KCl, and 1 mM DTT. In the presence of lipids, STARD4 was incubated with 15 mM total lipids, meant to mimic a cholesterol-rich donor membrane (31% POPC, 23% POPE, 23% POPS, and 23% cholesterol). Chromatograms are normalized to the major peak for each fractionation. The resulting chromatograms represent two independent experiments.
Iodoacetamide-NBD (IANBD) Labeling of STARD4
L124C and M206C STARD4 were labeled in 50 mM Tris (pH 7.2) and 150 mM KCl with a 100-fold molar excess of IANBD for 120 min at room temperature under agitation and protected from light. After incubation, excess dye was removed by passage through a Sephadex G-10 column that had been pre-equilibrated in HK buffer. The NBD concentration was determined by absorbance at 480 nm using a molar extinction coefficient of 25000 M–1 cm–1.39 The STARD4 concentration was determined using a bicinchoninic acid (BCA) assay, using BSA as a standard (Thermo Scientific Pierce). The labeling resulted in 0.8–0.9 mol of NBD/mol of protein.
Membrane Penetration
Membrane penetration assays were performed in HK buffer equilibrated at 37 °C on a SpectraMax M2 fluorometer (MDS Analytical Technologies, Sunnyvale, CA). NBD-labeled proteins were mixed with 100 μM total lipid cholesterol-rich donor membrane (31% POPC/Br-PC, 23% POPE, 23% POPS, and 23% cholesterol) in a 100 μL quartz cuvette. NBD was excited at 470 nm, and the emission spectra were collected from 490 to 630 nm and corrected for buffer, unlabeled protein, and lipids alone. The spectra were representative of at least two independent experiments (Figure 6).
Figure 6.

Ω1 loop does not insert into the bilayer. Membrane penetration assay of L124C and M206C STARD4 using POPC (A and B) or Br-PC (C and D) liposomes. The NBD fluorescence spectrum of each STARD4 mutant (0.5 μM) was collected in the absence or presence of liposomes. NBD was excited at 470 nm, and the emission spectra were collected from 490 to 630 nm and corrected for buffer, unlabeled protein, and lipids alone.
Maleimide-PEG Labeling
Maleimide-PEG 5 kDa (MP-5 kDa) modification experiments were performed in 50 mM Tris (pH 7.2) and 150 mM KCl. Wild-type and L124D STARD4 were incubated with a 50-fold molar excess of MP-5 kDa, and at various times, the reactions were quenched by the addition of 10 mM β-mercaptoethanol. Proteins were resolved on a 12% HEPES-Bicine gel and stained with Coomassie blue. As a positive control, wild-type and L124D STARD4 were denatured with 2% (w/v) SDS in the presence of MP-5 kDa and examined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE).
Far-UV and Thermal Melting Circular Dichroism (CD)
Far-UV and thermal melting circular dichroism experiments were performed on an AVIV Biomedical model 410 CD spectrometer using bundled software. Far-UV CD spectra from 200 to 250 nm, with a wavelength step of 0.1 nm and an averaging time of 1.7 s, were recorded at 25 °C on 100 μM STARD4 or 100 μM L124D STARD4. The cell path length was 0.02 cm, and four scans for each sample were averaged. Thermal melting curves were recorded at 222 nm in the temperature range from 5 to 95 °C with a heating rate of 1 °C/min on 100 μM STARD4, 100 μM L124D STARD4, or 100 μM STARD4–DHE complex. The raw millidegree values were transformed to mean residue molar ellipticity (degrees squqare centimeter per decimole).40 The CD spectra have been deposited in the Protein Circular Dichroism Data Bank (http://pcddb.cryst.bbk.ac.uk) as entries CD0004865000 and CD0004866000.
Formation of the STARD4–DHE Complex
STARD4 (1 μM) was mixed with 500 μM (total lipids) large sucrose-loaded liposomes [210 mM sucrose and 50 mM HEPES (pH 7.2)] for 15 min at 37 °C in HK buffer. The sample was centrifuged at 400000g for 20 min at 4 °C in a fixed-angle rotor (TLA 100.1, Beckman). The supernatant was recovered and concentrated to 100 μM for thermal melting circular dichroism. The DHE concentration was determined by absorbance using a molar extinction coefficient of 10000 M–1 cm–1 at 325 nm.41 The STARD4 concentration was determined using a BCA assay, using BSA as a standard (Thermo Scientific Pierce). Control centrifugations performed without liposomes indicated that STARD4 did not sediment. The molar concentration of DHE was 90–95% of the STARD4 concentration.
Results
Multiple Regions of STARD4 May Dictate Function
Previous molecular dynamics simulations of START proteins suggested that movement of the nonpolar Ω1 loop would be sufficient to allow sterol absorption and release. In addition, the crystal structure of STARD4 shows a cluster of positively charged residues (Figure 1A,B) that can interact with anionic lipids, such as phosphatidylserine, to facilitate membrane docking. Given the proximity of the polybasic patch (Figure 1A, blue) and the Ω1 loop (Figure 1A, green), it is possible that they work in tandem to ensure efficient sterol transfer to and from membranes. To understand the role of these regions, we characterized mutations in the polybasic patch and the Ω1 loop, using a liposomal sterol transfer assay depicted in Figure 2A. Donor liposomes (D) containing the fluorescent cholesterol analogue dehydroergosterol (DHE) and acceptor liposomes (A) containing dansyl-phosphatidylethanolamine are mixed with purified protein. The delivery of sterol from donor to acceptor membranes results in a sensitized Forster resonance energy transfer (FRET) signal when the two fluorescent lipids are in the same liposome. As previously reported, STARD4 is an efficient sterol transporter10 that is ∼1000-fold more effective than the cyclic heptasaccharide sterol carrier, methyl-β-cyclodextrin (Figure 2B).
Figure 2.

Mutation of STARD4 regions perturbs sterol transfer activity. (A) DHE transfer assay. The FRET signal was used to measure the transfer of DHE (red block) from donor (D) to acceptor (A) liposomes. (B) A total lipid concentration of 100 μM of donor and acceptor liposomes was incubated alone (no STARD4) or with 1 μM wild-type STARD4 at 37 °C. Methyl-β cyclodextrin (1 mM) was used as a positive control. (C) Representative DHE transfer assay traces using STARD4 point mutations in either the polybasic region (1 μM K49A/K52A or K219A) or Ω1 loop (1, 5, and 100 μM L124D). Donor liposomes contained POPC, POPE, POPS, and DHE, and acceptor liposomes contained POPC, POPE, POPS, PI, and dansyl-PE. (D) Quantification of the number of DHE molecules transferred per molecule of STARD4 per minute, as previously described.10 Error bars ± SEM from three experiments (**p < 0.005). The number above each bar is the average number of DHE molecules transferred per molecule of STARD4 per minute.
To determine the role of the polybasic patch and Ω1 loop (Figure 1A,B) in STARD4 activity, we initially characterized in more detail mutations that were previously described10,31 in which surface-exposed lysines (Figure 1A, blue) were replaced with alanines. Figure 2C shows that K49A/K52A and K219A mutations reduced the sterol transport activity compared to that of the wild type. The replacement of positive charge in the polybasic patch results in approximately 25 and 85% reductions in activity for K49A/K52A and K219A, respectively (Figure 2D).
To evaluate the role of the Ω1 loop in STARD4 sterol transport, we replaced a leucine with an aspartic acid residue, L124D, at the apex of the Ω1 loop (Figure 1A, green). From its nonpolar characteristic, we anticipated that the Ω1 loop might insert into the bilayer for sterol binding/release, and introduction of a negative charge should attenuate such activity. Panels C and D of Figure 2 show that the L124D mutation almost completely blocks STARD4 activity. These data indicate that the polybasic patch and Ω1 loop both contribute to STARD4 activity.
Reduced Activity of L124D STARD4 Is Not Due to Charge Repulsion
As previously reported, STARD4 activity is increased when sterol is transported between liposome populations that are enriched with anionic lipids.10 Because the assays in panels C and D of Figure 2 were performed with negatively charged liposomes, the reduced activity of L124D might result from charge–charge repulsion. We replaced the anionic lipids of donor and acceptor membranes with the zwitterionic lipid, phosphatidylcholine (PC), to generate neutral donor and acceptor liposomes (Figure 3). As expected,31 the activity of wild-type STARD4 was attenuated, resulting in a 50% reduction in activity (Figure 3). Interestingly, the absence of anionic lipids did not restore activity to L124D STARD4. This indicates that the reduction in sterol transport activity from the L124D mutation is not due to charge–charge repulsion.
Figure 3.

L124D STARD4 reduced activity is not due to charge repulsion. Anionic (red) or neutral (blue) liposomes (total lipid concentration of 100 μM) were incubated alone (no STARD4) or with 0.25–2 μM wild-type STARD4 at 37 °C. The number of DHE molecules transferred per molecule of STARD4 per minute was measured as described previously.10 Anionic liposomes have a composition identical to that of those used in Figure 2. In the nonanionic liposomes, POPE and POPE/PI were replaced by POPC. Error bars ± SEM from three experiments (*p < 0.05 compared to the wild-type value). The number above each bar is the average number of DHE molecules transferred per molecule of STARD4 per minute.
Crystal Structure of L124D STARD4
One potential explanation for L124D STARD4 reduced sterol transport activity would be protein misfolding. We investigated the secondary structure of the L124D and lysine to alanine mutations using UV circular dichroism (CD) (Figure S1 of the Supporting Information). The CD spectrum of all STARD4 mutants was similar to the wild-type spectrum, indicating that the mutations did not result in large structural perturbations. This did not eliminate the possibility that subtle local alterations could result in attenuated activity. Because the L124D mutation caused a large perturbation of sterol transfer activity, we obtained the crystal structure of L124D STARD4 by molecular replacement at 2.0 Å resolution (Table 1). The final refined structure had two molecules of L124D STARD4 per asymmetric unit with visible residues 13–222 in chain A and 13–223 in chain B. Both the N- and C-termini have one α-helix that encloses nine antiparallel β-sheets that comprise the protein core (Figure 4A,B). The root-mean-square deviation (rmsd) between the two molecules in the asymmetric unit is 0.061 Å2 over 180 residues. The rmsd between the wild-type and L124D mutant structures for chain A is 0.372 Å2 over 157 residues, while for chain B, it is 0.358 Å2 over 152 residues. Deviations in the structures are mainly in the loops, with the loops comprising residues 39–41, 75–79, 140–145, and 167–171 having the most significant shifts (Figure 4C). However, overall there are only minor differences observed between the wild-type and L124D structures (Figure 4B), indicating the attenuation of sterol transfer activity is not like to be the result of structural alteration.
Figure 4.

Crystal structure of L124D STARD4. (A) Ribbon representation of L124D STARD4 in an asymmetric unit with labeled N- and C-termini. (B) Ribbon representation of L124D (green) and wild-type STARD4 (red, PDB entry 1JSS(21)) overlay, with labeled N- and C-termini and Ω1 loop and L124D. (C) Plots of deviations for L124D STARD4 chains A and B as compared to wild-type STARD4. All calculations were conducted using wild-type STARD4 (PDB entry 1JSS) as the reference. rmsd comparison over residues 13–222 in chain A and residues 13–223 in chain B. Large deviations are observed in the loops, with the loop comprising residues 39–41, 75–79, 140–145, and 167–171 having significant shifts in rmsd values.
L124D STARD4 Has Weakened Interaction with Membranes
Because the crystal structure of L124D STARD4 did not show any significant alteration compared to the wild-type structure (Figure 4), we evaluated if mutation of the Ω1 loop perturbed the STARD4–membrane interaction. To do this, we utilized nuclear magnetic resonance (NMR) spectroscopy and collected 1H–15N heteronuclear single-quantum coherence (HSQC) spectra on mutant and wild-type STARD4 in the absence and presence of liposomes. In the absence of liposomes (black spectrum), both wild-type (Figure 5A) and L124D (Figure 5B) STARD4 have well-resolved 1H–15N HSQC spectra, indicating that both proteins are well-folded. In the presence of large unilamellar liposomes (red spectrum, Figure 5C), the wild-type STARD4 spectrum shows a significant loss of intensity among the amide cross-peaks compared to wild-type STARD4 in the absence of lipids (Figure 5A). The loss of signal is likely due to the formation of STARD4–liposome complexes, which are large and tumble too slowly to give rise to an NMR signal. Line broadening due to intermediate conformational exchange between lipid-free and lipid-bound states of the wild-type protein is unlikely because the full widths at half-maximum of peaks in the lipid-containing and lipid-free spectra were approximately the same. A decrease in intensity, as quantified by the ratio of peak intensity in the lipid-containing to lipid-free spectra, was relatively homogeneous throughout the protein sequence, suggesting that wild-type STARD4 binds liposomes as a concerted unit (data not shown). Strikingly, the amide cross-peaks in the spectrum for L124D in the presence of liposomes are not attenuated (red spectrum, Figure 5D). Assuming that all bound protein gives rise to no NMR signal and thus residual peak intensity arises from the fraction of free protein, the extent of binding can be estimated by calculating the ratio of peak intensities in the lipid-containing and lipid-free samples. The peak intensity ratios for 25 well-resolved resonances arising from residues throughout the primary sequence of both wild-type and L124D STARD4 were calculated and averaged (Figure 5E). In the presence of 6 mM total lipids, the intensity of the 50 μM wild-type STARD4 signal is reduced by approximately 80% while that of the 50 μM L124D signal is not affected, strongly suggesting that a majority of wild-type STARD4 molecules interact with the liposomes while those of L124D STARD4 do not.
Figure 5.

L124D STARD4 has weakened interactions with membranes. HSQC spectra of uniformly 15N-labeled wild-type STARD4 in the absence (A, black) and presence (C, red) of donor liposomes (total lipid concentration of 6 mM). HSQC spectra of L124D STARD4 in the absence (B, black) and presence (D, red) of 6 mM total donor lipids. (E) Ratio of signal intensity in the presence of 6 mM total donor lipids compared to the intensity in the absence of liposomes, averaged over 25 well-resolved amide resonances, for 50 μM wild-type and L124D STARD4. A loss of signal intensity in the presence of liposomes corresponds to binding to the liposomes, within the error of the NMR measurement. (F) Size exclusion chromatography plot for wild-type (left) or L124D (right) STARD4 in the absence (red) or presence (blue) of 15 mM total lipid.
To further examine STARD4–membrane interaction, we utilized size exclusion chromatography to monitor the elution of profile of 20 μM wild-type and L124D proteins in the absence and presence of 15 mM total lipids (Figure 5F).42 In the absence of lipids (red), both wild-type and L124D STARD4 elute as a prominent single peak around 20 mL, with L124D eluting later than the wild type. This elution profile is consistent with a protein monomer with a molecular weight of ∼25 kDa. Interestingly, after incubation with lipids (blue), the majority of the wild-type protein elutes at ∼10 mL, with a portion eluting at ∼20 mL, while there is only a small shift for the L124D mutant. This shift in the elution profile indicates that the majority of wild-type protein has interacted with the lipids and elutes earlier because of that interaction. However, this is not the case for L124D, which has only a minor shift in the elution profile, indicating that it may bind the liposomes weakly. Taken together, these results show that L124D STARD4 has a reduced capacity to interact with membranes.
The Ω1 Loop Does Not Insert into the Bilayer
One possibility is that the tip of the Ω1 loop directly inserts into the membrane bilayer, and the L124D mutation prevents this. To test this hypothesis, we monitored the fluorescence emission spectra of the environmentally sensitive fluorophore, (7-nitrobenz-2-oxa-1,3-diazol-4-yl)ethylenediamine (NBD) coupled to a cysteine at residue 124. To selectively label and monitor the Ω1 loop, we mutated all endogenous cysteines to serines and introduced a single cysteine at L124. L124C STARD4 is able to transfer sterol as efficiently as wild-type STARD4 (Figure S2A of the Supporting Information). Utilizing iodoacetamide-NBD (IANBD), we labeled L124C STARD4 and examined the fluorescence emission profile of L124C-IANBD in the absence and presence of liposomes (Figure 6A, red and blue). Interestingly, we did not observe any signal alteration. Additionally, L124C-IANBD STARD4 sterol transfer activity was 40% lower than those of L124C and wild-type STARD4, but with unaltered secondary structure (Figure S2A,B of the Supporting Information). This finding indicates that the Ω1 loop does not insert into the bilayer.
The carboxyl-terminal helix of STARD4 forms an amphipathic helix that might interact with the membrane.43−45 To investigate this, we mutated methionine 206 (Figure 1B, magenta) to a cysteine in the absence of other cysteines, labeled it with NBD, and asked if this M206C-IANBD inserted into the bilayer (Figure 6B). Similar to L124C, M206C is as active as wild-type STARD4 in sterol transfer (Figure S2A of the Supporting Information). In the absence of lipids, M206C-IANBD has an emission spectrum similar to that of L124C-IANBD (Figure 6B, green). However, following the introduction of liposomes, the emission spectrum intensity of M206C-IANBD increases ∼3-fold, indicating insertion of the NBD moiety into the nonpolar environment of the membrane (Figure 6B, black). However, while M206C-IANBD STARD4 shows insertion of the NBD moiety into the bilayer, it is properly folded but the sterol transfer activity was greatly reduced (Figure S2A,C of the Supporting Information). This is likely due to occlusion of the sterol binding pocket upon coupling of the cysteine at position 206 with the NBD.
To confirm that the alternation of NBD fluorescence was the result of insertion into the membrane bilayer rather than interactions in the sterol binding site, we replaced POPC with brominated PC (Br-PC) and monitored the fluorescence spectrum. As bromine is a collisional quencher of NBD,46 insertion of the NBD moiety will result in quenching of the fluorescence. Similar to POPC lipids (Figure 6A, red and blue), incubation of L124C-IANBD with Br-PC lipids did not alter the NBD signal (Figure 6C, red and blue). However, with M206C-IANBD, the presence of Br-PC prevented the increase in fluorescence upon interaction with liposomes, indicating membrane insertion (Figure 6D, green and black). Taken together, these findings indicate that the carboxyl-terminal helix and not the Ω1 loop inserts into the bilayer.
Mutation of the Ω1 Loop in STARD4 Stabilizes a Closed Conformation
To determine whether the L124D mutation affected the dynamics of the protein in solution, we analyzed the cysteine accessibility of wild-type and L124D STARD4 using a maleimide-PEG molecular weight 5 kDa (MP-5 kDa) probe (Figure 7A).47,48 Of the five cysteines in STARD4 (C112, C113, C148, C169, and C173), only C112 and C113 are solvent accessible.21 Maleimide probes form a thioether linkage and can be quenched using excess reducing agent, such as β-mercaptoethanol.49,50 STARD4 cysteine modification by MP-5 kDa results in a molecular weight shift that is dependent on the number of alkylations per STARD4. To assess total cysteine accessibility, STARD4 was denatured with 2% SDS followed by incubation with MP-5 kDa (Figure 7B, right lanes). Time course examination of either wild-type or L124D STARD4 with MP-5 kDa shows several molecular weight shifts indicative of alkylation (Figure 7B). However, the L124D STARD4 alkylation pattern shows a slower rate of cysteine accessibility, which is evident in the delay in the formation of high-molecular weight species. These observations suggest that wild-type STARD4 may more readily adopt an “open” conformation in which movement of the protein facilitates access of the solvent to the sterol binding pocket, while L124D STARD4 may more stably adopt a closed or inactive conformation.
Figure 7.

Mutation of the Ω1 loop in STARD4 stabilizes a closed conformation. (A) Ribbon representation of STARD4 with the five cysteines colored magenta. (B) SDS–PAGE gel of the maleimide–PEG 5 kDa reaction with wild-type and L124D STARD4. Molecular weight shifts are the result of the nonhydrolyzable linkage between maleimide and the STARD4 thiol side chain. Reactions were conducted at 37 °C and quenched with β-mercaptoethanol prior to denaturing and electrophoresis. As a positive control, wild-type and L124D STARD4 were denatured with SDS and incubated for 60 min with maleimide-PEG 10 kDa.
Thermal Melting CD Spectroscopy of L124D Reveals Changes in Protein Stability
To validate that the L124D mutation stabilizes the protein’s conformation, we evaluated the thermal stability of STARD4 by determining the thermal melting (Tm) point for wild-type and L124D STARD4. The Tm of L124D STARD4 is increased compared to that of wild-type STARD4 (Figure 8A). This indicates that the attenuated activity of L124D STARD4 may be associated with a more stable conformation that cannot interact with the membrane to bind or release sterol. As there is a direct relationship between the stabilization free energy provided by binding to a ligand forming a closed or protected conformation,51 we investigated the thermal stability of wild-type STARD4 complexed to DHE (Figure 8B). Interestingly, the wild-type STARD4–DHE complex results in a Tm similar to that of L124D STARD4, indicating an increase in the level of structural stabilization following ligand binding. Unfortunately, we were unable to load L124D STARD4 with sterol. Thermally induced denaturations of the wild type, L124D, and STARD4–DHE complex were irreversible. Mechanistically, these data demonstrate that a reduction in the level of dynamic movement of the Ω1 loop results in a conformation more thermostable than that of wild-type STARD4. Moreover, formation of the wild-type STARD4–DHE complex results in a shift in the Tm, resulting in a more stable conformation compared to apo wild-type STARD4 and similar to L124D STARD4.
Figure 8.

CD spectroscopy of L124D reveals changes in protein stability. (A) Thermal melting curve recorded at 222 nm for wild-type and L124D STARD4. (B) Thermal melting curve of the wild-type STARD4–DHE complex and apo wild-type STARD4. The concentratiosn of the wild type, L124D, and STARD4–DHE complex are 100 μM. The STARD4–DHE complex was in 0.9–0.95 mol of DHE/mol of protein.
Discussion
Elucidating the molecular mechanisms that mediate STARD4 protein–membrane interaction and sterol extraction is crucial for understanding how these proteins facilitate sterol/lipid transport among cellular organelles.52,53 Sterol entry and exit from START domain proteins has been suggested to be mediated by either reversible local unfolding of the C-terminal α-helix or conformational movement of the Ω1 loop.25,28 Our data are most consistent with a unifying model in which movement of both of these regions of STARD4 is required. This model reconciles several biochemical and biophysical observations.30,45,54
STARD4 transfer of sterol between membranes is enhanced by a surface-exposed basic patch that facilitates interactions with anionic lipids (Figure 1).10 Replacement of lysine residues within the basic patch attenuates sterol transfer activity (Figure 2). The electrostatic interaction between STARD4 and anionic lipid headgroups could orient the protein into an optimal position to bind and release sterol. We considered that the apex of the Ω1 loop (Figure 1) might also be oriented toward the anionic lipids, so we sought to attenuate this interaction by introducing a negative charge into the Ω1 loop. As expected, introduction of negative charge into the apex of the Ω1 loop, L124D, reduced sterol transfer activity approximately 100-fold (Figure 2). However, this reduction in activity could not be rescued by the removal of anionic lipids for membranes (Figure 3), indicating that the reduced activity was not due simply to charge–charge repulsion.
In comparing the structure of the wild-type and L124D STARD4 proteins, there are minimal alterations, with differences occurring primarily in flexible loops (Figure 4). This indicates that L124D STARD4 is well-folded, and mutation of the Ω1 loop did not greatly perturb the protein structure. We then investigated whether the mutation attenuated the ability of STARD4 to interact with membranes (Figure 5). On the basis of the loss of the NMR signal in the presence of liposomes and the change in elution volume when liposomes were present during size exclusion chromatography, wild-type STARD4 bound to membranes but L124D STARD4 had greatly weakened membrane interaction. These findings suggested that the loss of sterol transfer activity of L124D STARD4 results from weakened membrane interaction.
It seemed possible that L124 at the apex of the Ω1 loop would be involved in membrane insertion, so we investigated this using the environmentally sensitive dye NBD (Figure 6). Interestingly, when NBD was placed at position 124 in the Ω1 loop, we did not observe alterations in the emission spectra in the presence or absence of liposomes. This indicates that the NBD moiety and Ω1 loop do not insert into the bilayer. As an alternative region that could potentially interact with the membrane bilayer, the C-terminal α-helix has been suggested to be involved in membrane interaction and sterol binding.29,45 When NBD was conjugated on the C-terminal α-helix at M206C, the NBD emission spectrum was increased in the presence of POPC liposomes, and this increase was blocked by Br-PC in the liposomes (Figure 6). In the crystal structure, M206 points into the sterol binding pocket (Figure 1A, magenta) and away from the cytosol. Insertion of the NBD into the lipid indicates that the C-terminal α-helix needs to swing out to insert into the bilayer. This rearrangement could be mediated in part by movement of the flexible loop region at the amino terminus of the C-terminal α-helix and by its amphipathic nature. However, these findings do not explain how mutation of L124D results in attenuated sterol transport and membrane interaction.
One potential mechanism by which mutation of the Ω1 loop could attenuate activity is reduction of the extent of dynamic movement of the protein.45 Inspection of the 1H–15N HSQC spectrum of the L124D mutant shows that some peaks are much more intense than in the wild-type spectrum. This is an indication that the wild type may be undergoing conformational exchange among conformations. We investigated this using maleimide chemistry to probe the accessibility of endogenous cysteines of STARD4 (Figure 7). Interestingly, L124D STARD4 has reduced reactivity with the maleimide-PEG compared to that of wild-type STARD4, suggesting that L124D STARD4 had a reduced level of dynamic movement. Furthermore, this demonstrates that STARD4 undergoes structural transitions by which the maleimide-PEG gains access to the core of the protein to react with the cysteine residues and that this is greatly reduced with the L124D mutation. This stabilization was further verified using circular dichroism in which the L124D mutation resulted in an increase in thermal stability (Figure 8). Because the structure of the L124D mutant is nearly identical to that of the wild-type protein and because the L124 side chain is surface-exposed and forms no tertiary interactions, the overall stabilizing effect of this mutation may result from a destabilization of either the unfolded state of the protein or some transient but essential unfolding intermediate or transition state. Such an intermediate could indeed be key for allowing the protein to bind to membranes.
Taken together, these findings indicate that reductions in the level of STARD4–membrane interaction and sterol transfer activity of the L124D mutation are a consequence of a reduction in the conformationally flexibility of the protein. Crystallographic data from STARD4 and STARD11 have showed high B values for the Ω1 loop, indicating conformational flexibility.21,30 Additionally, there is a slight shift in the Ω1 loop between the wild-type and L124D STARD4 structures. Furthermore, a reduced level of maleimide-PEG labeling and the increased thermal stability of L124D STARD4 suggest that there is stabilization of the protein structure. This stabilization and subsequent attenuation of STARD4 activity suggest that dynamic movement of this region may be required for membrane interaction and sterol transfer. We propose that the initial STARD4–membrane interaction is mediated by electrostatic interactions of the basic patch with negative lipid headgroups that orient STARD4 for further rearrangement and membrane insertion. The C-terminal α-helix of STARD4 undergoes rearrangement and insertion into the bilayer. Apparently, rigidifying the protein structure by the L124D mutation in the Ω1 loop results in weak, transient membrane interactions through the basic patch and a greatly reduced level of sterol transfer. Understanding how the L124D mutant stabilizes the STARD4 structure and affects movement of the C-terminal helix will require further study.
Consistent with this interpretation, mutation of the Ω1 loop in STARD11 resulted in reduced levels of ceramide transfer and membrane interaction.30 We propose that hydrophobic or aromatic residues55−58 in the Ω1 loop introduce local dynamic instability that facilitates movement of the Ω1 loop that is required for rearrangement and membrane insertion of the C-terminal α-helix rather than acting as membrane-anchoring motifs.57,58
Movement of the C-terminal helix and insertion into the bilayer would facilitate exposure of the sterol binding pocket for transfer of cholesterol to or from the lipid bilayer. Interestingly, early work involving STARD1 showed that the C-terminal α-helix was protected from proteolysis when small unilamellar vesicles were present, indicating partitioning into the bilayer.45 Consistent with this model, cross-linking of the C-terminal α-helix of STARD1 reduced activity in vitro, suggesting that conformational movement of this region is required for activity.29
Our data are consistent with a model in which membrane interaction and sterol binding require a surface basic patch as well as an Ω1 loop that facilitates subsequent rearrangement and insertion of the C-terminal α-helix into the membrane.25,28 Mutation of the Ω1 loop results in a reduced level of dynamic movement accompanied by attenuated membrane interaction and activity. As such, it can be envisioned that conformational changes in the Ω1 loop and C-terminal α-helix stabilized by interaction with the bilayer would allow enough space and time for the sterol or lipid to diffuse, polar component first, into the ligand binding pocket. Following ligand binding, the protein can disengage from the membrane to transfer the sterol or lipid to another cellular organelle.
Acknowledgments
We thank Dr. Olga Boudker and Dr. Bruno Mesmin for useful discussion and critical reading of the manuscript.
Glossary
Abbreviations
- START
steroidogenic acute regulatory protein-related lipid transfer
- ERC
endocytic recycling compartment
- ER
endoplasmic reticulum
- StAR
steroidogenic acute regulatory
- STARD
START domain
- ACAT
acetyl-CoA:cholesterol acyl-transferase
- TCEP
tris(2-carboxyethyl)phosphine
- DHE
dehydroergosterol
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine
- POPE
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine
- POPS
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylserine
- PI
phosphatidylinositol
- PE
phosphatidylethanolamine
- FRET
fluorescent resonance energy transfer
- CD
circular dichroism
- rmsd
root-mean-square deviation
- NMR
nuclear magnetic resonance
- HSQC
heteronuclear single-quantum coherence
- NBD
(7-nitrobenz-2-oxa-1,3-diazol-4-yl)ethylenediamine
- IANBD
iodoacetamide-NBD
- MP-5 kDa
maleimide-PEG molecular weight 5 kDa
- PC
phosphatidylcholine.
Supporting Information Available
Far-UV CD spectroscopy of wild-type and L124D STARD4 (Figure S1) and evidence that cysteine mutations of STARD4 do not alter function (Figure S2). The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00618.
This work was supported by National Institutes of Health Grants F31-DK104631 (D.B.I.), R01-AG025440 (D.E.), R37-AG019391 (D.E.), and R37-DK27083 (F.R.M.). This work was also supported by the Irma T. Hirschl Foundation (D.E.) and by a gift from Herbert and Ann Siegel (D.E.).
The authors declare no competing financial interest.
Supplementary Material
References
- van Meer G.; Voelker D. R.; Feigenson G. W. (2008) Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield F. R.; van Meer G. (2010) Cholesterol, the central lipid of mammalian cells. Curr. Opin. Cell Biol. 22, 422–429 10.1016/j.ceb.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonen E. (2008) Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 9, 125–138 10.1038/nrm2336. [DOI] [PubMed] [Google Scholar]
- Maxfield F. R.; Menon A. K. (2006) Intracellular sterol transport and distribution. Curr. Opin. Cell Biol. 18, 379–385 10.1016/j.ceb.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Hao M.; Lin S. X.; Karylowski O. J.; Wustner D.; McGraw T. E.; Maxfield F. R. (2002) Vesicular and non-vesicular sterol transport in living cells. The endocytic recycling compartment is a major sterol storage organelle. J. Biol. Chem. 277, 609–617 10.1074/jbc.M108861200. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan A.; Goldstein J. L.; McDonald J. G.; Brown M. S. (2008) Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab. 8, 512–521 10.1016/j.cmet.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesmin B.; Maxfield F. R. (2009) Intracellular sterol dynamics. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1791, 636–645 10.1016/j.bbalip.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaea D. B.; Maxfield F. R. (2015) Cholesterol trafficking and distribution. Essays Biochem. 57, 43–55 10.1042/bse0570043. [DOI] [PubMed] [Google Scholar]
- Iaea D. B., Mao S., and Maxfield F. R. (2014) Steroidogenic Acute Regulatory Protein-related Lipid Transfer (START) Proteins in Non-vesicular Cholesterol Transport. In Cholesterol Transporters of the START Domain Protein Family in Health and Disease (Clark B. J., and Stocco D. M., Eds.) pp 173–188, Springer, New York. [Google Scholar]
- Mesmin B.; Pipalia N. H.; Lund F. W.; Ramlall T. F.; Sokolov A.; Eliezer D.; Maxfield F. R. (2011) STARD4 abundance regulates sterol transport and sensing. Mol. Biol. Cell 22, 4004–4015 10.1091/mbc.E11-04-0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo G.; Vicinanza M.; De Matteis M. A. (2008) Lipid-transfer proteins in biosynthetic pathways. Curr. Opin. Cell Biol. 20, 360–370 10.1016/j.ceb.2008.03.013. [DOI] [PubMed] [Google Scholar]
- Clark B. J. (2012) The mammalian START domain protein family in lipid transport in health and disease. J. Endocrinol. 212, 257–275 10.1530/JOE-11-0313. [DOI] [PubMed] [Google Scholar]
- Soccio R. E.; Breslow J. L. (2003) StAR-related lipid transfer (START) proteins: mediators of intracellular lipid metabolism. J. Biol. Chem. 278, 22183–22186 10.1074/jbc.R300003200. [DOI] [PubMed] [Google Scholar]
- Schrick K.; Nguyen D.; Karlowski W. M.; Mayer K. F. (2004) START lipid/sterol-binding domains are amplified in plants and are predominantly associated with homeodomain transcription factors. Genome Biol. 5, R41. 10.1186/gb-2004-5-6-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting C. P.; Aravind L. (1999) START: a lipid binding domain in StAR, HD-ZIP and signalling proteins. Trends Biochem. Sci. 24, 130–132 10.1016/S0968-0004(99)01362-6. [DOI] [PubMed] [Google Scholar]
- Alpy F.; Tomasetto C. (2005) Give lipids a START: the StAR-related lipid transfer (START) domain in mammals. J. Cell Sci. 118, 2791–2801 10.1242/jcs.02485. [DOI] [PubMed] [Google Scholar]
- Soccio R. E.; Adams R. M.; Romanowski M. J.; Sehayek E.; Burley S. K.; Breslow J. L. (2002) The cholesterol-regulated StarD4 gene encodes a StAR-related lipid transfer protein with two closely related homologues, StarD5 and StarD6. Proc. Natl. Acad. Sci. U. S. A. 99, 6943–6948 10.1073/pnas.052143799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Agudo D.; Calderon-Dominguez M.; Ren S.; Marques D.; Redford K.; Medina-Torres M. A.; Hylemon P.; Gil G.; Pandak W. M. (2011) Subcellular localization and regulation of StarD4 protein in macrophages and fibroblasts. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1811, 597–606 10.1016/j.bbalip.2011.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbarino J.; Pan M.; Chin H. F.; Lund F. W.; Maxfield F. R.; Breslow J. L. (2012) STARD4 knockdown in HepG2 cells disrupts cholesterol trafficking associated with the plasma membrane, ER, and ERC. J. Lipid Res. 53, 2716–2725 10.1194/jlr.M032227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujishita Y.; Hurley J. H. (2000) Structure and lipid transport mechanism of a StAR-related domain. Nat. Struct. Biol. 7, 408–414 10.1038/75192. [DOI] [PubMed] [Google Scholar]
- Romanowski M. J.; Soccio R. E.; Breslow J. L.; Burley S. K. (2002) Crystal structure of the Mus musculus cholesterol-regulated START protein 4 (StarD4) containing a StAR-related lipid transfer domain. Proc. Natl. Acad. Sci. U. S. A. 99, 6949–6954 10.1073/pnas.052140699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roderick S. L.; Chan W. W.; Agate D. S.; Olsen L. R.; Vetting M. W.; Rajashankar K. R.; Cohen D. E. (2002) Structure of human phosphatidylcholine transfer protein in complex with its ligand. Nat. Struct. Biol. 9, 507–511 10.1038/nsb812. [DOI] [PubMed] [Google Scholar]
- Iyer L. M.; Koonin E. V.; Aravind L. (2001) Adaptations of the helix-grip fold for ligand binding and catalysis in the START domain superfamily. Proteins: Struct., Funct., Genet. 43, 134–144. [DOI] [PubMed] [Google Scholar]
- Mathieu A. P.; Fleury A.; Ducharme L.; Lavigne P.; LeHoux J. G. (2002) Insights into steroidogenic acute regulatory protein (StAR)-dependent cholesterol transfer in mitochondria: evidence from molecular modeling and structure-based thermodynamics supporting the existence of partially unfolded states of StAR. J. Mol. Endocrinol. 29, 327–345 10.1677/jme.0.0290327. [DOI] [PubMed] [Google Scholar]
- Murcia M.; Faraldo-Gomez J. D.; Maxfield F. R.; Roux B. (2006) Modeling the structure of the StART domains of MLN64 and StAR proteins in complex with cholesterol. J. Lipid Res. 47, 2614–2630 10.1194/jlr.M600232-JLR200. [DOI] [PubMed] [Google Scholar]
- Thorsell A. G.; Lee W. H.; Persson C.; Siponen M. I.; Nilsson M.; Busam R. D.; Kotenyova T.; Schuler H.; Lehtio L. (2011) Comparative structural analysis of lipid binding START domains. PLoS One 6, e19521. 10.1371/journal.pone.0019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roostaee A.; Barbar E.; Lehoux J. G.; Lavigne P. (2008) Cholesterol binding is a prerequisite for the activity of the steroidogenic acute regulatory protein (StAR). Biochem. J. 412, 553–562 10.1042/BJ20071264. [DOI] [PubMed] [Google Scholar]
- Roostaee A.; Barbar E.; Lavigne P.; LeHoux J. G. (2009) The mechanism of specific binding of free cholesterol by the steroidogenic acute regulatory protein: evidence for a role of the C-terminal alpha-helix in the gating of the binding site. Biosci. Rep. 29, 89–101 10.1042/BSR20080111. [DOI] [PubMed] [Google Scholar]
- Baker B. Y.; Yaworsky D. C.; Miller W. L. (2005) A pH-dependent molten globule transition is required for activity of the steroidogenic acute regulatory protein, StAR. J. Biol. Chem. 280, 41753–41760 10.1074/jbc.M510241200. [DOI] [PubMed] [Google Scholar]
- Kudo N.; Kumagai K.; Matsubara R.; Kobayashi S.; Hanada K.; Wakatsuki S.; Kato R. (2010) Crystal structures of the CERT START domain with inhibitors provide insights into the mechanism of ceramide transfer. J. Mol. Biol. 396, 245–251 10.1016/j.jmb.2009.12.029. [DOI] [PubMed] [Google Scholar]
- Dikiy I.; Ramlall T. F.; Eliezer D. (2013) (1)H, (1)(3)C, and (1)(5)N backbone resonance assignments of the L124D mutant of StAR-related lipid transfer domain protein 4 (StARD4). Biomol. NMR Assignments 7, 245–248 10.1007/s12104-012-9419-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. In Methods in Enzymology (Carter C. W., Jr., Ed.) pp 307–326, Academic Press, San Diego. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P. V.; Grosse-Kunstleve R. W.; Echols N.; Headd J. J.; Moriarty N. W.; Mustyakimov M.; Terwilliger T. C.; Urzhumtsev A.; Zwart P. H.; Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr., Sect. D: Biol. Crystallogr. 68, 352–367 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marley J.; Lu M.; Bracken C. (2001) A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR 20, 71–75 10.1023/A:1011254402785. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Johnson B. A.; Blevins R. A. (1994) NMR View: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- Rosenfeld S. S.; Taylor E. W. (1985) Kinetic studies of calcium and magnesium binding to troponin C. J. Biol. Chem. 260, 242–251. [PubMed] [Google Scholar]
- Letourneau D.; Lorin A.; Lefebvre A.; Cabana J.; Lavigne P.; Lehoux J. G. (2013) Thermodynamic and solution state NMR characterization of the binding of secondary and conjugated bile acids to STARD5. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1831, 1589–1599 10.1016/j.bbalip.2013.07.005. [DOI] [PubMed] [Google Scholar]
- Smutzer G.; Crawford B. F.; Yeagle P. L. (1986) Physical properties of the fluorescent sterol probe dehydroergosterol. Biochim. Biophys. Acta, Biomembr. 862, 361–371 10.1016/0005-2736(86)90239-7. [DOI] [PubMed] [Google Scholar]
- Hummel J. P.; Dreyer W. J. (1962) Measurement of protein-binding phenomena by gel filtration. Biochim. Biophys. Acta 63, 530–532 10.1016/0006-3002(62)90124-5. [DOI] [PubMed] [Google Scholar]
- Wragg R. T.; Snead D.; Dong Y.; Ramlall T. F.; Menon I.; Bai J.; Eliezer D.; Dittman J. S. (2013) Synaptic vesicles position complexin to block spontaneous fusion. Neuron 77, 323–334 10.1016/j.neuron.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier R.; Douguet D.; Antonny B.; Drin G. (2008) HELIQUEST: a web server to screen sequences with specific alpha-helical properties. Bioinformatics 24, 2101–2102 10.1093/bioinformatics/btn392. [DOI] [PubMed] [Google Scholar]
- Yaworsky D. C.; Baker B. Y.; Bose H. S.; Best K. B.; Jensen L. B.; Bell J. D.; Baldwin M. A.; Miller W. L. (2004) pH-dependent Interactions of the carboxyl-terminal helix of steroidogenic acute regulatory protein with synthetic membranes. J. Biol. Chem. 280, 2045–2054 10.1074/jbc.M410937200. [DOI] [PubMed] [Google Scholar]
- Ben-Efraim I.; Shai Y. (1996) Secondary structure, membrane localization, and coassembly within phospholipid membranes of synthetic segments derived from the N- and C-termini regions of the ROMK1 K+ channel. Protein Sci. 5, 2287–2297 10.1002/pro.5560051115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akabas M. H.; Stauffer D. A.; Xu M.; Karlin A. (1992) Acetylcholine receptor channel structure probed in cysteine-substitution mutants. Science 258, 307–310 10.1126/science.1384130. [DOI] [PubMed] [Google Scholar]
- Javitch J. A. (1998) Probing structure of neurotransmitter transporters by substituted-cysteine accessibility method. Methods Enzymol. 296, 331–346. [DOI] [PubMed] [Google Scholar]
- Voeltz G. K.; Prinz W. A.; Shibata Y.; Rist J. M.; Rapoport T. A. (2006) A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 124, 573–586 10.1016/j.cell.2005.11.047. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Ho S. O.; Gassman N. R.; Korlann Y.; Landorf E. V.; Collart F. R.; Weiss S. (2008) Efficient site-specific labeling of proteins via cysteines. Bioconjugate Chem. 19, 786–791 10.1021/bc7002499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layton C. J.; Hellinga H. W. (2010) Thermodynamic analysis of ligand-induced changes in protein thermal unfolding applied to high-throughput determination of ligand affinities with extrinsic fluorescent dyes. Biochemistry 49, 10831–10841 10.1021/bi101414z. [DOI] [PubMed] [Google Scholar]
- Lev S. (2010) Non-vesicular lipid transport by lipid-transfer proteins and beyond. Nat. Rev. Mol. Cell Biol. 11, 739–750 10.1038/nrm2971. [DOI] [PubMed] [Google Scholar]
- Lavigne P.; Najmanivich R.; Lehoux J. G. (2010) Mammalian StAR-related lipid transfer (START) domains with specificity for cholesterol: structural conservation and mechanism of reversible binding. Subcell. Biochem 51, 425–437 10.1007/978-90-481-8622-8_15. [DOI] [PubMed] [Google Scholar]
- Lehoux J. G.; Mathieu A.; Lavigne P.; Fleury A. (2003) Adrenocorticotropin regulation of steroidogenic acute regulatory protein. Microsc. Res. Tech. 61, 288–299 10.1002/jemt.10338. [DOI] [PubMed] [Google Scholar]
- Wimley W. C.; White S. H. (1996) Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 3, 842–848 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- White S. H.; Wimley W. C. (1998) Hydrophobic interactions of peptides with membrane interfaces. Biochim. Biophys. Acta, Rev. Biomembr. 1376, 339–352 10.1016/S0304-4157(98)00021-5. [DOI] [PubMed] [Google Scholar]
- Lomize M. A.; Pogozheva I. D.; Joo H.; Mosberg H. I.; Lomize A. L. (2012) OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–376 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomize A. L.; Pogozheva I. D.; Lomize M. A.; Mosberg H. I. (2007) The role of hydrophobic interactions in positioning of peripheral proteins in membranes. BMC Struct. Biol. 7, 44. 10.1186/1472-6807-7-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.